Introduction
Congenital nystagmus (CN) is a group of hereditary
eye diseases, although the cause of the diseases has yet to be
elucidated. It can be divided into sensory defective nystagmus and
motor defective nystagmus according to its etiology. Sensory
defective nystagmus can be originated from insufficiency image
stimulus of macular region induced by the disease of anterior
visual pathway, or the function loss of the fovea induced by
organic diseases of macula, retina or optic nerve (1,2).
Usually, sensory defective nystagmus can occur in a variety of
diseases, such as congenital cataracts, aniridia, Peters' anomaly,
oculocutaneous albinism, achromatopsia, cone rod dystrophy, macular
defects, congenital stationary night blindness, Leber congenital
amaurosis and optic nerve hypoplasia (1,2).
Motor defective nystagmus may be originated from the central
nervous system or the abnormal pathway controlling the eye
movements. Patients with motor defective nystagmus don't have other
ocular abnormalities (3,4). Congenital motor nystagmus (CMN), also
known as congenital idiopathic nystagmus, is an isolated form of
nystagmus, consisting of involuntary oscillations of the eyes. It
is characterized by an absence of other ocular pathologies. A
variety of genetic modes, such as autosomal dominant, autosomal
recessive and X linked, have been proven to be associated with CMN
(5–8).
In 1999, two X-linked CMN loci were reported,
demonstrating that this form of inheritance is also genetically
heterogeneous with a locus for X-linked irregularly dominant CMN,
as reported by Kerrison et al (9) at Xq26-q27 (NYS1) and by Cabot et
al (10) at
Xp11.4-p11.3.8.
However, only one gene has been identified to be
associated with X-linked CMN. In 2006, Tarpey et al
(4) first identified
nystagmus-causing mutations in the FRMD7 gene within the Xq26-q27
interval. In the present study, FRMD7 mutation analysis and
detailed clinical evaluation were performed to identify novel
mutations and characterize new clinical features of the Chinese
population with CMN.
Materials and methods
Patients and clinical data
The patients presenting CMN were referred to the
Pediatric and Genetic Clinic in the Eye Hospital of the Zhongshan
Ophthalmic Center (Guangzhou, China). Written informed consent was
obtained from each participant prior to the study. The present
study was approved by the Ethics Committee of the Zhongshan
Ophthalmic Center (Guangzhou, China) and was performed according to
the tenets of the Declaration of Helsinki. Medical and ophthalmic
histories were obtained. A complete general physical examination
and detailed ophthalmological examinations, including anterior
segment observation with slit-lamp microscopy, fundus photography
and optical coherence tomography, were conducted to identify the
clinical features of CMN.
Mutation screening
Genomic DNA was prepared from venous blood. All of
the primers (Takara Bio, Inc., Otsu, Japan) for FRMD7 (Table I) were used to amplify coding exons
(exon1 to exon 12 of FRMD7) and the adjacent intronic sequence of
the gene (NCBI human genome build 36.3, NC_000023.10 for gDNA,
NM_194277.2 for cDNA and NP_919253.1 for protein of FRMD7). The PCR
reaction was performed in a thermocycler (Biometra GmbH, Göttingen,
Germany) under the following two conditions: The first was an
initial denaturation at 94°C for 5 min followed by 35 to 37 cycles
of 94°C for 30 sec, proper annealing temperature for 30 sec, and
72°C for 30 sec with a final extension cycle of 72°C for 5 min. The
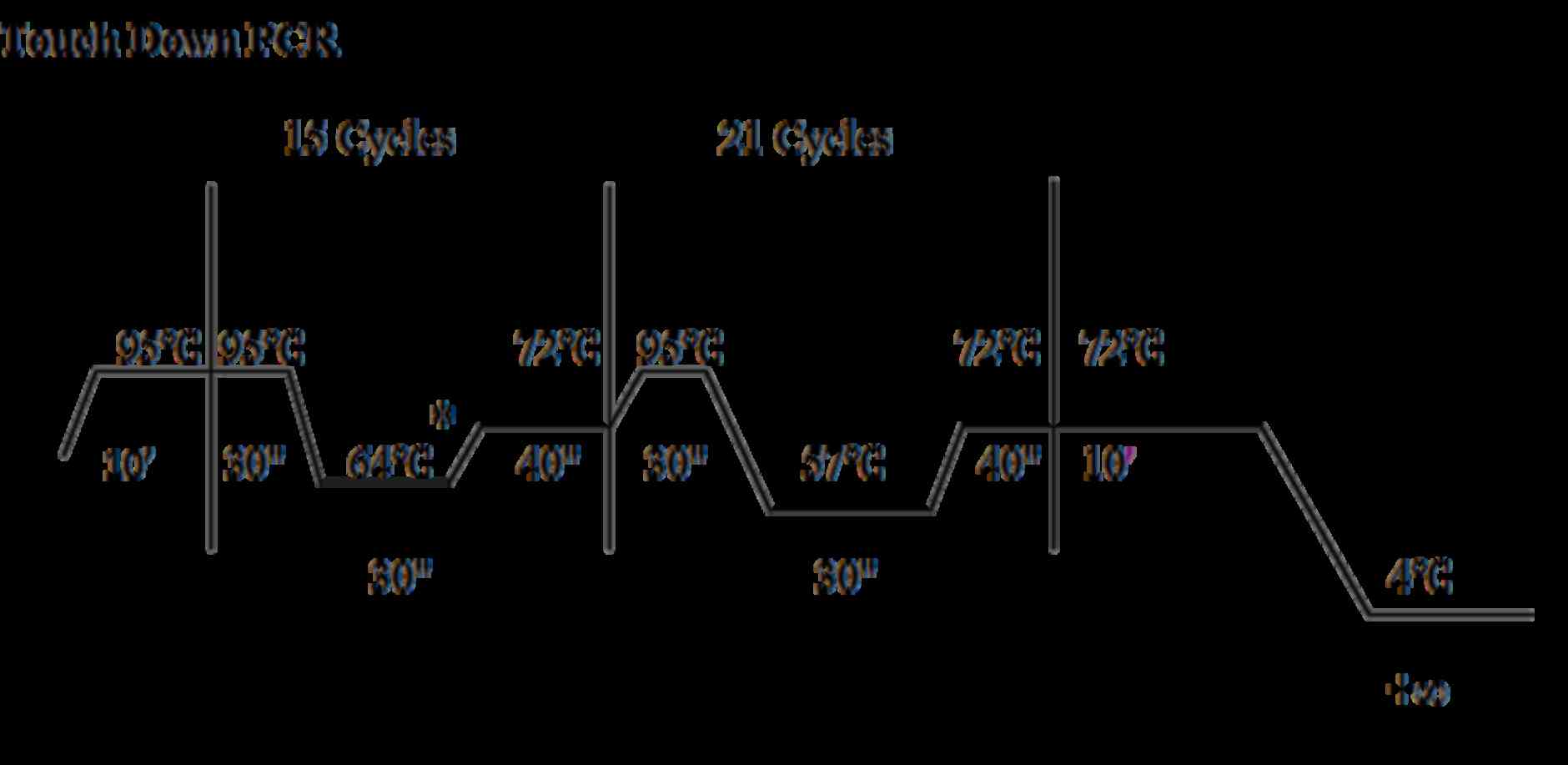
other condition was the touch down PCR program (Fig. 1). The PCR products of the exons and
adjacent intronic sequences for the patients were sequenced with
the ABI BigDye Terminator direct sequencing kit (version, 3.1;
Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) according to the manufacturer's recommendations and using a
3100 sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.)
confirmed by the authors. Sequencing results from patients, as well
as consensus sequences of FRMD7 from the NCBI human genome database
(NM_001604.3) were imported into the SeqManII program of the
Lasergene Package (DNASTAR, Madison, WI, USA) and aligned to
identify variations. Each mutation was confirmed by bidirectional
sequencing. Mutation was named according to the nomenclature
recommended by the Human Genomic Variation Society.
 | Table I.Oligonucleotides used for FRMD7
amplification. |
Table I.
Oligonucleotides used for FRMD7
amplification.
| Exon | Sequences
(5′-3′) | Size of PCR products
(bp) | Annealing temperature
(°C) |
|---|
| FRMD7-exon1-f |
AGGAAGTCCAGTTAGATTTG | 428 | 58.7 |
| FRMD7-exon1-r |
ACAGTCCTCCTTCATTCAGT |
|
|
| FRMD7-exon2-f |
ATGCAGGTCCTCTAAACAGT | 320 | 58.7 |
| FRMD7-exon2-r |
GGAATTGAACCCTACATACC |
|
|
| FRMD7-exon3-f |
GAAAATATAAGGGGGCAGAT | 368 | 54.4 |
| FRMD7-exon3-r |
TGGATGTATGAAGGGTTGAA |
|
|
| FRMD7-exon4-f |
GAGGGGACGGAAGAGGAGA | 287 | 61.6 |
| FRMD7-exon4-r |
TGAGAATGGCCAGAAGCACT |
|
|
| FRMD7-exon5-f |
CCCCAAAAAGGCATCTGA | 339 | 57.3 |
| FRMD7-exon5-r |
TCTCCCCTGTAAACCCTAAC |
|
|
| FRMD7-exon6-f |
GATGGAGGACAAGGGTATGC | 393 | 58.7 |
| FRMD7-exon6-r |
GCCACTGAAAGGGGAAAGAA |
|
|
| FRMD7-exon7-f |
AGCAAGCCCTTAAACCTGAG | 391 | 58.7 |
| FRMD7-exon7-r |
CCCTTTCTGGCTGGTGATAA |
|
|
| FRMD7-exon8-f |
AATGCCTTCTTTGACCACAGC | 365 | 62.9 |
| FRMD7-exon8-r |
GCCAGCCGGCTTTTACAAT |
|
|
| FRMD7-exon9-f |
AGTGGCCCTGTCTATTCCTC | 551 | 62.9 |
| FRMD7-exon9-r |
GGTGCCCCCATCTTCCTC |
|
|
| FRMD7-exon10,
exon11-f |
CTGCCTGGTCCTTGAATAAG | 582 | 57.3 |
| FRMD7-exon10,
exon11-r |
TCCCCAGGAAGCTAACCTA |
|
|
| FRMD7-exon12a-f |
ATGGATCTTGTTAAATGACTT | 541 | 51.1 |
| FRMD7-exon12a-r |
ACCAACCTGCTGACCTGTA |
|
|
| FRMD7-exon12b-f |
TCCCACATTGCTACATCAGTC | 520 | 58.7 |
| FRMD7-exon12b-r |
CAAATTTGGGTCTTCCTCTTC |
|
|
| FRMD7-exon12c-f |
ATGTGCCCTATATTCCTTGTA | 592 | 58.7 |
| FRMD7-exon12c-r |
ATGGGTGACCTTATTTCTTTG |
|
|
| FRMD7-exon12d-f |
TCCCAGAGCCAATCAGACAT | 435 | 58.7 |
| FRMD7-exon12d-r |
TTTCTGCCTAAGTCGGTAACA |
|
|
| FRMD7-mutation
(c.1403G>A/c.1492-1493intA)-f |
CAAGCTGTAAGTTTTCTGGTAATC | 213 | a |
| FRMD7-mutation
(c.1403G>A/c.1492-1493intA)-r |
GACTTGTCCTTTCCTCTGCTC |
|
|
| FRMD7-mutation
(c.473T>A)-f |
TGCTCCATTGCTAAGTTCCTCA | 234 | 56.8 |
| FRMD7-mutation
(c.473T>A)-r |
TCTGTCCCCAATTTTAGTGTTCTC |
|
|
| FRMD7-mutation
(c.605T>A)-f |
CATTCTTGAGGCATTTATTAGG | 173 | a |
| FRMD7-mutation
(c.605T>A)-r |
GTGAGCAACAGCCAGGTGA |
|
|
| FRMD7-mutation
(c.580G>T)-f |
GAGCTCTCAGGGTGGAAATGTCAT | 246 | 61.1 |
| FRMD7-mutation
(c.580G>T)-r |
GCTGAAGGGCTTGAAAGGAA |
|
|
| FRMD7-mutation
(c.811T>A)-f |
GTTTGGAAGGCATTGGGATTTGAA | 203 | 63.2 |
| FRMD7-mutation
(c.811T>A)-r |
TTTGGGCTTTGATTTGGGCTCTT |
|
|
| FRMD7-mutation
(c.57+1G>A)-f |
CCTCGCTGAGAATGCTAC | 189 | a |
| FRMD7-mutation
(c.57+1G>A)-r |
ATCACAGTCCTCCTTCAT |
|
|
Determination of changes in genetic
information
The nucleotide sequences containing base variation
and the standard sequences from NCBI human genome database were
inputted into the MapDraw program of the Lasergene package to
identify the impact of amino acid coding. Protein sequences of
different species were identified from the NCBI website and entered
into the MegAlign program of the Lasergene package, in order to
compare the protein sequences of different species and estimate the
conservation of mutation sites in the ClustalW program. The
differences in amino acid changes and the possible pathogenicity of
the mutation were evaluated through the Blosum 62 (http://www.uky.edu/Classes/BIO/520/BIO520WWW/blosum62.htm)
and PolyPhen (http://genetics.bwh.harvard.edu/pph/) (11) analytical programs respectively.
Heteroduplex single-strand
conformation polymorphism (SSCP) analysis
The variations detected in the gene were further
evaluated in 96 normal controls (informed consent, in accordance
with the Declaration of Helsinki, was obtained from the
participating individuals before the study) by using
heteroduplex-SSCP analysis as previously described in literature
(12–14). DNA fragments of mutation sites were
PCR-amplified according to Table
I. PCR products were mixed with an equal volume of gel-loading
buffer (95% formamide, 20 mM EDTA and 0.05% bromophenol blue, 0.05%
xylene cyanol) and denatured at 95°C for 5 min and immediately
placed on ice for 5 min. The samples were loaded directly onto 8%
polyacrylamide gels and run 8 h at room temperature at 40 W in a
solution of 0.5X TBE (Takara Bio, Inc.).
Results
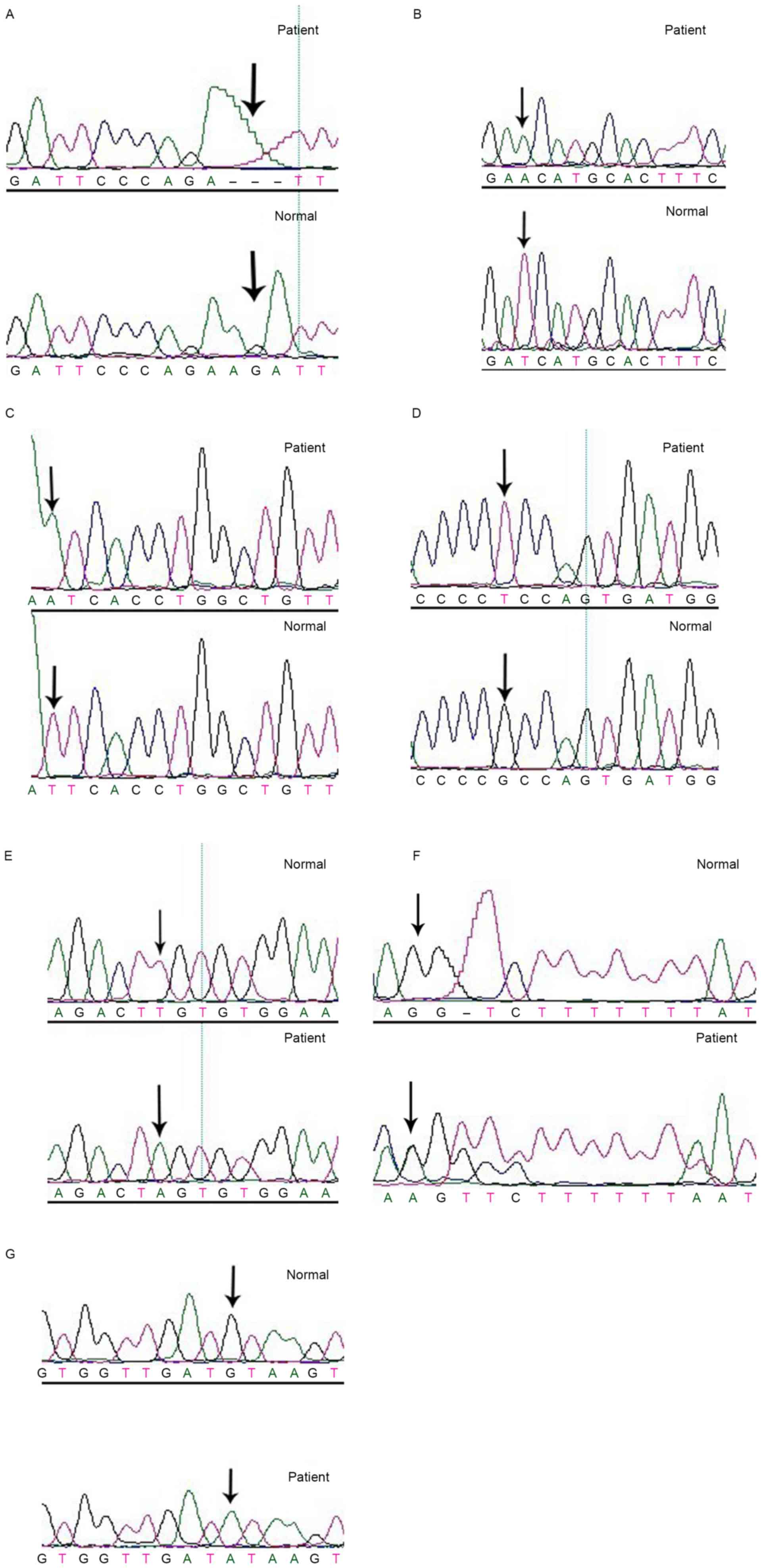
In 8 patients with CMN, there were 7 FRMD7 gene
mutations (6 new mutations). The screening rate was 38.89%,
including c.41_43delAGA (p.13-15delK); c.473T>A (p.I158N);
c.605T>A (p.I202N); c.580G>T (p.A194S); c.811T>A
(p.C271S); c.1493insA (p.Y498X); c.57+1G>A (slice mutation;
Fig. 2).
The mutation identified in the present study
resulted in a change at the protein level with a residue
substitution weight by Blosum 62, and a ‘probable damaging’ effect
identified by PolyPhen (Table
II). The conservation of the protein in this position was
identified by analyzing seven orthologs from different mammalian
species (Fig. 3). These mutations
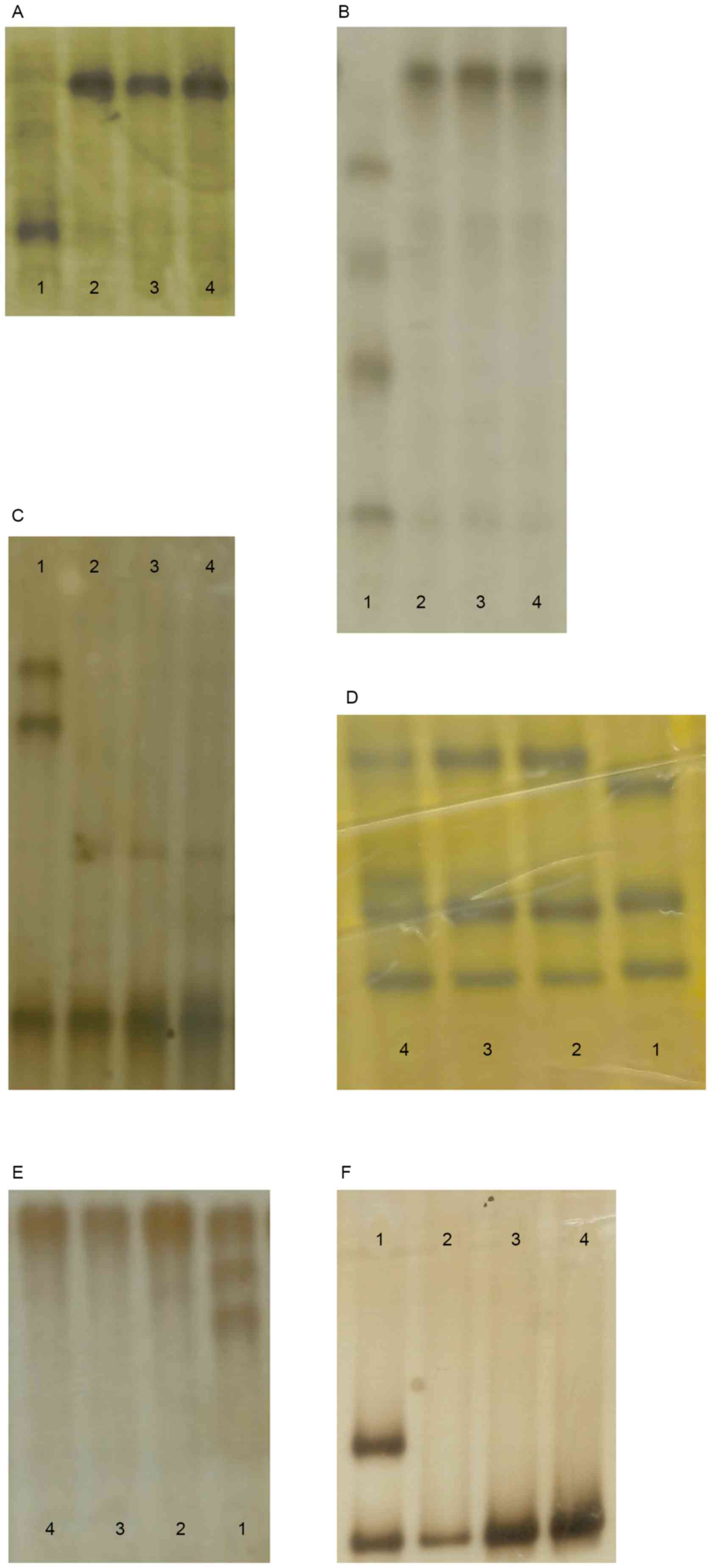
were also investigated in 96 unaffected control individuals
(Fig. 4) by heteroduplex-SSCP, but
none were identified.
| Table II.Analysis by Blosum62 and PolyPhen. |
Table II.
Analysis by Blosum62 and PolyPhen.
| Gene | Mutation | Blosum62 | PolyPhen | Score |
|---|
| FRMD7 | I158N | 4→-3 | Possibly
damaging | 0.998 |
| FRMD7 | I202N | 4→-3 | Possibly
damaging | 0.998 |
| FRMD7 | A194S | 4→1 | Benign | 0.993 |
| FRMD7 | C271S | 9→-1 | Possibly
damaging | 0.997 |
In the current study, all patients with idiopathic
nystagmus were male. Nystagmus usually manifests as a midrange
decline in visual acuity. The uncorrected visual acuity ranged from
0.1 to 0.4 and corrected visual acuity ranged from 0.4 to 0.9
(Table III). The results of
customary slit lamp and fundus examinations were normal.
| Table III.The visual acuity of patients with
CMN. |
Table III.
The visual acuity of patients with
CMN.
| Patient | Age (years) | Uncorrected visual
acuity | Corrected
vision |
|---|
| QT276 | 18 |
0.3+/0.5− | NA |
| QT321 | 12 | 0.2/0.1 | 0.4/0.4 |
| QT370 | 0.3 | Perception of
light | ND |
| QT385 | 28 |
0.7+/0.8 |
0.7+/0.8 |
| QT423 | 8 | 0.1/0.1 | 0.3/0.3 |
| QT439 | 12 | 1.0/0.7 | 1.0/0.7 |
| QT474 | 10 |
0.5/0.4+ | 0.7/0.8 |
| QT552 | 3.5 | 0.25/0.2 | ND |
| QT596 | 5 | 0.3/0.3 | 0.4/0.4 |
| QT664 | 3 | 0.4/0.4 | 0.5/0.5 |
| QT669 | 3.5 | ND | ND |
| QT684 | 5 | 0.3/0.4 |
0.7/0.7+ |
| QT712 | 3 | Perception of
light | ND |
| QT719 | 29 | 0.1/0.1 | 0.9/0.9 |
| QT726 | 8 | 0.5/0.5 | ND |
| QT756 | 5.5 |
0.4+/0.4+ | 0.7/0.6 |
| QT762 | 6 | 0.3/0.3 | 0.4/0.4 |
| QT835 | 1 | Perception of
light | ND |
Discussion
CMN is an ocular motor disorder characterized by
involuntary oscillation of the eyes that occurs in the first 6
months of life. It is speculated that the disease may be associated
with the abnormal development of senior center monitoring the
abnormal eye movements and eye gazing. The prevalence of CMN in the
population is estimated to be 24 per 10,000 (15). It is known to be genetically
heterogeneous as autosomal dominant, autosomal recessive and
X-linked patterns of inheritance. X-linked inheritance with
incomplete penetrance is the most common form (4–8).
However, the majority of patients of CMN are sporadic cases.
Until now, FRMD7 is the only one gene that has been
identified for X-linked CMN. It was demonstrated that FRMD7
expression is spatially and temporally regulated in the developing
human embryonic cortex. FRMD7 is expressed in most adult human
tissues, and has been detected in the developing neuroretina and
regions of the embryonic brain known to control eye movement
(16). FRMD7 consists of 12 exons
and encodes a polypeptide with 714 residues polypeptide, spanning
approximately ~51 kb on chromosome Xq26-q27. The FRMD7 protein has
B41, FERM-N, FERM-M and FERM-C domains. The conserved domains are
at the B41 and FERM-C domains. The B41 domain is located between
residues 1–192, and the FERM-C domain is located between residues
186–279.
Many mutations in FRMD7 have been reported in
Chinese families with CMN. In the current study, the authors
attempted to identify FRMD7 mutations and investigate the clinical
phenotype of sporadic cases with CMN in the Chinese population. In
the present study, there were seven FRMD7 gene mutations, six of
which 6 gene mutations are newly-identified in 7 patients with CMN.
The screening rate is was 38.89%, which is consistent with the
previous studies (17,18).
Through the MegAlign program, the protein sequences
of different species were compared and mutation sites were
identified (p.I158N, p.I202N, p.A194S and p.C271S), possessing
relative conservation. Though further analysis of missense
mutations, it was demonstrated that mutations of p.I158N, p.I202N
and p.C271S mutations are possibly damaging with scores ranging
from 0.997 to 0.998. The mutation of p.A194S is was benign, but
with a score of 0.993.
FRMD7 is highly expressed in regions of the
developing brain that are involved in oculomotor control, as well
as in the retina (16). FRMD7 may
serve a role in development of the oculomotor neural circuitry,
previous studies indicate that nystagmus-associated mutations in
the FERM and FA domains are likely to be critical to FRMD7 function
(19). The mutations of
c.41_43delAGA (p.13-15delK), c.473T>A (p.I158N), c.605T>A
(p.I202N), c.580G>T (p.A194S) and c.811T>A (p.C271S) are in
the conserved region of FRMD. Of them, the mutations of
c.41_43delAGA (p.13-15delK), c.473T>A (p.I158N), c.580G>T
(p.A194S) are in the B41 domain. The mutations of c.605T>A
(p.I202N) and c.811T>A (p.C271S) are in the FERM-C domain.
CMN-associated missense mutations within the N-terminal region of
the protein indicate that mutations can disrupt the interaction
with other proteins, preventing their co-localization at the plasma
membrane and impairing neurite formation (20).
The mutation site of p.Y498X is in the FERM-adjacent
(FA) domain. FA is identified in a subset of FERM domain proteins,
and which has been indicated to regulate protein function through
modifications such as phosphorylation (21).
In summary, CMN is a genetically heterogeneous
ocular movement disease. The presented result expands the mutation
spectrum of FRMD7 and provides evidence for future functional
studies, clinical diagnosis, differential diagnosis and genetic
counseling. In conclusion, these results enriched the gene mutation
spectrum of FRMD7. The authors systematically investigated the
clinical phenotype of congenital motor nystagmus in the Chinese
population, and provided further evidence for clinical diagnosis
and differential diagnosis and genetic counseling.
Acknowledgements
The authors would like to thank the patients and
family members for their participation. The present study was
supported in part by the National Natural Science Foundation of
China (grant no. 81170847).
References
|
1
|
Stayte M, Reeves B and Wortham C: Ocular
and vision defects in preschool children. Br J Ophthalmol.
77:228–232. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forssman B and Ringnér B: Prevalence and
inheritance of congenital nystagmus in a Swedish population. Ann
Hum Genet. 35:139–147. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jacobs JB and Dell'Osso LF: Congenital
nystagmus: Hypotheses for its genesis and complex waveforms within
a behavioral ocular motor system model. J Vis. 4:604–625. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tarpey P, Thomas S, Sarvananthan N, Mallya
U, Lisgo S, Talbot CJ, Roberts EO, Awan M, Surendran M, McLean RJ,
et al: Mutations in FRMD7, a newly identified member of the FERM
family, cause X-linked idiopathic congenital nystagmus. Nat Genet.
38:1242–1244. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schorderet DF, Tiab L, Gaillard MC, Lorenz
B, Klainguti G, Kerrison JB, Traboulsi EI and Munier FL: Novel
mutations in FRMD7 in X-linked congenital nystagmus. Mutation in
brief #963. Online. Hum Mutat. 28:5252007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Self JE, Shawkat F, Malpas CT, Thomas NS,
Harris CM, Hodgkins PR, Chen X, Trump D and Lotery AJ: Allelic
variation of the FRMD7 gene in congenital idiopathic nystagmus.
Arch Ophthalmol. 125:1255–1263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang B, Liu Z, Zhao G, Xie X, Yin X, Hu
Z, Xu S, Li Q, Song F, Tian J, et al: Novel mutations of the FRMD7
gene in X-linked congenital motor nystagmus. Mol Vis. 13:1674–1679.
2007.PubMed/NCBI
|
|
8
|
Zhang Q, Xiao X, Li S and Guo X: FRMD7
mutations in Chinese families with X-linked congenital motor
nystagmus. Mol Vis. 13:1375–1378. 2007.PubMed/NCBI
|
|
9
|
Kerrison JB, Vagefi MR, Barmada MM and
Maumenee IH: Congenital motor nystagmus linked to Xq26-q27. Am J
Hum Genet. 64:600–607. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cabot A, Rozet JM, Gerber S, Perrault I,
Ducroq D, Smahi A, Souied E, Munnich A and Kaplan J: A gene for
X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome
Xp11.4-p11.3. Am J Hum Genet. 64:1141–1146. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sale MM, Craig JE, Charlesworth JC,
FitzGerald LM, Hanson IM, Dickinson JL, Matthews SJ, Heyningen Vv,
Fingert JH and Mackey DA: Broad phenotypic variability in a single
pedigree with a novel 1410delC mutation in the PST domain of the
PAX6 gene. Hum Mutat. 20:3222002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stone EM: Leber congenital amaurosis-a
model for efficient genetic testing of heterogeneous disorders:
LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 144:791–811.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Q and Minoda K: Detection of
congenital color vision defects using heteroduplex-SSCP analysis.
Jpn J Ophthalmol. 40:79–85. 1996.PubMed/NCBI
|
|
15
|
Sarvananthan N, Surendran M, Roberts EO,
Jain S, Thomas S, Shah N, Proudlock FA, Thompson JR, McLean RJ,
Degg C, et al: The prevalence of nystagmus: The Leicestershire
nystagmus survey. Invest Ophthalmol Vis Sci. 50:5201–5206. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li N, Wang X, Wang Y, Wang L, Ying M, Han
R, Liu Y and Zhao K: Investigation of the gene mutations in two
Chinese families with X-linked infantile nystagmus. Mol Vis.
17:461–468. 2011.PubMed/NCBI
|
|
17
|
Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L,
Dai X, Zhang M, Wang L, Liu M and Wang QK: Identification of a
novel GPR143 mutation in a large Chinese family with congenital
nystagmus as the most prominent and consistent manifestation. J Hum
Genet. 52:565–570. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Ge X, Yu Y, Zhang Y, Wu Y, Luan
Y, Sun J, Qu J, Jin ZB and Gu F: Identification of three novel
mutations in the FRMD7 gene for X-linked idiopathic congenital
nystagmus. Sci Rep. 4:37452014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Watkins RJ, Thomas MG, Talbot CJ, Gottlob
I and Shackleton S: The role of FRMD7 in idiopathic infantile
nystagmus. J Ophthalmol. 2012:4609562012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koyano Y, Kawamoto T, Shen M, Yan W,
Noshiro M, Fujii K and Kato Y: Molecular cloning and
characterization of CDEP, a novel human protein containing the
ezrin-like domain of the band 4.1 superfamily and the Dbl homology
domain of Rho guanine nucleotide exchange factors. Biochem Biophys
Res Commun. 241:369–375. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baines AJ: A FERM-adjacent (FA) region
defines a subset of the 4.1 superfamily and is a potential
regulator of FERM domain function. BMC Genomics. 7:852006.
View Article : Google Scholar : PubMed/NCBI
|