Introduction
Cardiovascular disease remains the leading cause of
morbidity and mortality in patients with chronic kidney disease
(CKD), particularly in patients who elect to undergo dialysis
treatment (1,2). Indoxyl sulfate (IS) is a typical
protein-bound uremic toxin, which is challenging to remove using
current dialysis techniques, and serves a central role in cardiac
damage among patients with CKD (3). The authors' previous clinical
research suggested that IS is associated with heart failure in
patients undergoing hemodialysis (4). However, the mechanism underlying
IS-induced cardiac tissue damage has yet to be fully revealed.
Research has revealed that IS promotes cardiomyocyte hypertrophy
and cardiac fibrosis via enhanced oxidative stress (5), and suppresses anti-oxidative
mechanisms (6). Studies have also
demonstrated that prolonged endoplasmic reticulum (ER) stress may
serve an important role in cardiomyocyte hypertrophy and apoptosis
(7–9). The ER is the cellular compartment in
which proteins are synthesized and acquire the correct
three-dimensional configuration needed for biological activity.
When cells synthesize secretory proteins in quantities that exceed
the capacity of the ER, unfolded proteins accumulate in the ER,
inducing ER stress (ERS). In response to ERS, various ER
chaperones, including glucose-regulated protein 78 (GRP78), are
upregulated. When ERS is excessive and/or prolonged, the initiation
of apoptosis is promoted by the transcriptional induction of
CCAAT-enhancer-binding homologous protein (CHOP), and/or by the
activation of c-Jun N-terminal kinase (JNK)-dependent pathways
(10).
In the current study, the authors sought to examine
whether IS causes cell apoptosis, and whether ERS participates in
the molecular mechanism underlying IS-induced cardiac toxicity.
Materials and methods
Cells, culture and reagents
H9C2 cardiomyocytes (Type Culture Collection of the
Chinese Academy of Sciences, Shanghai, China) were taken from
liquid nitrogen and cultivated in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), and maintained at 37°C in a humidified
atmosphere containing 5% CO2. For different experiments,
cells were seeded into 6-cm dishes or 6-well plates, or onto
12-mm-diameter glass coverslips in 24-well plates. IS and
4-phenylbutyric acid (4-PBA) were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany).
Experimental groups
To evaluate whether IS induces apoptosis in H9C2
cardiomyocytes, cells were exposed to increasing concentrations of
IS (500, 1,000, and 2,000 µM) for 24 h, and apoptosis was detected
by flow cytometry. To determine whether IS-induced apoptosis is
associated with ERS, cells were divided into 4 groups: i) Control
group (n=4): Cells were treated with serum-free DMEM for 48 h,
culture medium was changed after the first 24 h; ii) PBA group
(n=4): Cells were treated with PBA (1 mM) for 24 h, and then
cultured with DMEM for 24 h; iii) IS group (n=4): Cells were
treated with DMEM for 24 h, and then treated with IS (1 mM) for 24
h; and iv) PBA+IS group (n=4): Cells were pretreated with PBA (1
mM) for 24 h, prior to IS (1 mM) treatment for 24 h. Serum-free
DMEM was used in all experimental groups. The present study
determined 1 mM to be the optimal concentration of PBA, according
to a previous study (11), and
determined 1 mM to be the optimal concentration of IS with respect
to its induction of apoptosis and ERS, which is presented in the
results.
Flow cytometry
An apoptosis kit with Annexin-V Alexa Fluor™ 488 and
propidium iodide (PI; Thermo Fisher Scientific, Inc.) was used to
evaluate cell apoptosis. Cells were washed with cold PBS and
binding buffer, and then stained with AnnexinV-FITC for 20 min at
4°C, followed by PI for 5 min per sample. Data were acquired using
an Attune NxT Flow Cytometer (Thermo Fisher Scientific, Inc.), and
FlowJo software (v.14.0; TreeStar, Inc. San Carlos, CA, USA) was
used for data analysis.
Western blot assay
Protein samples from H9C2 cells were lysed in radio
immunoprecipitation assay buffer (catalog no. KGP702, Keygen,
Jiangsu, China). Lysates were centrifuged at 12,000 × g (4°C) for 5
min. Supernatants were collected, and equal amount of proteins (50
µg per sample) were subjected to 10% SDS-PAGE, followed by
transference to Immobilon-PVDF membranes (Merck KGaA, Darmstadt,
Germany) for 90 min at 110 V on ice. Membranes were incubated in 5%
non-fat milk for 60 min at room temperature, and then reacted
overnight at 4°C with specific antibodies: Anti-GRP78 (1:1,000,
catalog no. 3183), anti-CHOP (1:1,000, catalog no. 2895),
anti-phospho (p)-stress activated protein kinase (SAPK)/JNK
(1:1,000, catalog no. 9255), anti-SAPK/JNK (1:1,000, catalog no.
9252), anti-cleaved-caspase 3 (1:1,000, catalog no. 9664) and
anti-caspase3 (1:1,000, catalog no. 9662) were purchased from Cell
Signaling Technology, Inc. Danvers, MA, USA. The membranes were
rinsed in PBS 3 times (10 min each time), then incubated with the
peroxidase-conjugated AffiniPure Goat anti-rabbit IgG antibody
(1:10,000, catalog no. 111-035-003, Jackson ImmunoResearch
Laboratories Inc., PA, USA) and peroxidase-conjugated AffiniPure
Goat anti-mouse IgG antibody (1:10,000, catalog no. 115-035-003,
Jackson ImmunoResearch Laboratories Inc., PA, USA) for 1 h at room
temperature. Then, the membranes were rinsed 3 times (10 min each
time) in PBS. Proteins were visualized by enhanced chemiluminescent
reagents (Thermo Scientific, Inc.), and band intensity was analyzed
using program Image-J software (v.1.4.3.67; National Center for
Biotechnology Information).
Statistical analysis
All experiments were performed at least in
triplicate. Data are presented as the mean ± standard deviation.
Statistical differences were determined using analysis of variance
coupled with Dunnett test or Tuckey's test. Statistical analysis
was performed using SPSS software, version 22.0 (IBM SPSS, Armonk,
NY, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
IS stimulation induces apoptosis and
ERS
Treatment of H9C2 cells was conducted using an IS
concentration range of 0–2,000 µM, and cells were collected for
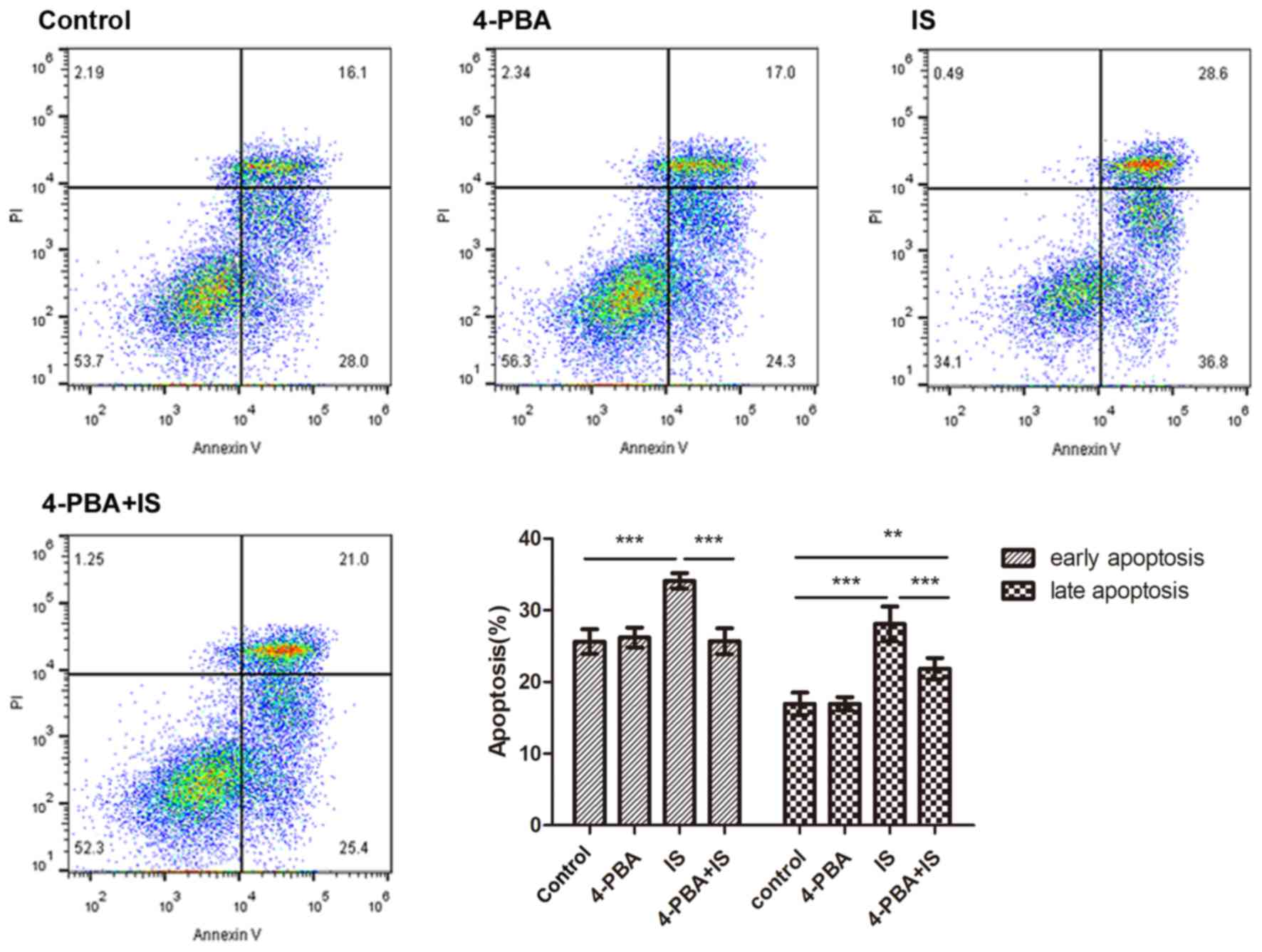
apoptosis evaluation. IS (1,000–2,000 µM) significantly stimulated
both early and late apoptosis (Fig.
1). Treatment of H9C2 cells occurred at IS concentrations
ranging from 0–1,000 µM, and cells were collected for western blot
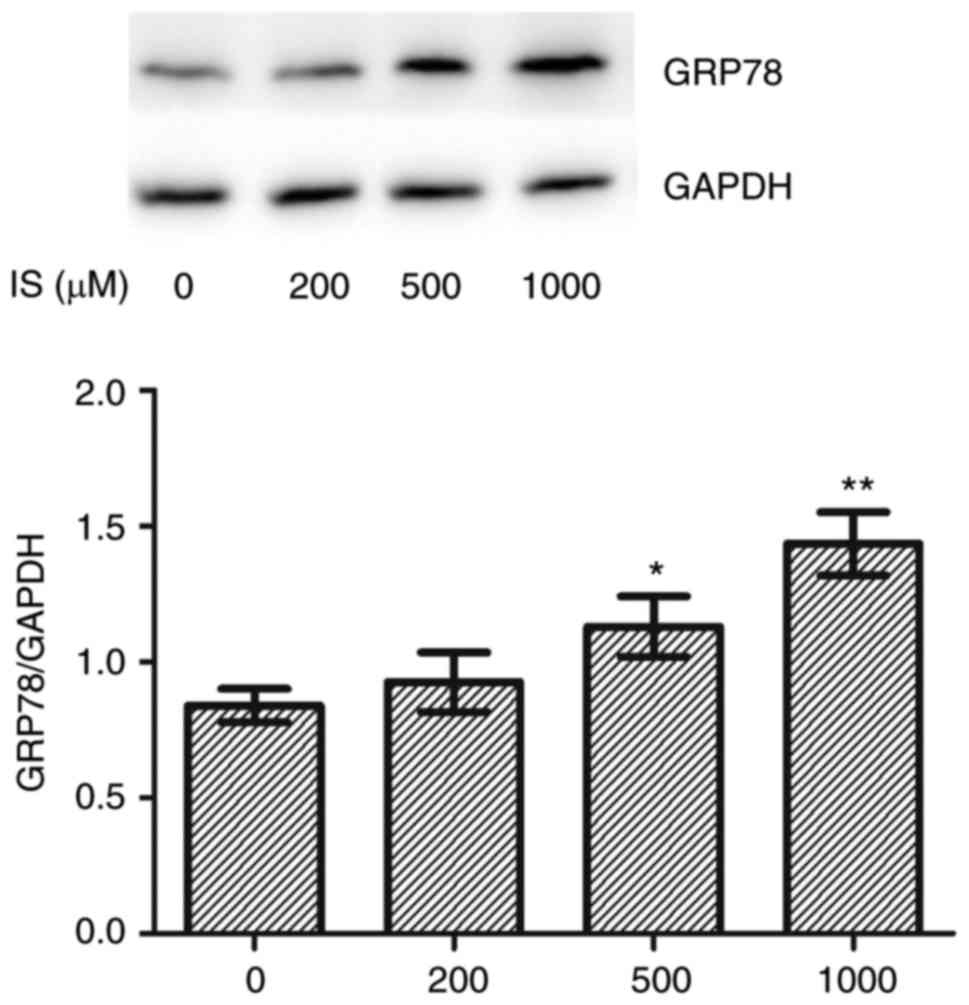
analysis. Compared with the control group, the expression of the ER
chaperone GRP78 was significantly increased in the IS 500 µΜ
(P<0.05) and 1,000 µM (P<0.01) treated groups (Fig. 2). IS induced both apoptosis and ERS
at a concentration of 1,000 µM. Therefore, 1,000 µM IS was used for
subsequent experiments.
Apoptosis of H9C2 is associated with
ERS in the context of IS stimulation
H9C2 cardiomyocytes were divided into 4 groups: i)
Control group; ii) 4-PBA group; iii) IS group; and iv) 4-PBA+IS
group. Compared with the control group, early apoptosis was
significantly increased in the IS group (P<0.001), and early
apoptosis was significantly attenuated in the 4-PBA+IS group, when
compared with the IS group (P<0.001). A similar trend was
observed with the late apoptosis phase, as presented in Fig. 3. Compared with the control group,
the expression of cleaved-caspase-3 in the IS and 4-PBA+IS groups
was significantly increased. Compared with IS group, the expression
of cleaved-caspase-3 protein is significantly lower in 4-PBA+IS
group (Fig. 4).
 | Figure 4.Protein expression levels of GRP78,
CHOP, p-JNK and cleaved-caspase 3. The protein expression levels of
GRP78, CHOP, p-JNK and cleaved-caspase 3 were determined in H9C2
cardiomyocytes in groups 1 to 4 (control, 4-PBA, IS, and 4-PBA+IS)
(n=4). *P<0.05, **P<0.01, ***P<0.001. IS, indoxyl sulfate;
4-PBA, 4-phenylbutyric acid; GRP78, glucose-regulated protein 78;
p, phosphorylated; CHOP, CCAAT-enhancer-binding protein homologous
protein; JNK, c-Jun N-terminal kinase. |
JNK-CHOP signaling pathway activity
increases under IS stimulation
The protein expression levels of GRP78, CHOP and
p-JNK in each group are presented in Fig. 4. Compared with the control group,
the expression levels of GRP78 and CHOP in the IS and 4-PBA+IS
groups were significantly increased, whereas the phosphorylation of
JNK was only increased in the IS group. Compared with the IS group,
the protein expression levels of GRP78 and CHOP were significantly
decreased in the 4-PBA+IS group, as was the phosphorylation of JNK.
No statistically significant difference was observed in the
expression of these proteins between the control and the 4-PBA
groups.
Discussion
The mechanisms of cardiovascular disease in
hemodialysis patients are complex. Conventional risk factors, such
as hypertension and volume overload, are considered to play
prominent roles in uremic cardiomyopathy (12). However, previous research
demonstrated that a reduction of blood pressure has little effect
in preventing the development of uremic cardiac hypertrophy
(13). The authors hypothesized
that non-established risk factors, including uremic toxins, have
been underestimated and are important in the progression of
cardiovascular disease in CKD. IS is a typical protein-bound uremic
toxin that cannot be removed by current hemodialysis techniques,
and has been demonstrated to have a critical effect on the
prognosis of cardiovascular disease induced by CKD, both in
pre-clinical (5,14–16)
and clinical (4,17–19)
research. The conventional mechanism of pathological cardiac
hypertrophy involves β-adrenergic receptor-, α-adrenergic
receptor-, and Ca2+/calmodulin-related pathways, while
the key mechanism of IS-induced cardiac hypertrophy is the direct
induction of oxidative stress (5).
It is important to further investigate the mechanism of IS-induced
cardiomyocyte injury, and to explore potential therapeutic
targets.
There is plenty of evidence to suggest that the key
mechanism of IS cardiotoxicity is the induction of oxidative stress
(5,16,20).
ERS and oxidative stress are closely related in many physiological
and pathological conditions (21).
Protein misfolding in ERS conditions results in reactive oxygen
species production, and oxidative stress disturbs the ER redox
state and promotes protein misfolding (22). Therefore, the authors hypothesized
that ERS may also play an important role in the harmful effects of
IS. GRP78 is an ER molecular chaperone, and a specific marker of
ERS (10). The present study
analyzed the rate of apoptosis in H9C2 cardiomyocytes, and GRP78
protein expression, and it was demonstrated that IS
dose-dependently induced ERS, and promoted the apoptosis of H9C2
cardiomyocytes. Previous studies demonstrated that, Indoxyl sulfate
induces apoptosis of kidney mesangial cells (23), renal tubular cells (24,25)
and osteoblast cells (26), which
are in accordance with the present results. Importantly, the study
showed that the ERS modulator 4-PBA attenuated the effect of IS on
ERS and apoptosis induction, which indicated that the apoptosis
induced by IS treatment is likely via ERS induction.
It was further demonstrated that IS increased the
protein expression of GRP78, CHOP, and p-JNK, which could be
suppressed by the ER inhibitor 4-PBA, and this indicating that the
CHOP and JNK pathways may be associated with IS-induced ERS and
apoptosis. The results identified ERS-related apoptosis as a novel
mechanism in IS-induced cardiomyocyte toxicity, providing a
potentially valuable therapeutic avenue for the treatment of
IS-induced cardiovascular diseases.
However, various limitations of the present study
should be mentioned. It is difficult to accurately recapitulate the
conditions of CKD in vitro, as CKD is a long-term and
complicated pathological progress, and the authors only treated
cells with IS for 24 h in the present study. In addition, the
concentration of IS used in the study is higher than would be
expected in clinical circumstances, as a low level of IS would not
cause damage in cardiomyocytes in the timeframe of the experiment.
The authors intend to establish an animal model to further
investigate the role of IS in uremic cardiomyopathy and its
mechanism, particularly the effect on apoptosis and ERS.
In conclusion, the results suggested that IS, a
protein-bound uremic toxin, induces ERS-related apoptosis in H9C2
cardiomyocytes via the CHOP and JNK pathways, and the effects were
attenuated by the ERS modulator 4-PBA. The findings will aid the
further understanding of the mechanisms of IS cardiotoxicity. As it
is difficult to remove IS with current dialysis techniques, further
findings regarding the mechanism may contribute to the development
of a novel therapeutic strategy, particularly focused on ERS and
apoptosis reduction.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and
Technology Commission of Shanghai Municipal (grant no. 14DZ2260200,
the project of Shanghai Key Laboratory of Kidney and Blood
Purification); Shanghai Committee of Science and Technology, China
(grant no. 15DZ0503402); and the Shanghai Municipal Commission of
Health and Family Planning (grant no. 2014 ZY3-CCCX-3-3043).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XT and XSC performed the western blotting and flow
cytometry experiments. PZ contributed to the cell culture. FFX and
JT participated in experiment design and edited the manuscript. JZZ
and XQD designed the present study.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Herzog CA, Asinger RW, Berger AK, Charytan
DM, Diez J, Hart RG, Eckardt KU, Kasiske BL, McCullough PA, Passman
RS, et al: Cardiovascular disease in chronic kidney disease. A
clinical update from Kidney Disease: Improving global outcomes
(KDIGO). Kidney Int. 80:572–586. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Granata A, Clementi A, Virzi GM, Brocca A,
de Cal M, Scarfia VR, Zanoli L, Ronco C, Corrao S and Malatino L:
Cardiorenal syndrome type 4: From chronic kidney disease to
cardiovascular impairment. Eur J Intern Med. 30:1–6. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tan X, Cao X, Zou J, Shen B, Zhang X, Liu
Z, Lv W, Teng J and Ding X: Indoxyl sulfate, a valuable biomarker
in chronic kidney disease and dialysis. Hemodial Int. 21:161–167.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cao XS, Chen J, Zou JZ, Zhong YH, Teng J,
Ji J, Chen ZW, Liu ZH, Shen B, Nie YX, et al: Association of
indoxyl sulfate with heart failure among patients on hemodialysis.
Clin J Am Soc Nephrol. 10:111–119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yisireyili M, Shimizu H, Saito S, Enomoto
A, Nishijima F and Niwa T: Indoxyl sulfate promotes cardiac
fibrosis with enhanced oxidative stress in hypertensive rats. Life
Sci. 92:1180–1185. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin CY, Hsu YJ, Hsu SC, Chen Y, Lee HS,
Lin SH, Huang SM, Tsai CS and Shih CC: CB1 cannabinoid receptor
antagonist attenuates left ventricular hypertrophy and Akt-mediated
cardiac fibrosis in experimental uremia. J Mol Cell Cardiol.
85:249–261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rani S, Sreenivasaiah PK, Cho C and Kim
DH: Salubrinal alleviates pressure overload-induced cardiac
hypertrophy by inhibiting endoplasmic reticulum stress pathway. Mol
Cells. 40:66–72. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lu WW, Zhao L, Zhang JS, Hou YL, Yu YR,
Jia MZ, Tang CS and Qi YF: Intermedin1-53 protects against cardiac
hypertrophy by inhibiting endoplasmic reticulum stress via
activating AMP-activated protein kinase. J Hypertens. 33:1676–1687.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Logue SE, Cleary P, Saveljeva S and Samali
A: New directions in ER stress-induced cell death. Apoptosis.
18:537–546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshida H: ER stress and diseases. FEBS J.
274:630–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gao Y, Jia P, Shu W and Jia D: The
protective effect of lycopene on hypoxia/reoxygenation-induced
endoplasmic reticulum stress in H9C2 cardiomyocytes. Eur J
Pharmacol. 774:71–79. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taddei S, Nami R, Bruno RM, Quatrini I and
Nuti R: Hypertension, left ventricular hypertrophy and chronic
kidney disease. Heart Fail Rev. 16:615–620. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Siedlecki AM, Jin X and Muslin AJ: Uremic
cardiac hypertrophy is reversed by rapamycin but not by lowering of
blood pressure. Kidney Int. 75:800–808. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang K, Wang C, Nie L, Zhao X, Gu J and
Guan X: Klotho protects against indoxyl sulphate-induced myocardial
hypertrophy. J Am Soc Nephrol. 26:2434–2446. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adijiang A, Goto S, Uramoto S, Nishijima F
and Niwa T: Indoxyl sulphate promotes aortic calcification with
expression of osteoblast-specific proteins in hypertensive rats.
Nephrol Dial Transplant. 23:1892–1901. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang K, Xu X, Nie L, Xiao T, Guan X, He T,
Yu Y, Liu L, Huang Y, Zhang J and Zhao J: Indoxyl sulfate induces
oxidative stress and hypertrophy in cardiomyocytes by inhibiting
the AMPK/UCP2 signaling pathway. Toxicol Lett. 234:110–119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lin CJ, Liu HL, Pan CF, Chuang CK,
Jayakumar T, Wang TJ, Chen HH and Wu CJ: Indoxyl sulfate predicts
cardiovascular disease and renal function deterioration in advanced
chronic kidney disease. Arch Med Res. 43:451–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu M, Kim YJ and Kang DH: Indoxyl
sulfate-induced endothelial dysfunction in patients with chronic
kidney disease via an induction of oxidative stress. Clin J Am Soc
Nephrol. 6:30–39. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin CJ, Pan CF, Liu HL, Chuang CK,
Jayakumar T and Wang TJ: The role of protein-bound uremic toxins on
peripheral artery disease and vascular access failure in patients
on hemodialysis. Atherosclerosis. 225:173–179. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fujii H, Nishijima F, Goto S, Sugano M,
Yamato H, Kitazawa R, Kitazawa S and Fukagawa M: Oral charcoal
adsorbent (AST-120) prevents progression of cardiac damage in
chronic kidney disease through suppression of oxidative stress.
Nephrol Dial Transplant. 24:2089–2095. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kupsco A and Schlenk D: Oxidative stress,
unfolded protein response, and apoptosis in developmental toxicity.
Int Rev Cell Mol Biol. 317:1–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang WJ, Cheng MH, Sun MF, Hsu SF and Weng
CS: Indoxyl sulfate induces renin release and apoptosis of kidney
mesangial cells. J Toxicol Sci. 39:637–643. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ellis RJ, Small DM, Ng KL, Vesey DA,
Vitetta L and Francis RS: Indoxyl sulfate induces apoptosis and
hypertrophy in human kidney proximal tubular cells. Toxicol Pathol.
46:449–459. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim SH, Yu MA, Ryu ES, Jang YH and Kang
DH: Indoxyl sulfate-induced epithelial-to-mesenchymal transition
and apoptosis of renal tubular cells as novel mechanisms of
progression of renal disease. Lab Invest. 92:488–498. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim YH, Kwak KA, Gil HW, Song HY and Hong
SY: Indoxyl sulfate promotes apoptosis in cultured osteoblast
cells. BMC Pharmacol Toxicol. 14:602013. View Article : Google Scholar : PubMed/NCBI
|