Introduction
In 2001, Zhou et al (1) associated the mutations in
Pantothenate kinase 2 (PANK2) gene with a heterogenic
neurologic disorder known as Hallervorden Spatz disease, later
renamed as Pantothenate Kinase Associated Neurodegeneration (PKAN).
It is a rare, autosomal recessive, neurodegenerative disease
presenting iron deposition in the globus pallidus (2,3) and
included in the Neurodegeneration with Brain Iron Accumulation
(NBIA) category (4). Based on age
of onset and rate of progression, PKAN can be divided in a more
common, early onset, classical form and a late onset, atypical one.
While extrapyramidal signs such as dystonia, dysarthria and
choreoatethosis, together with spasticity, cognitive decline and
pigmentary retinopathy are common features of the classical form,
parkinsonism and neuropsychiatric symptoms are usually observed in
the atypical form. Magnetic resonance imaging is extremely useful
at orienting the diagnosis since it usually reveals a sign known as
‘eye of the tiger’ that corresponds to iron deposition and tissue
damage at the globus pallidus (5).
PanKs catalyze the phosphorylation of vitamin B5 in
the first and essential step of coenzyme A (CoA) biosynthesis
(6). Mammalians cells express up
to four different forms of PanKs (7) and therefore it is not straightforward
to understand the specific biochemical connections between defects
in PANK2 functioning and the neurodegenerative process.
Localization in mitochondria (8–11)
together with specific biochemical properties may confer to the
enzyme a pivotal role as a sensor for cellular CoA requirements
(12,13). Even though it is ubiquitously
expressed, it is present at high relative levels in the brain
(1,12), thus suggesting a relevant role in
neural cells. Studies performed in different experimental settings
(14,15) point to a reduction in cellular CoA
level as the main consequence of PANK2 defects. This hypothesis is
robustly supported by two facts: a) The rescue potential of
molecules capable of bypassing the blockage in CoA biosynthesis,
such as pantethine (16,17), 4P-pantetheine (18,19)
and CoA itself (16,20); b) the recent identification of
patients with clinical features largely overlapping to those of
PKAN and carrying mutations in COASY gene, encoding the last
enzyme of the same biochemical pathway (21,22)
and now linked to a new form of NBIA named COASY protein-associated
Neurodegeneration (CoPAN).
We recently developed zebrafish models of PKAN and
CoPAN by microinjection of gene specific morpholinos (16). We described an essential role of
pank2 in neuronal development, with pank2 morphants
showing severe perturbation of brain morphology and drastic
reduction in the expression of fundamental transcription factors
governing neuronal cell development and differentiation.
Surprisingly, we also observed defects in the developing
vasculature, with clear reduction in the intersegmental vessels
(IVS) formation, structural alteration of the caudal plexus and
caudal edema. We detected similar abnormalities also in
coasy morphants, where we could also measure a reduction of
CoA levels (16,20). More importantly, all these features
were efficiently rescued by providing panthetine (for pank2
morphants) or CoA (for both models) to fish water, thus confirming
the specificity of the phenotype. The data let infer a selective
sensibility of endothelial cells to pank2 and CoA depletion.
At the moment, there is no evidence indicating a role for
vasculature defects in the pathogenesis of PKAN or CoPAN. These
alterations were never investigated or documented in any other
animal models. To evaluate the relevance of this data in mammalian
settings, we analyzed the effects of PANK2 downregulation on
the angiogenic properties of human umbilical vascular endothelial
cells (HUVECs). Different experimental approaches clearly indicated
that PANK2 is essential for HUVEC angiogenic activity, which in
turn advocate a potential mechanism contributing to the development
of PKAN pathology.
Materials and methods
Cells isolation, maintenance and
transfection
HUVECs were previously isolated from umbilical veins
according to an established protocol (23). Briefly, cord was washed with PBS
containing penicillin 100 U/ml and streptomicin 100 µg/ml. An
injection needle was inserted into the umbilical vein and clamped;
vein was washed twice with PBS using a 50cc syringe to remove
residual blood clots and check for leaks. Then, a solution
containing 0.1 U/ml of collagenase type I and 0.8 U/ml of dispase I
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was injected in the
vein and the cord was incubated at 37°C. After 30′, the liquid was
collected and the vein was washed twice with PBS to collect all the
detached cells. After centrifugation, the supernatant was removed;
cells were suspended in growth medium and seeded on a collagen
coated dish. Cells from 3 different cords were pooled, eventually
frozen, and used at early passages (III–VII). HUVECs were cultured
in EGM-2™ Bullet Kit™ (Lonza, Walkersville,
MD, USA) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), penicillin 100 U/ml,
streptomicin 100 µg/ml (BioSera, Nuaille, France) on collagen
coated dishes. Cells were maintained at 37°C under 5%
CO2.
One set of three different 27-mer siRNAs duplexes
specific for PANK2 gene was obtained from Origene (ID 80025;
OriGene Technologies, Inc., Rockville, MD, USA). Out of them, the
siRNA SR312773A was the most efficient and hence selected for all
described experiments. We used a universal scrambled negative
control siRNA duplex (SCR) from the same company as a negative
control in all transfection experiments.
For transfection, 15×103
HUVECs/cm2 were seeded in 12-well or 6-wells plates.
After 16 h they were incubated with specific and control siRNAs (1
nM final concentration) and 3 or 7.5 µl of
Lipofectamine® RNAiMax Transfection Reagent (Thermo
Fisher Scientific, Inc.). After 5 h of incubation, the transfection
medium was replaced by standard growth medium with or without 25 µM
CoA (Sigma-Aldrich; Merck KGaA). Mock-treated cells received only
the transfection reagent without the siRNA. After the transfection,
cells were collected and analyzed or treated as described in the
following section.
Biochemical and immunological
methods
Equal amounts of protein extracts (50 µg) obtained
from HUVECs 48 h after the transfection were separated on SDS-PAGE
and transferred to PVDF membrane. The membranes were incubated at
4°C for 16 h with a monoclonal mouse anti-PANK2 (1:500, no.
TA501321; OriGene Technologies, Inc.) and a monoclonal mouse
anti-actin (1:1,000, no. TA811000; OriGene Technologies, Inc.).
Membranes were washed three times with TBS 0,05% Tween and
incubated for 1 h with HRP-conjugated anti-mouse IgG (1:5,000;
Pierce; Thermo Fisher Scientific, Inc.). Immunocomplexes were
detected by a chemiluminescence detection kit (Protein Detection
System; Genespin, Milan, Italy) with Odissey® Imaging
System (LiCor Bioscience, Lincoln, NE, USA). The densitometric
analysis was performed with the Kodak 1D 3.6 program.
Cell viability/proliferation
At 24 h after the transfection, 15×103
HUVECs/cm2 were seeded in 12-well plates in triplicates.
After 48 h the medium was collected and analyzed for the presence
of dead cells. Adherent cells were detached with trypsin 0.1%/EDTA,
washed and stained with trypan blue to check for vitality.
MTT assay
48 h after the transfection, 5,000/well HUVECs were
seeded in 96-well plates in triplicates. The day after, cells were
washed and incubated with 0.5 mg/ml methyl-thiazol-tetrazolium
(Sigma-Aldrich; Merck KGaA) in cell medium for 3.5 h. The
supernatant was then removed, the insoluble formazan blue dissolved
in 75 µl DMSO, and absorbance was measure at 540 nm with the
EnSight Multimode Plate Reader (PerkinElmer, Inc., Waltham, MA,
USA).
ATP measurement
ATP concentration was measured using the ATP
luminescence assay kit (CellTiter-Glo Luminescent Cell Viability
Assay; Promega Corporation, Madison, WI). 24 h after the
transfection, 25,000 cells/well were transferred in a 96-well plate
in triplicate and equilibrated at room temperature for 30 min. 100
µl of reagent were added in each well of the plate and the
luminescent signal measured by the EnSight Multimode Plate
Reader.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
HUVECs were collected 48 h after the transfection
and RNA was purified by using TriReagent solution (Sigma-Aldrich;
Merck KGaA). 1 µg of total RNA was used to synthesize the first
strand of cDNA with the Improm-II Reverse Transcriptase (Promega
Corporation), using oligo-dT 4 µM as primer in a total volume of 20
µl. 1 µl of the reaction was used as template for the real-time
amplification. We set up triplicate reactions in a total volume of
10 µl, with GoTaq® qPCR Master Mix (Promega Corporation)
and 0,5 µM of the specific primers. The reaction was run on Eco 48
PCR Real-time Machine (PCR max, Staffordshire, ST15 0SA, UK) for 40
cycles of 95°C for 10 sec and 60°C for 20 sec. Hypoxanthine guanine
phosphoribosyl transferase 1 (HPRT1) mRNA was used as endogenous
reference for the relative quantification, which was performed by
the ΔΔCq method, as previously described (24,25).
For human PANK2 mRNA amplification we used a
PrimeTime® qPCR Assay primers mix located on exon 5–6
(IDT Technologies Inc., Coralville, IA, USA). The primers for the
other genes are shown in Table I.
For the comparison of the amount of different PanK mRNA isoforms in
HUVECs, the ratio between the quantification cycle (Cq) of each
PanK gene and the Cq of HPRT1 gene was calculated. Since the Cq
inversely correlates with the amount of target mRNA and the HPRT1
Cq value is the same for all samples, the highest ratio value
corresponds to the lowest level of target mRNA.
 | Table I.Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Sequences of primers used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| HPRT1 |
TGTTTTCCTTGGTCAGGCAG |
AAGCTTGCGACCTTGACCAT |
| PANK1 |
AGGTGTCAGCATTCTAGCCG |
GGTCTCACAACCAGTCAGCA |
| PANK3 |
GCACACAAGCTGACAAGCTG |
ACATTCGTGCCACAGAACCA |
| VE-CADHERIN |
GCTCCCCTCCAAAGACGGTCG |
AACAGATCGTGCCTGTGGGCTGAGG |
| VEGFR2 |
GAACATTTGGGAAATCTCTTGC |
CGGAAGAACAATGTAGTCTTTGC |
| CD31 |
TCCACATCAGCCCCACCGGA |
TGGGCCACAATCGCCTTGTCC |
| FGFR1 |
GGGCTGGAATACTGCTACAA |
GCCAAAGTCTGCTATCTTCATC |
Cell motility assay
Cell motility was assessed by time-lapse
video-microscopy. Briefly, 5,000 transfected cells were seeded in
12-well plate and stimulated with 30 ng/ml VEGF or 2 and 10% FBS.
Constant temperature (37°C) and 5% CO2 were maintained
throughout the experimental period by means of a heatable stage and
climate chamber. Cells were observed under an inverted
photomicroscope (Zeiss Axiovert 200 M) and phase-contrast snap
photographs (one every 10 min) were digitally recorded for 5 h.
Cell paths (20–30 cells per experimental point) were generated from
centroid positions and migration parameters were analysed with the
‘Chemotaxis and Migration Tool’ of ImageJ Software (rsbweb.nih.gov/ij).
Wound healing scratch assay
HUVECs were transfected as described earlier. After
48 h (when confluency was reached), wounds were created in the
cellular monolayers with a 1.0-mm wide rubber policeman. An image
was captured and the surface area of the wound was delimited by a
rectangular shape with ImageJ Software. Then, cells were incubated
in fresh medium with 5% FCS. After 10 h, cell monolayers were fixed
with 2.5% glutaradehyde, stained with crystal violet 0.1% and
photographed. Endothelial cells invading the rectangular area (same
size in all samples) delimiting the original wound were counted by
computer-assisted analysis of the digitalized images with ImageJ
Software (rsbweb.nih.gov/ij).
Morphogenesis on three-dimensional
gels
10 µl/well of Geltrex™ (Thermo Fisher
Scientific Inc.) was used to coat 15-well µ-Slide Angiogenesis
plates (IBIDI, Martinsried, Germany) at 4°C. After gelification at
37°C, 10,000 transfected HUVECs were seeded at least in triplicate
onto coated dishes in 5% FBS cell growth medium. Newly formed
endothelial cell ‘cords’ and ‘tubes’ were photographed after 20 h
at 40X magnification (Olympus IX51 inverted microscope) and empty
areas were counted by two investigators without knowledge of the
samples tested.
Statistical analyses
One-way analysis of variance with Dunnett's post hoc
test were applied to compare the different groups. The results were
analyzed by GraphPad Prism 6.01 software (GraphPad Software, Inc.,
La Jolla, CA, USA) and P<0.05 was considered to indicate a
statistically significant difference. Data were presented as the
mean ± standard deviation of at least 3 independent experiments,
unless otherwise stated in the figure legend.
Results
We first analyzed the expression of different
PANK isoforms in HUVECs by RT-qPCR. PANK2 mRNA level
was the highest, together with PANK3 mRNA, while
PANK1 was expressed at lower level (Fig. 1A). This condition is also observed
in human brain (12). To
characterize the role of PANK2 in HUVECs we downregulated
its expression by siRNA technology. We selected the best performing
siRNA out of three different molecules and identified the
concentration of 1 nM as the one providing the best ratio between
silencing efficiency and absence of off target effects (data not
shown). At this low dose of specific siRNA, we obtained a
significant downregulation of PANK2 mRNA levels
(approximately 75%) as measured by RT-qPCR 48 h after the
transfection (Fig. 1B). We did not
observe any change in PANK1 and PANK3 mRNAs amount in
these cells and transfection with an unrelated, control siRNA (SCR)
resulted in no modification of the expression of any of the
PANK genes (Fig. 1B). The
immunoblotting performed with protein extracts collected 48 h after
the transfection confirmed the decrease in the synthesis of PANK2
protein, with a reduction of the bands corresponding to precursor
(63 kDa), immature (59 kDa) and mature forms (48 kDa) of the
protein. Altogether, PANK2 protein level was decreased by
more than 60% in the silenced cells as compared to mock- and
SCR-transfected cells (Fig. 1C).
To verify the biochemical consequences of PANK2 silencing,
we analyzed parameters known to be perturbed by downregulation of
the gene in other cell types (26,27),
such as cell proliferation and ATP content. The same number of
cells was plated, exposed to the silencing reaction and counted
after 72 h. Silenced cells were approximately 25% less than mock-
and SCR-transfected ones (P<0.01; Fig. 1D). Since we did not find signs of
cell death in any sample, the difference should represent a
reduction of the proliferation rate. To assess vitality and
energetic profile of silenced cells we performed the MTT assay and
measured ATP content. PANK2-silenced cell showed significant
reduction of the dehydrogenase activity and of ATP content
(P<0.05; Fig. 1E and F). All
the features associated with PANK2 downregulation were
prevented by the addition of 25 µM CoA to the cell medium 5 h after
the transfection. This indirectly suggests that PANK2
silencing leads to depletion of cellular CoA levels and confirms
the specificity of our experimental setting. Altogether this set of
analyses shows that the reduction of PANK2 expression in
HUVECs induces well known features of energetic and mitochondrial
defects and reduced proliferation.
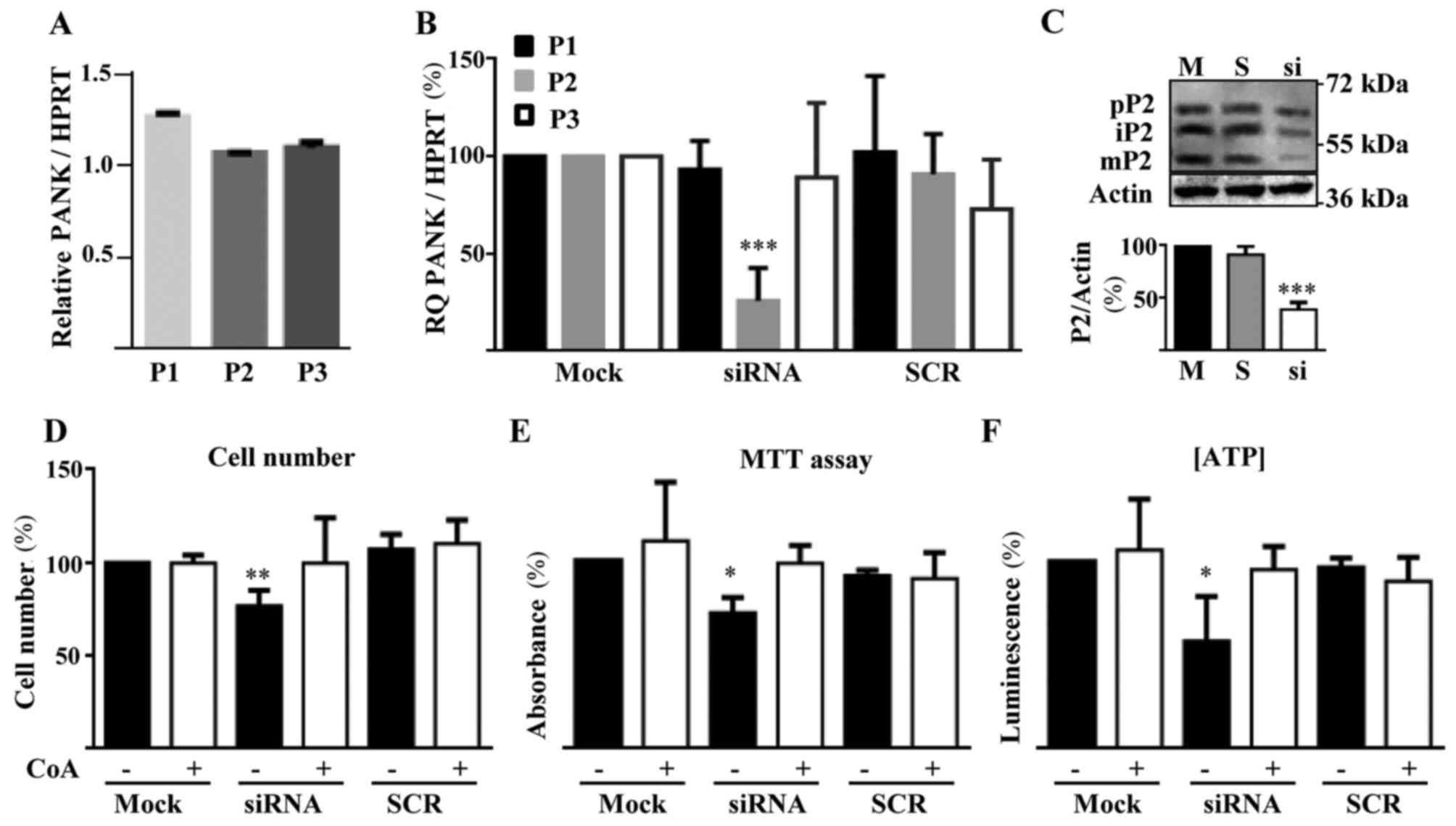 | Figure 1.PANK2 expression and silencing
in HUVECs. (A) RT-qPCR quantification of the expression of
different PanKs isoforms in HUVECs. The Y-axis presents the ratio
between the relative expression of each PanK and HPRT1 gene. (B)
RT-qPCR evaluation of efficiency and specificity of PANK2
siRNA silencing. (C) Representative immunoblotting (n=3) and
quantification of human PANK2 protein in control and silenced
HUVECs. (D) Quantification of cell number, (E) viability (MTT
Assay), and (F) ATP content in HUVECs transfected with PANK2
and control (SCR group) siRNA. *P<0.05, **P<0.01 and
***P<0.001 vs. Mock. P1, P2, and P3 indicate PANK1,
PANK2, and PANK3, respectively; pP2, iP2, and mP2
indicate precursor, immature, and mature PANK2, respectively. SCR,
scrambled negative control siRNA duplex; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; PANK2,
pantothenate kinase 2; HUVECs, human umbilical vascular endothelial
cells; ATP, adenosine triphosphate; HPRT1, hypoxanthine guanine
phosphoribosyl transferase 1; siRNA, small interfering RNA; CoA,
Coenzyme A. |
Then, we moved to the analysis of the angiogenic
properties of control and silenced HUVECs. Firstly, we evaluated
single cell motility by time-lapse video-microscopy. 24 h after the
transfection, 5,000 cells/well were seeded in triplicate in a
12-well plate, stimulated with either 30 ng/ml VEGF, or 10 or 2%
FBS and observed under an inverted photomicroscope. Phase-contrast
photographs were taken every 10 min for 5 h and the migration
pattern of at least 30 cells/well was determined by the ‘Chemotaxis
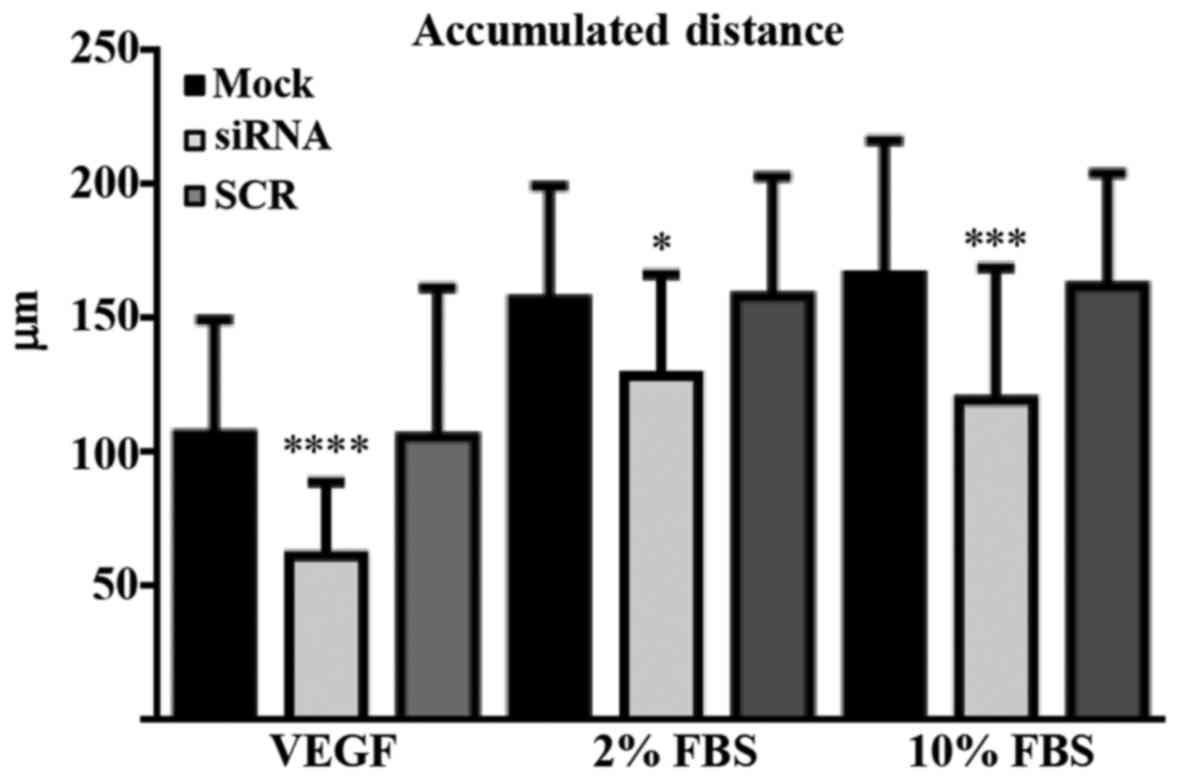
and Migration Tool’ of ImageJ Software. As shown in Fig. 2A, the accumulated distance of
PANK2-silenced HUVECs treated with VEGF was greatly reduced
(58%) as compared to those of mock- and SCR-transfected cells
(P<0.001). Similar results were obtained when cells were treated
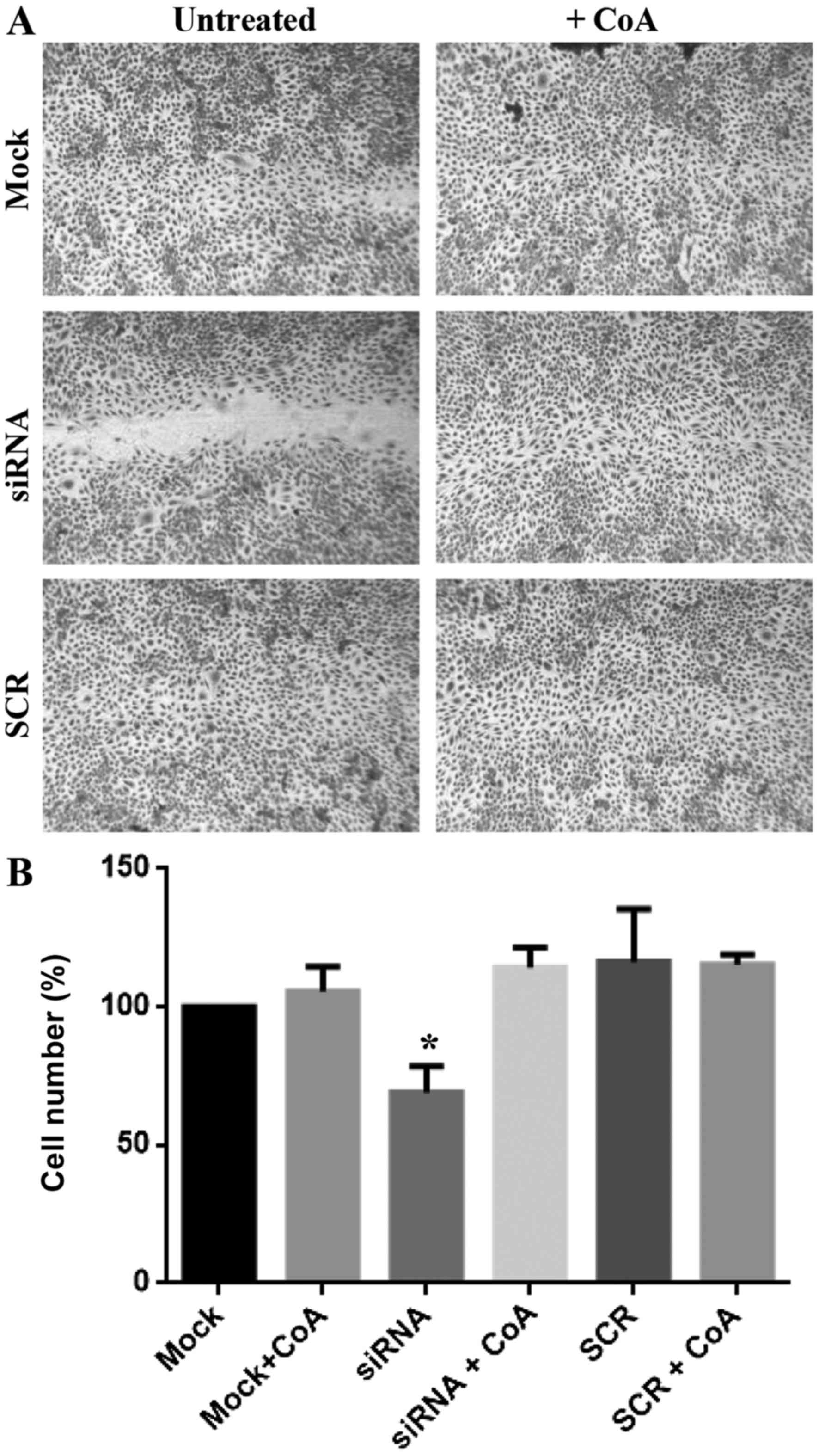
with 10 or 2% FBS instead of VEGF (Fig. 2). Subsequently, we analyzed the
collective migration performance of PANK2-deficient cells by the
wound healing scratch assay. As shown in Fig. 3A, the wound was hardly visible in
the wells with mock- and SCR-treated cells after 10 h of
incubation, whereas it was still evident in the wells of
PANK2-silenced HUVECs. We counted the cells migrated towards
the wound and found that much fewer cells (reduction of
approximately 30%) invaded the region of the wound at the end of
the incubation when silenced for PANK2 in comparison to
mock- and SCR-treated cells (Fig.
3B). The treatment with 25 µM CoA restored the normal migration
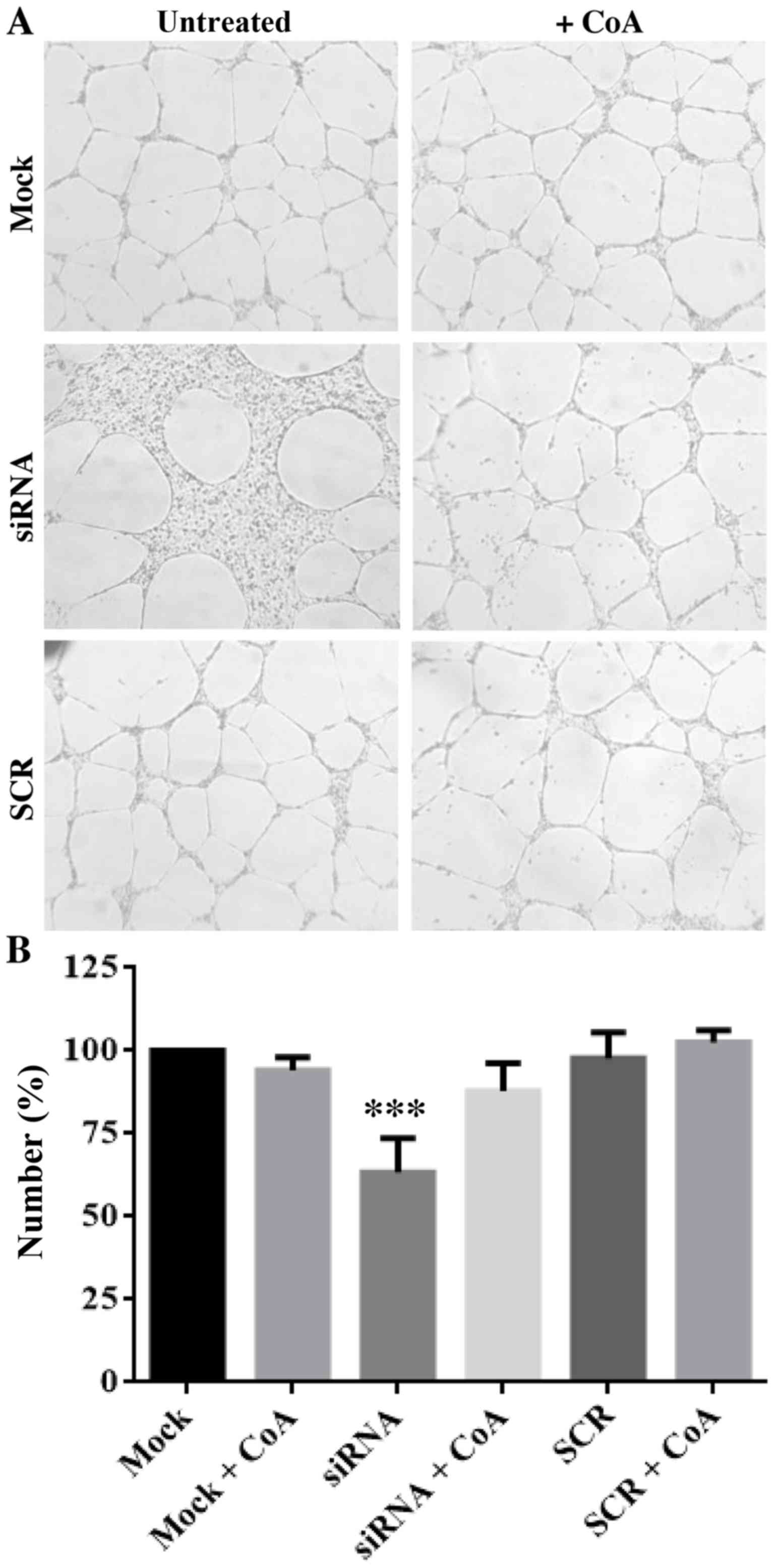
also in this case. Finally, we evaluated the capacity of the cells
to form capillary-like structures in a three-dimensional gel (tube
formation assay). We seeded 10,000 cells in triplicate in wells
coated with Matrigel and counted the number of areas fully
delimited by cell cords/tubes (empty areas) after 20 h of
incubation at 37°C. Once again, we observed a net reduction
(approximately 40%, P<0.001) in the number of empty areas
delimited by PANK2-silenced cells, suggesting that
downregulation of PANK2 inhibits not only the motility of
HUVECs, but also their ability to reorganize in tube-like
structures. Again, the addition of CoA to the medium restored the
values observed in control and SCR-treated cells (Fig. 4). To investigate possible molecular
mechanism linking the silencing of PANK2 with the defects of
the angiogenic properties of HUVECs, we analyzed the expression
level of different genes known to play a major role in the
proliferative/migration response of endothelial cells by using
RT-qPCR. We did not detect any difference in the level of CD31, a
typical marker of endothelial cells (Fig. 5A), thus indicating the conservation
of main endothelial features in transfected cells. The relative
mRNA amount of two major receptors involved in the response to
proliferative stimuli, Vascular Endothelial Growth Factor Receptor
2 (VEGFR2) and Fibroblast Growth Factor receptor 1 (FGFR1) was also
unchanged (Fig. 5C and D).
Interestingly, we detected a moderate decrease of VE-CADHERIN
expression level (Fig. 5B), which
was approximately 60% of that of mock- and SCR-treated cells
(P<0.05).
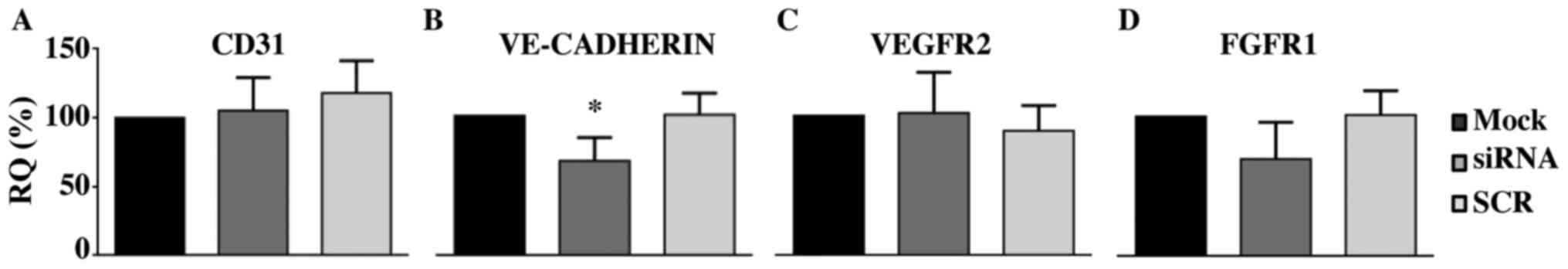 | Figure 5.Silencing of PANK2 decreases
VE-CADHERIN expression in human umbilical vascular endothelial
cells. Silenced cells were collected 48 h post-transfection, RNA
was extracted and the expression of endothelial markers and
proliferative factors were analyzed by reverse
transcription-quantitative polymerase chain reaction. Hypoxanthine
guanine phosphoribosyl transferase 1 was used the reference gene.
The relative expression of each target mRNA was calculated as a
percentage of that obtained in the mock-treated cells. (A) CD31,
(B) VE-CADHERIN, (C) VEGFR2 and (D) FGFR1. Data are presented as
the mean ± standard deviation of three independent experiments.
*P<0.05 vs. Mock. PANK2, pantothenate kinase 2; siRNA, small
interfering RNA; SCR, scrambled negative control siRNA duplex;
CD31, cluster of differentiation; VE-CADHERIN, vascular endothelial
cadherin; VEGFR2, vascular endothelial growth factor receptor 2;
FGFR1, fibroblast growth factor receptor 1; RQ, relative
quantification. |
Discussion
Mutations in PANK2 lead to the development of
PKAN, a movement disorder included in the NBIA category. It is
highly probable that a shortage of CoA represents the initial
pathogenic trigger, even though direct measurements in patients are
still missing. CoA and its acyl-derivatives are fundamental
cofactors for a large number of biochemical reactions in all types
of cells and tissues; it is remarkable and poorly understood that
the clinical features of PKAN are essentially limited to the
central nervous system. When we observed severe perturbations of
vasculature development in zebrafish embryos with downregulation of
pank2 expression, we reasoned that it could be a feature
limited to this experimental model. Zebrafish embryos develop
externally and essentially rely on the inherited pool of vitamin B5
and CoA for many days of growth. Minor perturbations in this
delicate balance may induce more severe consequences in Danio
rerio embryos than in other systems, where external supply of
these metabolites may support compensatory mechanisms. At the same
time, assuming that human and zebrafish pantothenate kinases have
similar biochemical properties, the relevance of pank2 in CoA
biosynthesis could be increased in zebrafish embryos due to a
limited reservoir of acetyl-CoA, a potent inhibitor of PANK2
activity (13). Nonetheless, we
investigated the effects of PANK2 silencing in mammalian
cells and found that the reduction of PANK2 protein severely
affects the angiogenetic properties of HUVECs. Different types of
analysis evidenced defects in migration and cord formation
capability in PANK2-silenced cells as compared to control.
This was associated with features, such as lower proliferation rate
and energetic defects, which are commonly observed in other cells
upon PANK2 suppression (16,26,28).
The addition of CoA to the medium immediately after the
transfection prevented all these alterations efficiently,
indirectly indicating a selective decrease in CoA availability. It
could well be that the defect in HUVECs angiogenic properties is a
direct consequence of the mitochondrial dysfunction (MTT assay) and
the lower amount of ATP, but recent studies pinpoint a more direct
role of acetyl-CoA and fatty acid oxidation (FAO) in controlling
angiogenesis and cell differentiation, even in the absence of
significant energetic defects. HUVECs cultured in charcoal-stripped
serum (CSS) showed impaired proliferation and angiogenic
properties, which were largely restored by treatment with palmitic
acid or acetyl-CoA precursor acetate (29). The same molecules reestablished
normal cell proliferation and sprouting activity in ECs (30) and lymphangiogenesis in lymphatic
ECs (31), suffering from
pharmacological or genetic blockage of FAO. Thus, it seems
plausible that acetyl-CoA (and indirectly CoA), exerts a control on
cell fate and activity through the direct connection with fatty
acid metabolism and independent of energetic changes. Since PANK2
represents an important link between acetyl-CoA levels and FAO
(13), its loss of function may
have direct consequences on cellular programs controlling
proliferation and differentiation. Mechanisms such as altered
nucleotide production (30) and
modification of histone acetylation (31) may contribute to this phenomenon. We
documented a reduction of VE-CADHERIN mRNA levels in silenced
HUVECs. We observed a similar reduction, together with that of
fli1, in pank2 and coasy morphants (17,20).
It is known that the decrease of cellular CoA availability affects
histones 3 and 4 acetylation (27); this could result in modification of
transcriptional processes and lower mRNA level of VE-CADHERIN. A
thorough analysis of the transcriptional changes associated with
PANK2 defects could shed more light onto downstream, synergistic
processes participating in disease development.
Alterations in angiogenesis may concur to
neurodegenerative processes (32)
and represent a potential target for therapeutic strategies
(33). Our data seem to support
such an involvement in PKAN or CoPAN. The replication in more
appropriate experimental models for brain microvasculature
endothelial cells as well as in other systems such as
Pank2−/− mice and endothelial cells derived from
patients induced Pluripotent Stem cells could be ways to further
investigate this hypothesis. Albeit limited by the extreme paucity
of cases, the histological analysis of brain samples from patients
could provide more definitive arguments. Noteworthy, the globus
pallidus of PKAN patients presents numerous proteinaceous
aggregates enriched with ubiquitin and apolipoprotein E and most
probably representing remnants of degenerated neurons (34). Similar lesions were detected in
non-PKAN brains at and around areas of infarct involving large
GABAergic neurons, such as Purkinje cells or those found in the
globus pallidus and the substantia nigra, pars reticulata. These
aggregates represent ischemic injuries and may suggest the
involvement of hypoxia in PKAN pathogenesis. Metabolic changes in
both neuronal and endothelial cells with altered CoA biosynthesis
and eventually microvasculature defects may concur to the
establishment of a hypoxic condition that leads to neuronal cell
death and neurodegeneration. Both cellular (large GABAergic neurons
with long axonal extension) and tissue [poor vascularization
(35), high iron content] features
may contribute to the specific susceptibility of the globus
pallidus to reduced CoA availability due to PANK2 mutations.
Beside this initial indication regarding the
implication for vasculature development and maintenance, our
analysis strengthens once again the essential role of PANK2 in
cellular CoA homeostasis. Like neurons, HUVECs express different
isoforms of PANK, with PANK2 and PANK3 being the most abundant at
the mRNA level. With the intent to limit any possible off-target
effects, we applied low doses of siRNA and obtained approximately
75 and 60% reduction in mRNA and protein level, respectively. This
is clearly a limit of our approach: the residual enzymatic activity
could mitigate the severity of the phenotype and facilitate the
rescue efficiency of CoA. Nonetheless, the phenotypic changes we
observed were highly reproducible, suggesting that neither PANK3
nor PANK1 can compensate for PANK2 defects in HUVECs. Even though
partial explanation comes from the specific localization of PANK2
in the mitochondria and the differences in the regulatory
properties of the enzyme (13),
this aspect remains poorly understood and its comprehension could
provide more insight into the specific neuronal vulnerability and
the mechanisms underpinning the neurodegenerative process. To this
aim, we are editing the genome of mammalian cells and fish to
generate models with PANK2 null alleles. They will be
important tools to investigate cellular mechanisms of CoA
deficiency and possible correction strategies.
Acknowledgements
The authors are grateful to Dr Elisabetta Crescini
(University of Brescia, Brescia, Italy) for their technical
assistance with the isolation and culture of HUVECs, and to
Professor Patrizia Dell'Era (University of Brescia) for assisting
with some of the experiments.
Funding
The present study was supported by Health and Wealth
(ZEBRACA) and ex60% funds from University of Brescia.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding authors on reasonable
request.
Authors' contributions
FP designed and performed the experiments, analyzed
the data, and wrote and revised the manuscript. AT, DK, EG and SM
assisted with the study design, performed some of the experiments
and revised the manuscript. DZ and DF conceived and designed the
experiments, analyzed the data, prepared the figures, and wrote and
revised the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zhou B, Westaway SK, Levinson B, Johnson
MA, Gitschier J and Hayflick SJ: A novel pantothenate kinase gene
(pank2) is defective in hallervorden-spatz syndrome. Nat Genet.
28:345–349. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Angelini L, Nardocci N, Rumi V, Zorzi C,
Strada L and Savoiardo M: Nhallervorden-spatz disease: Clinical and
mri study of 11 cases diagnosed in life. J Neurol. 239:417–425.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swaiman KF: Hallervorden-Spatz syndrome
and brain iron metabolism. Arch Neurol. 48:1285–1293. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hayflick SJ, Westaway SK, Levinson B, Zhou
B, Johnson MA, Ching KH and Gitschier J: Genetic, clinical, and
radiographic delineation of hallervorden-Spatz syndrome. N Engl J
Med. 348:33–40. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hayflick SJ, Hartman M, Coryell J,
Gitschier J and Rowley H: Brain MRI in neurodegeneration with brain
iron accumulation with and without PANK2 mutations. Ajnr Am J
Neuroradiol. 27:1230–1233. 2006.PubMed/NCBI
|
|
6
|
Abiko Y: Investigations on pantothenic
acid and its related compounds. IX. Biochemical studies.4.
Separation and substrate specificity of pantothenate kinase and
phosphopantothenoylcysteine synthetase. J Biochem. 61:290–299.
1967. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dansie LE, Reeves S, Miller K, Zano SP,
Frank M, Pate C, Wang J and Jackowski S: Physiological roles of the
pantothenate kinases. Biochem Soc Trans. 42:1033–1036. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hörtnagel K, Prokisch H and Meitinger T:
An isoform of hPANK2, deficient in pantothenate kinase-associated
neurodegeneration, localizes to mitochondria. Hum Mol Genet.
12:321–327. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brunetti D, Dusi S, Morbin M, Uggetti A,
Moda F, D'Amato I, Giordano C, d'Amati G, Cozzi A, Levi S, et al:
Pantothenate kinase-associated neurodegeneration: Altered
mitochondria membrane potential and defective respiration in Pank2
knock-out mouse model. Hum Mol Genet. 21:5294–5305. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alfonso-Pecchio A, Garcia M, Leonardi R
and Jackowski S: Compartmentalization of mammalian pantothenate
kinases. PLoS One. 7:e495092012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kotzbauer PT, Truax AC, Trojanowski JQ and
Lee VM: Altered neuronal mitochondrial coenzyme A synthesis in
neurodegeneration with brain iron accumulation caused by abnormal
processing, stability, and catalytic activity of mutant
pantothenate kinase 2. J Neurosci. 25:689–698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leonardi R, Zhang YM, Lykidis A, Rock CO
and Jackowski S: Localization and regulation of mouse pantothenate
kinase 2. FEBS Lett. 581:4639–4644. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leonardi R, Rock CO, Jackowski S and Zhang
YM: Activation of human mitochondrial pantothenate kinase 2 by
palmitoylcarnitine. Proc Natl Acad Sci USA. 104:1494–1499. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levi S and Finazzi D: Neurodegeneration
with brain iron accumulation: Update on pathogenic mechanisms.
Front Pharmacol. 5:992014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aoun M and Tiranti V: Mitochondria: A
crossroads for lipid metabolism defect in neurodegeneration with
brain iron accumulation diseases. Int J Biochem Cell Biol.
63:25–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zizioli D, Tiso N, Guglielmi A, Saraceno
C, Busolin G, Giuliani R, Khatri D, Monti E, Borsani G, Argenton F
and Finazzi D: KNock-down of pantothenate kinase 2 severely affects
the development of the nervous and vascular system in zebrafish,
providing new insights into PKAN disease. Neurobiol Dis. 85:35–48.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rana A, Seinen E, Siudeja K, Muntendam R,
Srinivasan B, van der Want JJ, Hayflick S, Reijngoud DJ, Kayser O
and Sibon OC: Pantethine rescues a Drosophila model for
pantothenate kinase-associated neurodegeneration. Proc Natl Acad
Sci USA. 107:6988–6993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Srinivasan B, Baratashvili M, van der
Zwaag M, Kanon B, Colombelli C, Lambrechts RA, Schaap O, Nollen EA,
Podgoršek A, Kosec G, et al: Extracellular 4′-phosphopantetheine is
a source for intracellular coenzyme A synthesis. Nat Chem Biol.
11:784–792. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Di Meo I, Colombelli C, Srinivasan B, de
Villiers M, Hamada J, Jeong SY, Fox R, Woltjer RL, Tepper PG,
Lahaye LL, et al: Acetyl-4′-phosphopantetheine is stable in serum
and prevents phenotypes induced by pantothenate kinase deficiency.
Sci Rep. 7:112602017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Khatri D, Zizioli D, Tiso N, Facchinello
N, Vezzoli S, Gianoncelli A, Memo M, Monti E, Borsani G and Finazzi
D: Down-regulation of coasy, the gene associated with NBIA-VI,
reduces Bmp signaling, perturbs dorso-ventral patterning and alters
neuronal development in zebrafish. Sci Rep. 6:376602016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dusi S, Valletta L, Haack TB, Tsuchiya Y,
Venco P, Pasqualato S, Goffrini P, Tigano M, Demchenko N, Wieland
T, et al: Exome sequence reveals mutations in CoA synthase as a
cause of neurodegeneration with brain iron accumulation. Am J Hum
Genet. 94:11–22. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Annesi G, Gagliardi M, Iannello G and
Quattrone A, Iannello G and Quattrone A: Mutational analysis of
COASY in an italian patient with NBIA. Parkinsonism Relat Disord.
28:150–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grillo E, Ravelli C, Corsini M,
Ballmer-Hofer K, Zammataro L, Oreste P, Zoppetti G, Tobia C, Ronca
R, Presta M and Mitola S: Monomeric gremlin is a novel vascular
endothelial growth factor receptor-2 antagonist. Oncotarget.
7:35353–35368. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gatta LB, Vitali M, Verardi R, Arosio P
and Finazzi D: Inhibition of heme synthesis alters amyloid
precursor protein processing. J Neural Transm (Vienna). 116:79–88.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative pcr and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Poli M, Derosas M, Luscieti S, Cavadini P,
Campanella A, Verardi R, Finazzi D and Arosio P: Pantothenate
kinase-2 (Pank2) silencing causes cell growth reduction,
cell-specific ferroportin upregulation and iron deregulation.
Neurobiol Dis. 39:204–210. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Siudeja K, Srinivasan B, Xu L, Rana A, de
Jong J, Nollen EA, Jackowski S, Sanford L, Hayflick S and Sibon OC:
Impaired Coenzyme A metabolism affects histone and tubulin
acetylation in Drosophila and human cell models of pantothenate
kinase associated neurodegeneration. Embo Mol Med. 3:755–766. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Campanella A, Privitera D, Guaraldo M,
Rovelli E, Barzaghi C, Garavaglia B, Santambrogio P, Cozzi A and
Levi S: Skin fibroblasts from pantothenate kinase-associated
neurodegeneration patients show altered cellular oxidative status
and have defective iron-handling properties. Hum Mol Genet.
21:4049–4059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vanetti C, Bifari F, Vicentini LM and
Cattaneo MG: Fatty acids rather than hormones restore in vitro
angiogenesis in human male and female endothelial cells cultured in
charcoal-stripped serum. PLoS One. 12:e01895282017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schoors S, Bruning U, Missiaen R, Queiroz
KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia
J, et al: Fatty acid carbon is essential for dNTP synthesis in
endothelial cells. Nature. 520:192–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wong BW, Wang X, Zecchin A, Thienpont B,
Cornelissen I, Kalucka J, García-Caballero M, Missiaen R, Huang H,
Brüning U, et al: The role of fatty acid β-oxidation in
lymphangiogenesis. Nature. 542:49–54. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brown WR and Thore CR: Review: Cerebral
microvascular pathology in ageing and neurodegeneration.
Neuropathol Appl Neurobiol. 37:56–74. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Carmeliet P and Ruiz de Almodovar C: VEGF
ligands and receptors: Implications in neurodevelopment and
neurodegeneration. Cell Mol Life Sci. 70:1763–1778. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Woltjer RL, Reese LC, Richardson BE, Tran
H, Green S, Pham T, Chalupsky M, Gabriel I, Light T, Sanford L, et
al: Pallidal neuronal apolipoprotein E in pantothenate
kinase-associated neurodegeneration recapitulates ischemic injury
to the globus pallidus. Mol Genet Metab. 116:289–297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wolfram-Gabel R and Maillot C: Vascular
networks of the nucleus lentiformis. Surg Radiol Anat. 16:373–377.
1994. View Article : Google Scholar : PubMed/NCBI
|