Introduction
Acute promyelocytic leukemia (APL) is a specific
type of acute myeloid leukemia (AML), which exhibits an aggressive
clinical course. Patients with APL are prone to hemorrhage,
infection and concurrent disseminated intravascular coagulation,
and have a high rate of mortality. APL is treated with all-trans
retinoic acid (ATRA) in conjunction with other approaches,
including therapeutic regiments, have shown promising results, and
there are examples of the successful treatment of tumors using
agents, which induce cellular differentiation (1). Retinoic acid inducible gene I (RIG-I)
was originally identified in the ATRA-induced terminal granulocytic
differentiation of the NB4 APL cell line (1–3). In
a previous study in 2004, RIG-I was cloned in double-stranded
RNA-induced innate immunity. Structural analysis revealed that
RIG-I is a RNA helicase enzyme with a DExD/H box motif consisting
of 925 amino acid residues, with two CARD domains at the N terminus
and one RNA helicase domain at the C terminus (4). Following these observations,
investigations of RIG-I as an important pattern recognition
receptor in the innate immune response have become increasingly
popular (5–9).
Although the role of RIG-I in the anti-virus innate
immune response has been clearly defined, it was originally
identified using a system, which did not use extraneous virus,
namely during the ATRA-induced terminal granulocytic
differentiation of APL cells, suggesting it may have other inherent
activities. Analysis performed using RIG-I gene-knockout mice
showed that RIG-I was an essential negative regulatory factor of
myelogenous hyperplasia (3).
Additionally, RIG-I was shown to promote the differentiation of
leukemia cells by stimulating activity of the signal transducer and
activator of transcription 1 signaling pathway (10). Previous studies have also indicated
that RIG-I attenuates the proliferation of U937 AML cells by
inhibiting the AKT-mammalian target of rapamycin (mTOR) pathway
(11). In addition to mTOR, the
Forkhead Box (FOX)O3A transcription factor is another important AKT
downstream target protein, which assists in the control of cell
proliferation, differentiation, apoptosis and cell cycle. FOXO3A is
located in the nucleus, and when combined with DNA, induces the
protein expression of p27, p130-Rb2 and cyclinD1/2 (cell-cycle
regulation), and the expression of tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) and B cell lymphoma-2-interacting
mediator of cell death in apoptosis. When AKT is phosphorylated,
the Thr32 and Ser253 phosphorylation sites of FOXO3A are directly
phosphorylated by AKT. FOXO3A then interacts with 14-3-3 proteins,
leading to the transfer of FOXO3A from the nucleus into the
cytoplasm. Therefore, the retention of FOXO3A in the cytoplasm
inhibits its associated transcription, and results in cell
proliferation and survival (12).
It has been reported that ATRA induces APL cell differentiation and
apoptosis by activating the transcription factor, FOXO3A (13). The above-mentioned studies suggest
a potential association between the expression of RIG-I during
ATRA-induced terminal granulocytic differentiation of the NB4 APL
cell line and the AKT-FOXO3A signaling pathway.
In the present study, the lentivirus method was used
to knock down the expression of RIG-I in ATRA-induced NB4 cells
in vitro, and the resulting effects on NB4 cell
proliferation, the cell cycle and apoptosis were examined, as were
the roles of RIG-I and the AKT-FOXO3A signaling pathway. The
results showed that the knockdown of RIG-I reduced cell
proliferation inhibition, cell cycle arrest and apoptosis in the
ATRA-induced NB4 cells by affecting the AKT-FOXO3A signaling
pathway.
Materials and methods
Cell culture
The NB4 APL cell line was grown in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (HyClone Laboratories; GE
Healthcare Life Sciences; Logan, UT, USA), 100 U/ml penicillin and
100 U/ml streptomycin (Beyotime Institute of Biotechnology,
Shanghai, China), in an incubator set at 37°C and containing 5%
CO2.
Lentivirus infection
Actively proliferating NB4 cells were inoculated in
a 12-well culture plate (1×105 cells/well) and
cultivated in 1 ml of 1640 medium without fetal bovine serum.
Subsequently, lentivirus containing green fluorescent protein (GFP)
(Shanghai Genepharma Co., Ltd, Shanghai, China) and polybrene
(final concentration of 8 µg/ml) were added to each well of NB4
cells, which were then centrifuged at 1,000 g for 90 min at room
temperature. Following centrifugation, the cells were placed in a
37°C incubator containing 5% CO2 for 5 h, following
which 10% fetal bovine serum was added. The NB4 cells infected with
control (Con)-small interfering (si)RNA
(5′-GCTCCCGTGAATTGGAATCCT-3′) were designated as LV-shCon cells,
and those infected with RIG-I-siRNA lentivirus
(5′-GGAATTTGGAACACAGAAATAG-3′) were designated as LV-shRIG-I cells.
The expression of GFP was detected using flow cytometry at 72 h
post-infection.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
At 0 and 72 h post-ATRA induction, total cellular
RNA was isolated from the NB4, LV-shCon and LV-shRIG-I cells using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) The
extracted RNA was analyzed using electrophoresis, and the
concentration of non-degraded RNA was quantified by measuring
optical density values. All samples were reverse transcribed to
cDNA using PrimeScript™ RT reagent kit (Takara
Biotechnology Co., Ltd., Dalian, China). Reaction systems were
established, including RNA (1.0 µg), 5X PrimeScript™
Buffer (2 µl), PrimeScript RT Enzyme Mix I (2 µl), oligo dT Primer
(0.5 µl) and random 6 mers (0.5 µl), with RNase-free
dH2O to make the final volume up to 10 µl. The RT
reaction conditions were as follows: 37°C for 15 min followed by
85°C for 5 sec. The primer sequences for qPCR were as follows:
RIG-I, sense 5′-GGACGTGGCAAAACAAATCAG-3′ and antisense
5′-GCAATGTCAATGCCTTCATCA-3′; β-actin, sense
5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and antisense
5′-CTGTCACCTTCACCGTTCCAGTTT-3′. The primers and cDNA was prepared
using SYBR Green Master mix (Takara Bio, Inc., Otsu, Japan)
according to the manufacturer's protocol. The reaction process was
performed at 50°C for 2 min, 95°C for 30 sec, followed by 40 cycles
of 95°C for 5 sec and 60°C for 30 sec, and a final extension step
at 72°C for 10 min. All RT-qPCR experiments were detected with an
ABI PRISM 7900HT Sequence Detection System (Applied Biosystems;
Thermo Fisher Scientific, Inc.). With β-actin as the internal
control, the relative gene expression levels of RIG-I were analyzed
using the 2−ΔΔCq method (14).
Western blot analysis
At 0 and 72 h post-ATRA induction, the cells were
washed with ice-cold phosphate-buffered saline (PBS) and lysed in
lysis buffer (Solarbio Science and Technology Co., Ltd., Beijing,
China) for 30 min. The cell lysate was then centrifuged at 8,000 g
for 5 min at 4°C, and the soluble protein fraction was boiled at
100°C for 10 min; following which, the protein concentration was
determined using a bicinchoninic acid protein assay kit (Solarbio
Science and Technology Co., Ltd.). A 30 µg sample of total soluble
protein was separated by electrophoresis on an 8–10% sodium dodecyl
sulfate polyacrylamide gel (Solarbio Science and Technology Co.,
Ltd.) at a constant voltage of 120 V. The separated protein bands
were then electrophoretically transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Bedford, MA, USA). The
membranes were blocked with 5% non-fat milk at room temperature for
1 h, and then immunoblotted at 4°C overnight with the following
antibodies: Monoclonal rabbit anti-human RIG-I (cat. no. 4200;
1:2,000; Cell Signaling Technology, Inc, Danvers, MA, USA),
polyclonal rabbit anti-human AKT (cat. no. 9272; 1:2,000; Cell
Signaling Technology, Inc.), monoclonal rabbit anti-human
phospho-AKT (Thr308; cat. no. 4056; 1:500; Cell Signaling
Technology, Inc.), polyclonal rabbit anti-human FOXO3A (cat. no.
2497; 1:2,000; Cell Signaling Technology, Inc.), monoclonal rabbit
anti-human phospho-FOXO3A (Thr32; cat. no. 2599; 1:1,000; Cell
Signaling Technology, Inc.), monoclonal rabbit anti-human p27 (cat.
no. 3686; 1:1,000; Cell Signaling Technology, Inc.) and monoclonal
rabbit anti-human TRAIL (cat. no. 3219; 1:1,000; Cell Signaling
Technology, Inc.). Following immunoblotting, the membranes were
incubated with goat anti-rabbit horseradish peroxidase-linked
secondary antibody (cat. no. 401315; 1:5,000; EMD Millipore) at
room temperature for 1 h. The immunostained protein bands were
visualized using SuperSignal West Pico stable peroxide solution
(Pierce; Thermo Fisher Scientific, Inc.). Quantitative analysis was
performed using Gel-Pro Analyzer software (version 4.5; Media
Cybernetics, Inc., Rockville, MD, USA). β-actin was used as a
loading control.
Detection of cell proliferative
activity
The NB4, LV-shCon and LV-shRIG-I cells were seeded
into separate wells of a 96-well plate (1×104
cells/well). ATRA was added to each well (1 µM final
concentration), and the plate was incubated at conditions of 37°C
and 5% CO2. Following incubation for 12, 24, 48 and 72
h, respectively, 20 µl of 0.5% MTT solution was added to each well.
The cells were then cultured under conventional conditions for 4 h,
following which the culture solution was centrifuged at 300 g for 5
min at room temperature and the supernatant removed. Subsequently,
150 µl of DMSO was added to each well, which was then oscillated
for 10 min. Following oscillation, a microplate reader was used to
measure the absorbance of each well at a wavelength of 490 nm. A
cell proliferation curve was created with time on the X-axis and
the A490 value on the Y-axis.
Cell cycle analysis using flow
cytometry
At 0 and 72 h post-ATRA induction, the NB4,
LV-shCon, and LV-shRIG-I cells were fixed in ice-cold 70% ethanol
at a density of 1×105 cells/ml, and then treated with
ribonuclease (200 µg/ml) for 30 min at 37°C. Propidium iodide was
subsequently added to the solution. The DNA content in each aliquot
of cells was then quantitated using flow cytometry (FACSCalibur; BD
Biosciences; Franklin Lakes, NJ, USA). The resultant data were
analyzed using CellQuest software (version 5.1; BD Biosciences)
software set on 10,000 events.
Analysis of apoptosis using flow
cytometry
Cellular apoptosis was analyzed according to the
manufacturer's protocol using an apoptosis detection kit (BD
Biosciences). At 0 and 72 h following ATRA induction, the NB4,
LV-shCon, and LV-shRIG-I cells were washed with cold PBS and
resuspended in 1X binding buffer at a concentration of
1×106 cells/ml. Subsequently, 100 µl of each solution
was transferred into a 5 ml culture tube, to which 5 µl of PE
Annexin V and 5 µl 7-AAD were added. The tubes were then gently
vortexed and incubated for 15 min at room temperature in the dark.
Following incubation, 400 µl of 1X binding buffer was added to each
tube, and the cells were analyzed using flow cytometry
(FACSCalibur; BD Biosciences).
Statistical analysis
All assays were performed independently in
triplicate and results are expressed as the mean ± standard
deviation. Data were analyzed using Student's t-test in SPSS
software (version 19.0; IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
ATRA-induced expression of RIG-I in
NB4 cells is knocked down by LV-mediated shRIG-I
At 72 h post-LV infection, the intensity of the
expression of GFP in the LV-shCon and LV-shRIG-I cells was measured
using flow cytometry. The LV-shCon and LV-shRIG-I cells were
>80% positive for GFP, indicating high infection efficiencies
(Fig. 1A). The background
expression of RIG-I in the NB4 cells was low, and ATRA was capable
of inducing the expression of RIG-I. To validate RIG-I-knockdown in
the LV-infected NB4 cells, the induction of RIG-I in the NB4,
LV-shCon and LV-shRIG-I cells at the mRNA level was analyzed using
RT-qPCR analysis 72 h following ATRA induction. The results showed
that, compared with the expression of RIG-I at 0 h, the mRNA
expression levels of RIG-I were significantly upregulated in the
NB4 and LV-shCon cells at 72 h post-ATRA induction (P<0.01;
Fig. 1B). However, at the same
time-point, the mRNA expression of RIG-I in the LV-shRIG-I cells
had decreased by 78 and 82%, compared to expression in the NB4 and
LV-shCon cells, respectively, (P<0.01; Fig. 1B). The results of the western blot
analysis showed that, compared with the expression at 0 h, the
protein expression levels of RIG-I in the NB4 and LV-shCon cells
were significantly upregulated 72 h following ATRA induction
(P<0.01; Fig. 1C and D).
Compared with its expression in NB4 and LV-shCon cells, the protein
expression of RIG-I in the LV-shRIG-I cells was significantly
knocked down at 72 h (P<0.01; Fig.
1C and D). These results suggested that the LV-mediated shRNA
had successfully infected the NB4 cells, and that the expression of
RIG-I in the ATRA-induced NB4 cells had been effectively knocked
down by RIG-I-specific shRNA at the mRNA and protein levels.
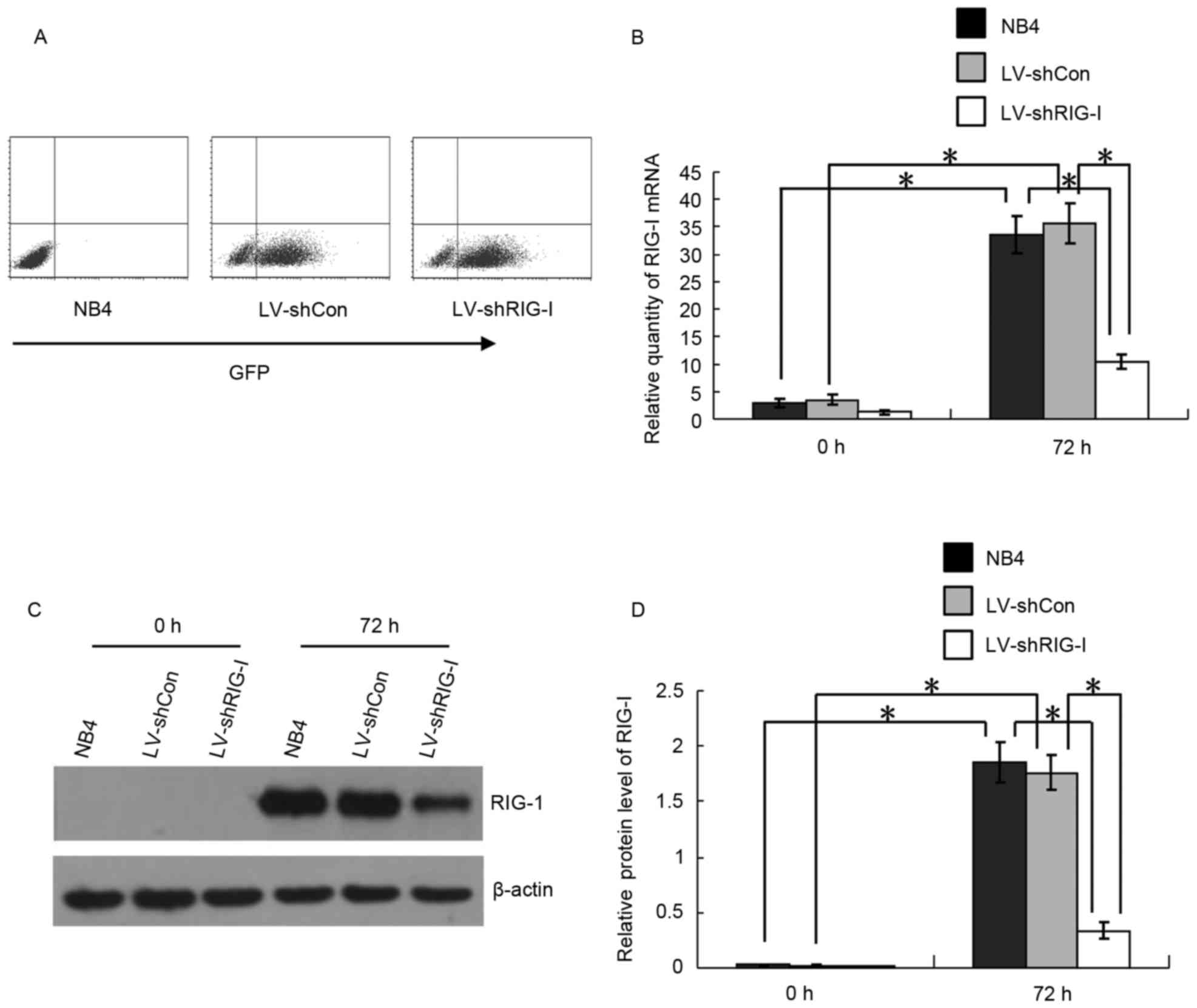 | Figure 1.Expression of RIG-I in ATRA-induced
NB4 cells is knocked down by LV-mediated shRIG-I. (A) Flow
cytometric detection of the expression of GFP in LV-shCon and
LV-shRIG-I cells at 72 h post-lentivirus infection. (B) Reverse
transcription-quantitative polymerase chain reaction analysis for
detection of the RNA expression of RIG-I in NB4, LV-shCon and
LV-shRIG-I cells at 0 and 72 h post-ATRA induction (n=3).
*P<0.01. (C) Western blot analysis of the protein expression of
RIG-I in NB4, LV-shCon and LV-shRIG-I cells at 0 and 72 h post-ATRA
induction. (D) Quantitative analysis of protein expression of RIG-I
in NB4, LV-shCon and LV-shRIG-I cells at 0 and 72 h post-ATRA
induction (n=3) *P<0.01. Data are presented as the mean ±
standard deviation. RIG-I, retinoic acid inducible gene I; ATRA,
all-trans retinoic acid; GFP, green fluorescent protein; LV,
lentivirus; Con, control; siRNA, small interfering RNA; LV-shCon,
NB4 cells infected with LV Con-siRNA; LV-shRIG-I, NB4 cells
infected with LV RIG-I-siRNA. |
RIG-I knockdown alleviates the
inhibition of proliferation and cell cycle arrest induced in NB4
cells by ATRA
The MTT method was used to analyze the proliferation
of NB4, LV-shCon and LV-shRIG-I cells at 12, 24, 48 and 72 h with
and without ATRA induction. The results showed that the NB4,
LV-shCon and LV-shRIG-I cells had similar rates of proliferation
without ATRA induction (Fig. 2A).
However, the NB4 and LV-shCon cells, which were induced with ATRA
showed significantly reduced proliferation at 72 h post-induction,
compared with proliferation in the NB4 and LV-shCon cells, which
were not induced with ATRA (P<0.01; Fig. 2A and B). Compared with the
ATRA-induced NB4 cells and LV-shCon cells, the ATRA-induced
LV-shRIG-I cells exhibited marked proliferation, particularly at 72
h post-induction (P<0.05; Fig.
2B), indicating that RIG-I knockdown alleviated the
ATRA-induced inhibition of NB4 cell proliferation. Further analysis
of the cell cycle using flow cytometry revealed that, compared with
the results obtained at 0 h, NB4 and LV-shCon cells were arrested
in the G1 phase at 72 h post-ATRA induction (P<0.05; Fig. 2C). However, at 72 h post-ATRA
induction, the percentage of LV-shRIG-I cells in the G1 phase
(53.28±4.10%) had decreased significantly, compared with the
percentages of NB4 and LV-shCon cells in the G1 phase (73.56±5.63
and 72.04±5.58%, respectively; P<0.05; Fig. 2C). These results suggested that
RIG-I knockdown alleviated ATRA-induced cell cycle arrest of the
NB4 cells at the G1 phase.
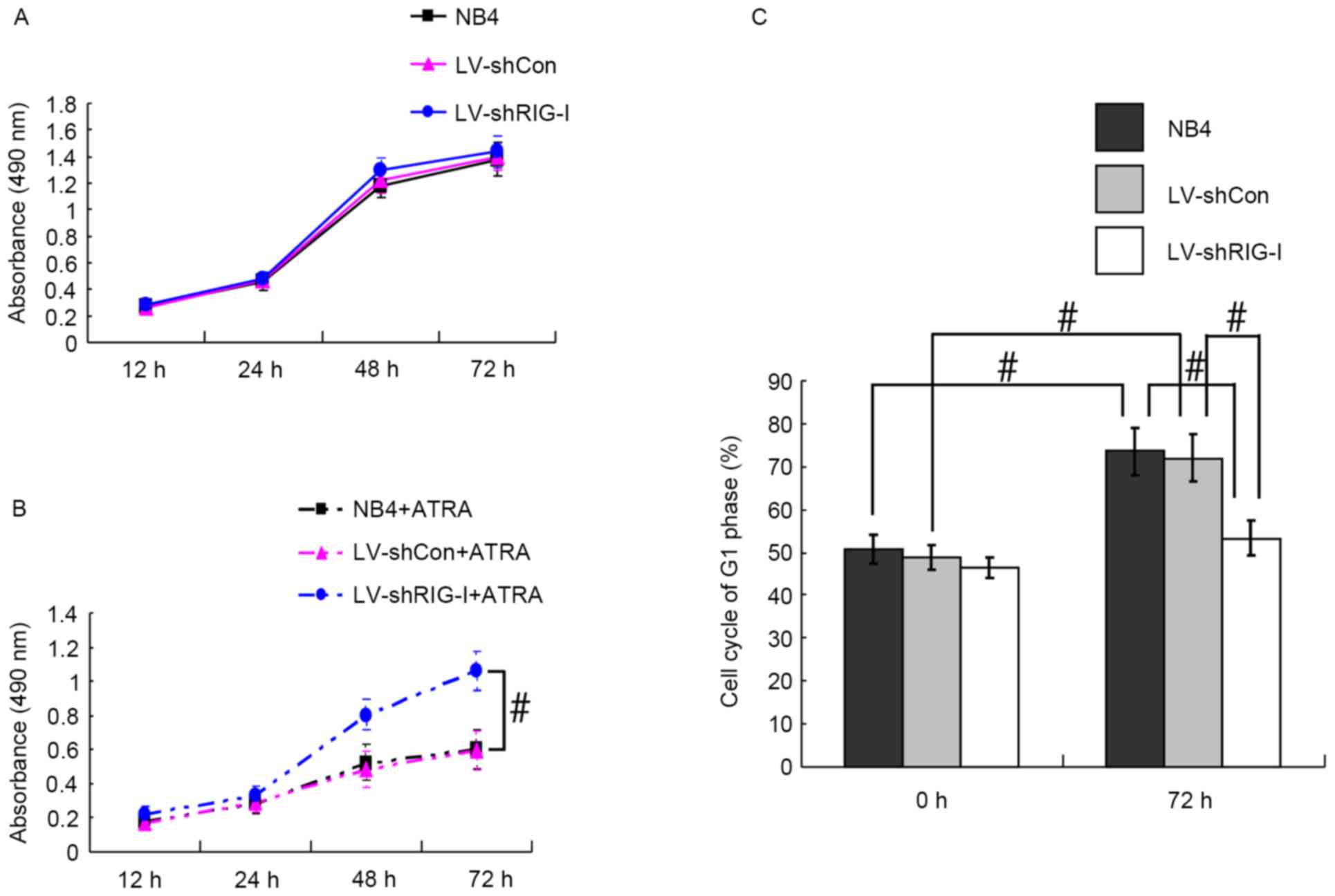 | Figure 2.RIG-I knockdown alleviates
ATRA-induced reduced proliferation and cell cycle arrest in NB4
cells. (A) MTT detection of NB4, LV-shCon and LV-shRIG-I cell
proliferation without ATRA induction at different time points. (B)
MTT detection of NB4, LV-shCon and LV-shRIG-I cell proliferation
with ATRA induction at different time points (n=3)
#P<0.05. (C) Flow cytometric detection of NB4,
LV-shCon and LV-shRIG-I cells in the G1 phase at 0 and 72 h
post-ATRA induction (n=3) #P<0.05. Data are presented
as the mean ± standard deviation. RIG-I, retinoic acid inducible
gene I; ATRA, all-trans retinoic acid; LV, lentivirus; Con,
control; siRNA, small interfering RNA; LV-shCon, NB4 cells infected
with lentivirus Con-siRNA; LV-shRIG-I, NB4 cells infected with
lentivirus RIG-I-siRNA. NB4 + ATRA, ATRA-induced NB4 cells;
LV-shCon + ATRA, ATRA-induced LV-shCon cells; LV-shRIG-I + ATRA,
ATRA-induced LV-shRIG-I cells. |
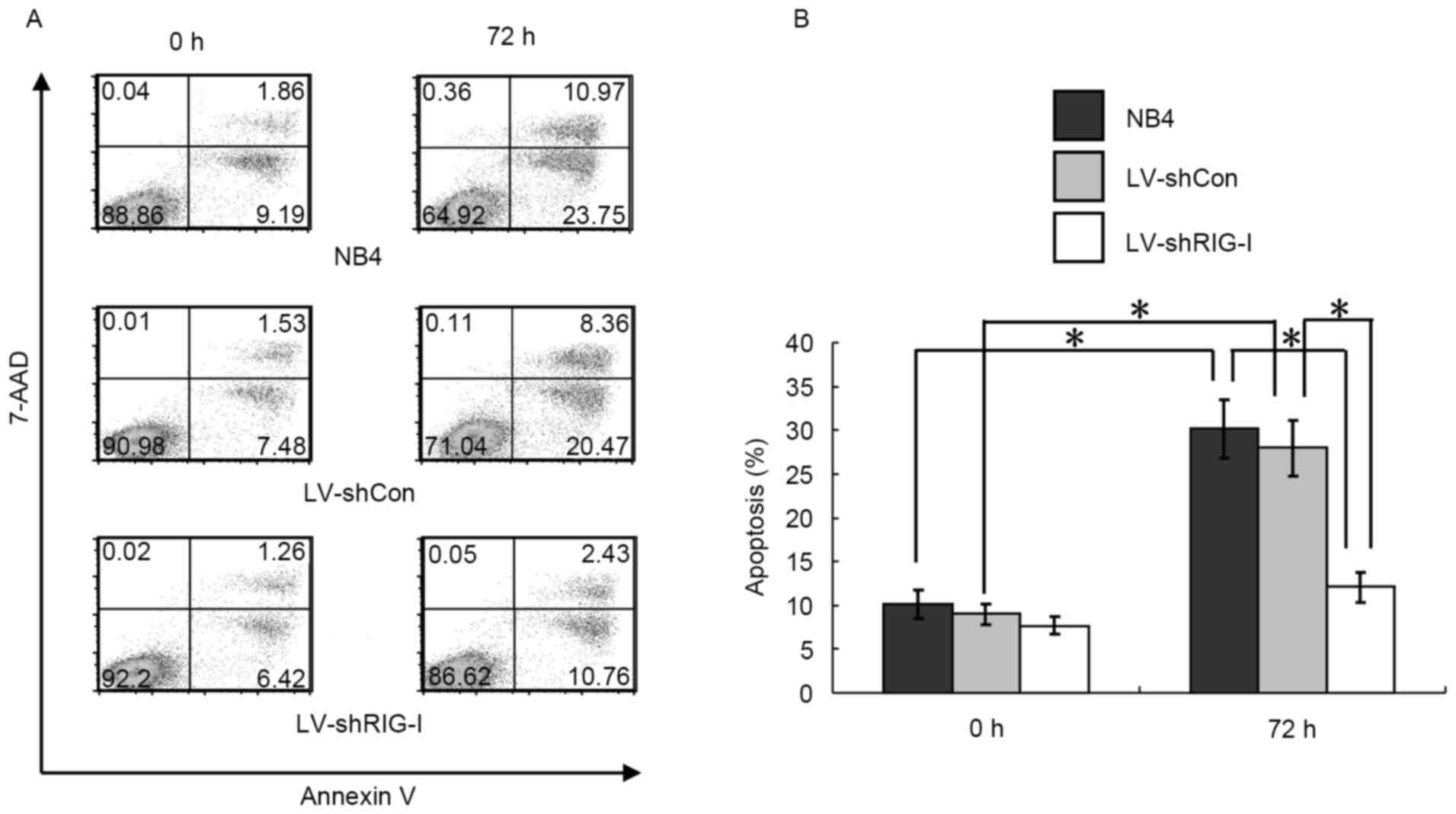
RIG-I knockdown inhibits ATRA-induced
apoptosis in NB4 cells
Flow cytometry was used to investigate the effect of
RIG-I-knockdown on ATRA-induced apoptosis in NB4 cells. The results
showed that, compared with the results obtained at 0 h, the NB4 and
LV-shCon cells exhibited signs of apoptosis at 72 h post-induction
with ATRA (P<0.01; Fig. 3A and
B). By contrast, a significantly lower proportion of LV-shRIG-I
cells (12.32±1.10%) exhibited signs of apoptosis at 72 h
post-induction, compared with those in the NB4 and LV-shCon cell
groups (32.51±2.39 and 30.60±2.53%, respectively; P<0.01,
Fig. 3A and B), suggesting that
RIG-I-knockdown inhibited the ATRA-induced apoptosis of NB4
cells.
RIG-I knockdown reduces cell
proliferation inhibition, cell cycle arrest and apoptosis in
ATRA-induced NB4 cells via the AKT-FOXO3A signaling pathway
To determine whether RIG-I knockdown reduced the
cell proliferation inhibition, cell cycle arrest and apoptosis in
ATRA-induced NB4 cells, western blot analysis was used to examine
the AKT-FOXO3A signaling pathway in the LV-shCon and LV-shRIG-I
cells at 0 and 72 h following ATRA induction. The results showed
that, compared with the levels of pAKT-Thr308 at 0 h, the levels of
pAKT-Thr308 in the LV-shCon cells at 72 h post-ATRA induction were
decreased with RIG-I-induced expression (P<0.01; Fig. 4A and B). Furthermore, pFOXO3A-Thr32
downstream AKT was downregulated (P<0.01; Fig. 4A and C), which resulted in
increased expression of protein p27, promoting G1 phase arrest
(P<0.01; Fig. 4A and D), and
TRAIL, promoting cell apoptosis (P<0.01, Fig. 4A and E), which are directly
transcribed by FOXO3A. At 72 h post-ATRA induction, compared with
the LV-shCon cells, the LV-shRIG-I cells with RIG-I-knockdown
showed increased levels of pAKT-Thr308 (P<0.01; Fig. 4A and B) and pFOXO3A-Thr32
(P<0.01; Fig. 4A and C), and
there was downregulated expression levels of protein p27,
(P<0.01; Fig. 4A and D) and
TRAIL (P<0.01; Fig. 4A and E).
These results showed that the knockdown of RIG-I reduced cell
proliferation inhibition, cell cycle arrest and apoptosis in the
ATRA-induced NB4 cells via the AKT-FOXO3A signaling pathway.
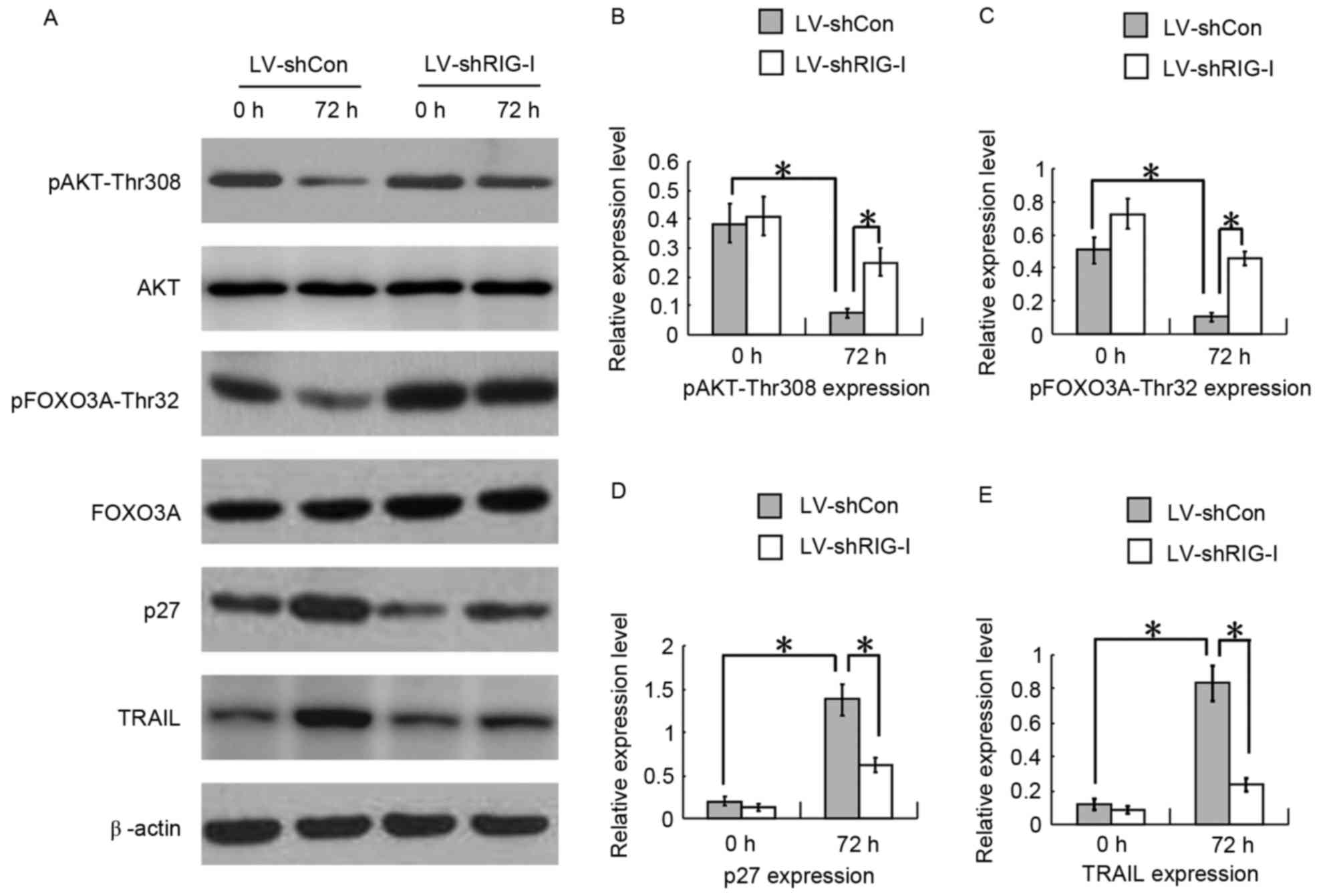 | Figure 4.RIG-I-knockdown reduces cell
proliferation inhibition, cell cycle arrest and apoptosis in
ATRA-induced NB4 cells via the AKT-FOXO3A signaling pathway. (A)
Western blot analyses of expression levels of pAKT-Thr308,
pFOXO3A-Thr32, p27 and TRAIL in LV-shCon and LV-shRIG-I cells at 0
and 72 h post-ATRA induction. (B) Quantitative analysis of protein
levels of pAKT-Thr308 in LV-shCon and LV-shRIG-I cells at 0 and 72
h post-ATRA induction (n=3) *P<0.01. (C) Quantitative analysis
of protein levels of pFOXO3A-Thr32 in LV-shCon and LV-shRIG-I cells
at 0 and 72 h post-ATRA induction (n=3) *P<0.01. (D)
Quantitative analysis of protein levels of p27 in LV-shCon and
LV-shRIG-I cells at 0 and 72 h post-ATRA induction (n=3)
*P<0.01. (E) Quantitative analysis of protein levels of TRAIL in
LV-shCon and LV-shRIG-I cells at 0 and 72 h post-ATRA induction
(n=3) *P<0.01. Data are presented as the mean ± standard
deviation. RIG-I, retinoic acid inducible gene I; ATRA, all-trans
retinoic acid; LV, lentivirus; Con, control; siRNA, small
interfering RNA; LV-shCon, NB4 cells infected with lentivirus
Con-siRNA; LV-shRIG-I, NB4 cells infected with lentivirus
RIG-I-siRNA. |
Discussion
In the present study, the lentivirus method was used
to knock down the gene expression of RIG-I in ATRA-induced NB4 APL
cells, and proliferation, cell cycle and apoptosis in the NB4 cells
were investigated. The association between RIG-I and the AKT-FOXO3A
signaling pathway was also examined. The results showed that the
siRNA-mediated knockdown of RIG-I reduced cell proliferation
inhibition, cell cycle arrest and apoptosis in the ATRA-induced NB4
cells via the AKT-FOXO3A signaling pathway.
RIG-I is upregulated during ATRA-induced terminal
granulocytic differentiation of the NB4 cell line of APL cells.
Previously, it was found that RIG-I gene-knockout mice had
significantly increased numbers of myeloid cells, indicating a
degree of differentiation disorder. The percentage of cells
undergoing apoptosis in RIG-I-deficient mice was significantly
decreased, whereas the numbers of cells entering their cell cycle
were increased, resulting in myeloproliferative disorder, and
suggesting that RIG-I is involved in regulating apoptosis and cell
cycle (3). Another study showed
that, on reducing the expression of RIG-I in HL60 cells, subsequent
ATRA-induced cell differentiation, cell cycle arrest and apoptosis
were suppressed (15). These
findings are analogous with the results of the present study, which
showed that RIG-I-knockdown reduced cell proliferation inhibition,
cell cycle arrest and apoptosis in ATRA-induced NB4 cells.
Translocation events in APL cells typically occur on
chromosomes 15 and 17 (16), and
result in fusion of the promyelocytic leukemia (PML) gene on
chromosome 15 with the retinoic acid receptor α (RARα) gene on
chromosome 17, to produce an abnormal PML-RARα fusion gene
(17). Previous studies have shown
that ATRA inhibits APL cell proliferation, and promotes cell
differentiation and apoptosis by stimulating proteinase dependent
PML-RARα decomposition and consequent PML-nuclear body (PML-NB)
formation (18,19). As tumor inhibitors, PML proteins
are an important component of PML-NBs. However, the PML-RARα fusion
protein in APL cells damages PML-NBs and inhibits endogenous PML
tumor inhibition function. Therefore, PML-RARα decomposition and
PML-NB formation are considered to be key responses, which occur in
cells exposed to ATRA (18–20).
It has been reported that PML-NBs, protein phosphatase 2A (PP2A),
nuclear AKT and the transcription factor, FOXO3A, jointly
constitute a tumor inhibitor network. PML proteins have been shown
to specifically collect PP2A into their PML-NBs and then exhibit
inhibited nuclear AKT phosphorylation activity, resulting in the
deactivation of AKT (21). As the
FOXO3A transcription factor can be deactivated by AKT, which is
activated by phosphorylation, PML-NB deficiency or its functional
deletion activates the phosphorylation of AKT, which results in
deficient FOXO3A transcription activity. Sakoe et al
(13) reported that phosphorylated
FOXO3A was located in the cytoplasm of APL-derived NB4 cells and
primary patient cells. Following ATRA treatment, the levels of
phosphorylated FOXO3A were reduced and FOXO3A entered the nucleus.
The mRNA and protein levels of TRAIL were also increased, and
transfection with an shRNA oligonucleotide specific for FOXO3A was
shown to significantly inhibit differentiation and apoptosis in
ATRA-induced NB4 cells. The AKT-FOXO3A signaling pathway is
essential in the process of ATRA-induced APL granulocyte
differentiation and apoptosis (13). In the present study, ATRA-induced
proliferation inhibition, cell cycle arrest and apoptosis of NB4
cells were accompanied by the expression of RIG-I and decreased
levels of phosphorylated AKT, resulting in the deactivation of AKT,
whereas the levels of phosphorylated FOXO3A regulated by AKT were
decreased, leading to its activation. These events led to increased
expression levels of the cell cycle arrest protein, p27, and
apoptosis protein, TRAIL, which are directly transcribed by FOXO3A.
By contrast, following the knockdown of ATRA-induced RIG-I, the
levels of phosphorylated AKT increased, AKT was activated, the
level of phosphorylated FOXO3A was increased, and FOXO3A was
deactivated. The protein expression levels of p27 and TRAIL
transcribed by FOXO3A were decreased, resulting in decreased cell
cycle arrest and apoptosis in the ATRA-induced NB4 cells. These
findings suggested that RIG-I-knockdown reduced cell proliferation
inhibition, cell cycle arrest and apoptosis of ATRA-induced NB4
cells via the AKT-FOXO3A signaling pathway. Based on the above, it
was hypothesized that ATRA-induced proteinase-dependent PML-RARα
decomposition, PML-NB formation and expression of RIG-I were
synergistically involved in the AKT-FOXO3A signaling pathway to
inhibit NB4 cell proliferation, arrest cell cycle and promote NB4
cell apoptosis.
In conclusion, RIG-I was shown to be important in
the events leading to the inhibition of cell proliferation, arrest
of the cell cycle and promotion of apoptosis in ATRA-induced NB4
cells. Lentivirus-mediated RIG-I-knockdown relieved cell
proliferation inhibition, cell cycle arrest and apoptosis in the
ATRA-induced NB4 cells via the AKT-FOXO3A signaling pathway.
Acknowledgements
The present study was supported by a grant from the
Natural Science Foundation of Tianjin Municipal Committee of
Science and Technology (grant no. 13JCYBJC21200).
References
|
1
|
Wang ZY and Chen Z: Acute promyelocytic
leukemia: From highly fatal to highly curable. Blood.
111:2505–2515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu TX, Zhang JW, Tao J, Zhang RB, Zhang
QH, Zhao CJ, Tong JH, Lanotte M, Waxman S, Chen SJ, et al: Gene
expression networks underlying retinoic acid-induced
differentiation of acute promyelocytic leukemia cells. Blood.
96:1496–1504. 2000.PubMed/NCBI
|
|
3
|
Zhang NN, Shen SH, Jiang LJ, Zhang W,
Zhang HX, Sun YP, Li XY, Huang QH, Ge BX, Chen SJ, et al: RIG-I
plays a critical role in negatively regulating granulocytic
proliferation. Proc Natl Acad Sci USA. 105:10553–10558. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoneyama M, Kikuchi M, Natsukawa T,
Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S and Fujita T:
The RNA helicase RIG-I has an essential function in double-stranded
RNA-induced innate antiviral responses. Nat Immunol. 5:730–737.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zeng W, Sun L, Jiang X, Chen X, Hou F,
Adhikari A, Xu M and Chen ZJ: Reconstitution of the RIG-I pathway
reveals a signaling role of unanchored polyubiquitin chains in
innate immunity. Cell. 141:315–330. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Loo YM and Gale M Jr: Immune signaling by
RIG-I-like receptors. Immunity. 34:680–692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
O'Neill LA and Bowie AG: The power stroke
and camshaft of the RIG-I antiviral RNA detection machine. Cell.
147:259–261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoo JS, Kato H and Fujita T: Sensing viral
invasion by RIG-I like receptors. Curr Opin Microbiol. 20:131–138.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chiang JJ, Davis ME and Gack MU:
Regulation of RIG-I-like receptor signaling by host and viral
proteins. Cytokine Growth Factor Rev. 25:491–505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang LJ, Zhang NN, Ding F, Li XY, Chen L,
Zhang HX, Zhang W, Chen SJ, Wang ZG, Li JM, et al: RA-inducible
gene-I induction augments STAT1 activation to inhibit leukemia cell
proliferation. Proc Natl Acad Sci USA. 108:1897–1902. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li XY, Jiang LJ, Chen L, Ding ML, Guo HZ,
Zhang W, Zhang HX, Ma XD, Liu XZ, Xi XD, et al: RIG-I modulating
Src-mediated AKT activation to restrain leukemic stemness. Mol
Cell. 53:407–419. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang H and Tindall DJ: Dynamic FoxO
transcription factors. J Cell Sci. 120:2479–2487. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakoe Y, Sakoe K, Kirito K, Ozawa K and
Komatsu N: FOXO3A as a key molecule for all-trans retinoic
acid-induced granulocytic dirreretiation and apoptosis in acute
promyelocytic leukemia. Blood. 115:3787–3795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang NN, Chen L, Zhang W, Li XY, Jiang
LJ, Ding F, Zhang HX, Chen SJ, Wang ZG, Chen Z and Zhu J: RIG-I
activates expression of interferon-stimulated genes (ISGs) and
inhibits the proliferation of acute myeloid leukemia cells. Blood.
112:28462008.
|
|
16
|
Rowley JD, Golomb HM and Dougherty C:
15/17 translocation, a consistent chromosomal change in acute
promyelocytic leukaemia. Lancet. 1:549–550. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kakizuka A, Miller WH Jr, Umesono K,
Warrell RP Jr, Frankel SR, Murty VV, Dmitrovsky E and Evans RM:
Chromosomal translocation t(15;17) in human acute promyelocytic
leukemia fuses RAR alpha with a novel putative transcription
factor, PML. Cell. 66:663–674. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Breitman TR, Collins SJ and Keene BR:
Terminal differentiation of human promyelocytic leukemic cells in
primary culture in response to retinoic acid. Blood. 57:1000–1004.
1981.PubMed/NCBI
|
|
19
|
Tallman MS, Andersen JW, Schiffer CA,
Appelbaum FR, Feusner JH, Woods WG, Ogden A, Weinstein H, Shepherd
L, Willman C, et al: All-trans retinoic acid in acute promyelocytic
leukemia: Long-term outcome and prognostic factor analysis from the
North American Intergroup protocol. Blood. 100:4298–4302. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Salomoni P and Bellodi C: New insights
into the cytoplasmic function of PML. Histol Histopathol.
22:937–946. 2007.PubMed/NCBI
|
|
21
|
Trotman LC, Alimonti A, Scaglioni PP,
Koutcher JA, Cordon-Cardo C and Pandolfi PP: Identification of a
tumor suppressor network opposing nuclear Akt function. Nature.
441:523–527. 2006. View Article : Google Scholar : PubMed/NCBI
|