Introduction
Numerous anticancer drugs have been used for cancer
chemotherapy, but safer and more effective molecular targeted
anticancer agents are needed. In the last decade, immune checkpoint
inhibitors and target therapies have been important and effective
therapies for many intractable malignant neoplasms including
progressive malignant melanoma. In 2002, it was demonstrated that
oncogenic BRAF mutations, predominantly at codon 600, are in
approximately 70% of cutaneous melanomas (1). The most common BRAF mutations
are V600E or V600K (2,3). These mutations lead to constitutive
activation of the mitogen-activated protein kinase (MAPK) pathway
and increased extracellular signal-related kinase (ERK) activation,
which drive the growth and differentiation of malignant cells
(4). BRAF inhibitors, such as
vemurafenib or dabrafenib, efficiently inhibit ERK activation and
tumour proliferation, which rapidly respond after the onset of
therapies (5,6). However, unfortunately, responses to
BRAF inhibitors are short-lived, with evidence of disease
progression within 6–8 months after the beginning of therapy
(7). To reduce resistance to BRAF
inhibitors, several combinatorial treatments using
mitogen-activated protein kinase kinase (MEK) inhibitors (8) or phosphoinositide 3-kinase (PI3K)
inhibitors (9) have proven
effective. In particular, the MEK inhibitor, trametinib, has been
clinically used with dabrafenib as a combination therapy. However,
more effective methods to treat BRAF inhibitor-resistant melanomas
are needed.
Sphingolipids are the main components of lipid rafts
and have crucial functions as signalling molecules. Sphingosine
1-phosphate (S1P) and ceramide regulate proliferation and apoptosis
(10–13). In response to various stimuli,
ceramide mediates cell death and apoptosis, whereas S1P abrogates
apoptosis and mediates cell proliferation and migration (14). Sphingosine kinase (SK) is the key
enzyme responsible for converting sphingosine to S1P. Signalling
pathways via the S1P receptor contribute to cancer cell survival
and proliferation (15), apoptosis
reduction (16), and oncogenic
transformation (17). Various
cancer cells have high levels of SK1 expression/activity, resulting
in their enhanced resistance to anticancer agents such as
anthracyclines, doxorubicin, and camptothecin (18,19).
Thus, SK1 may play an important role in the development and
proliferation of cancers such as melanoma (20–22).
Fingolimod (FTY720), an immune-suppressive drug developed by
chemical modification of myriocin and a metabolite of the fungus
Isaria sinclairii (23,24),
is phosphorylated by SK1 and SK2 (23). Phosphorylated FTY720 (FTY720-P) is
a structural analogue of S1P and binds to four S1P receptor
subtypes (S1P1, S1P3, S1P4, and
S1P5) (25). However,
persistent activation of S1P1 by FTY720-P causes its
internalization and degradation, thereby acting as a functional
antagonist in lymphocytes (26).
FTY720 also induces apoptosis in melanoma (27,28),
liver cancer (16), and breast
cancer (29) by direct inhibition
of SK1 (29). FTY720 has
predominantly been used for the treatment of multiple sclerosis
(30). Recent in vitro and
in vivo studies have shown that it is cytotoxic and
efficiently reduced the viability of ovarian (31), breast (32) and prostate (33) cancer cells.
We previously reported that combination treatment
with cisplatin and FTY720 has synergistic effects on apoptosis
induction in cisplatin-resistant melanoma cells (34). Therefore, in this study we
investigated the combined effects of FTY720 and vemurafenib in
established vemurafenib-resistant melanoma cells, as well as the
underlying molecular mechanisms.
Materials and methods
Chemical reagents
FTY720 was obtained from Cayman Chemical (Ann Arbor,
MI, USA). Vemurafenib, bovine serum albumin (BSA), Dulbecco's
modified Eagle's medium (DMEM), and Eagle's minimal essential
medium (EMEM) were purchased from Wako Pure Chemical Industries
(Osaka, Japan). RPMI-1640, non-essential amino acids (NEAA), and
dimethylsulfoxide (DMSO) were obtained from Sigma-Aldrich; Merck
KGaA (Darmstadt, Germany). Rabbit polyclonal antibodies against
SK1, p53, cleaved poly (adenosine diphosphate-ribose) polymerase
(PARP), PI3K, phosphorylated PI3K (p-PI3K), Akt, p-Akt, MEK, p-MEK,
ERK, p-ERK, S6 kinase (S6 K), mammalian target of rapamycin (mTOR),
p-mTOR, epidermal growth factor receptor (EGFR), and p-EGFR were
obtained from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Cell lines and cell viability
measurement
Human melanoma SK-Mel-28 cells were obtained from
JCRB Cell Bank (Osaka, Japan), and human melanoma cell lines (A375,
A2058, WM115) were purchased from the European Collection of Cell
Cultures. SK-Mel-28 and WM115 cells were cultured in RPMI-1640
containing 10% foetal bovine serum (FBS) and 0.1% tyrosine. A375
cells were cultured in DMEM containing 10% FBS. A2058 cells were
grown in EMEM containing 1% NEAA and 10% FBS. To assess viability,
melanoma cells were plated at a density of 5×103
cells/well in 96-well plates, and after 24 h vemurafenib and/or 3
µM FTY720 were added to the medium. Viability was analysed after
24, 48, 72 and 96 h using the standard
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliudm bromide
(MTT) assay, where viable cells with active metabolism, but not
dead cells, convert MTT into the corresponding purple-coloured
formazan. We found the most effective time to be 72 h on
vemurafenib in melanoma cells. When the determination of 50%
inhibitory concentrations (IC50) for vemurafenib was
performed in a 96-well plate, vemurafenib was added to the cells at
concentrations from 1 to 15 µM for 72 h in the presence or absence
of 3 µM FTY720. Cells treated with DMSO only were used as a
control, and IC50 values were calculated.
To prepare vemurafenib-resistant cells, SK-Mel-28
cells were repeatedly exposed to high doses of vemurafenib over a
period of 10 months as described by Stordal et al (35). When the cell cultures had undergone
approximately five doublings in the presence of 10 µM vemurafenib,
the proliferating cells were collected and transferred to drug-free
culture conditions for recovery and MTT assay was then performed.
The treatment was repeated at doses of 10 µM vemurafenib for 10
months and the most resistant clone, R-SK-Mel, was selected.
R-SK-Mel cells were grown in DMEM medium supplemented with 10 µM
vemurafenib, and before each experiment, the resistant cells were
cultured in the absence of the target drug for 24 h.
Western blot analysis
Cells were lysed by sonication in RIPA buffer [50 mM
Tris-HCL buffer (pH 7.6), 150 mM NaCl, 1% (w/v) NP-40, 0.5% (w/v)
sodium deoxycholate, protease inhibitor cocktail, and 0.1% (w/v)
sodium dodecyl sulfate (SDS)]. Total cell lysates (5 µg protein)
were separated by electrophoresis on 7.5–10% SDS-polyacrylamide
gels and transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked in 5%
BSA. Expression of each protein was measured by western blotting
with each antibody. Anti-β-actin antibody was used as the loading
control. After several washes, bound antibodies were detected using
the ECL western blotting detection system (Luminescent Image
Analyzer LAS-4000; Fujifilm, Tokyo, Japan). Protein band density
was determined with a densitometer (Multi gauge, version 3.1;
Fujifilm).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Microsoft Excel (Microsoft Corp., Redmond, WA, USA) was used to
calculate the correlation index. The statistical significance was
analysed by either Student's t-test or one-way factorial analysis
of variance with Fisher's protected least significant difference
post hoc test for multiple comparisons, using EZR version 1.35
software (Saitama Medical Center, Jichi Medical University,
Saitama, Japan). P<0.05 was considered to indicate a
statistically significant difference.
Results
Sensitivity of various melanoma cells
to vemurafenib
To examine the sensitivity of four melanoma cell
lines (SK-Mel-28, A375, A2058 and WM-115) to vemurafenib, cell
viability was measured after a 72 h incubation with vemurafenib.
The IC50 for vemurafenib was highest in WM-115 cells and
lowest in SK-Mel-28 cells (Fig.
1). The relative IC50 values for vemurafenib in
SK-Mel-28, A375, A2058 and WM-115 cells were 82±9.19, 145±24.7,
452±46.1 and 1227±336, respectively. Therefore, WM-115 and
SK-Mel-28 cells were used in subsequent experiments as
vemurafenib-resistant and vemurafenib-sensitive cell lines,
respectively.
Synergistic effects of FTY 720 and
vemurafenib combination treatment on apoptosis in
vemurafenib-resistant cells
To evaluate the effects of FTY720 on WM-115 cell
viability, we compared the viability of cells treated with
vemurafenib alone and the combination of vemurafenib and FTY720.
The IC50 of treatment with vemurafenib alone was reduced
by approximately 75% in the presence of 3 µM FTY720 (Fig. 2A). FTY720 alone at 3 µM had no
significant effects on viability (data not shown). These results
suggest that the addition of FTY720 to vemurafenib significantly
reduced viability compared to treatment with vemurafenib alone in
resistant WM-115 cells. The effects of FTY720 and vemurafenib on
the apoptosis of WM-115 cells were examined using western blotting
to measure the amount of PARP protein degradation, a marker of
apoptosis. In WM-115 cells, cleaved PARP protein was expressed by 2
µM vemurafenib alone, but not by 3 µM FTY720 alone (Fig. 2B). However, the combination
treatment with 2 µM vemurafenib and 3 µM FTY720 induced a
synergistic increase in PARP degradation (Fig. 2B). These results showed that the
addition of FTY720 to vemurafenib enhanced apoptosis compared with
vemurafenib alone in WM-115 cells.
 | Figure 2.(A) The effects of FTY720 on the
viability of WM-115 cells. WM-115 cells (1×104
cells/well) were incubated with various concentrations of
vemurafenib in the presence or absence of 3 µM FTY720 for 72 h,
then MTT assays were performed. Each condition was measured in
triplicate, and the data are expressed as the mean ± standard
deviation of three independent experiments. *P<0.05 and
**P<0.01 vs. vemurafenib+FTY720 at the corresponding
concentration. (B) The effects of FTY720 on the apoptosis of WM-115
cells. WM-115 cells were treated with 3 µM FTY720 and 2 µM
vemurafenib, alone or in combination for 48 h. Cell lysates were
subjected to western blot analysis with an antibody against cleaved
PARP, and the band intensity was measured. The results are
expressed as a percentage increase relative to the untreated
controls. The data are expressed as the mean ± standard deviation.
**P<0.01, as indicated. FTY720, fingolimod; PARP, poly
(adenosine diphosphate-ribose) polymerase; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliudm bromide. |
Changes in the expression of proteins
in cell signaling pathways and SK1 by combination treatment with
vemurafenib and FTY720 in vemurafenib-resistant cells
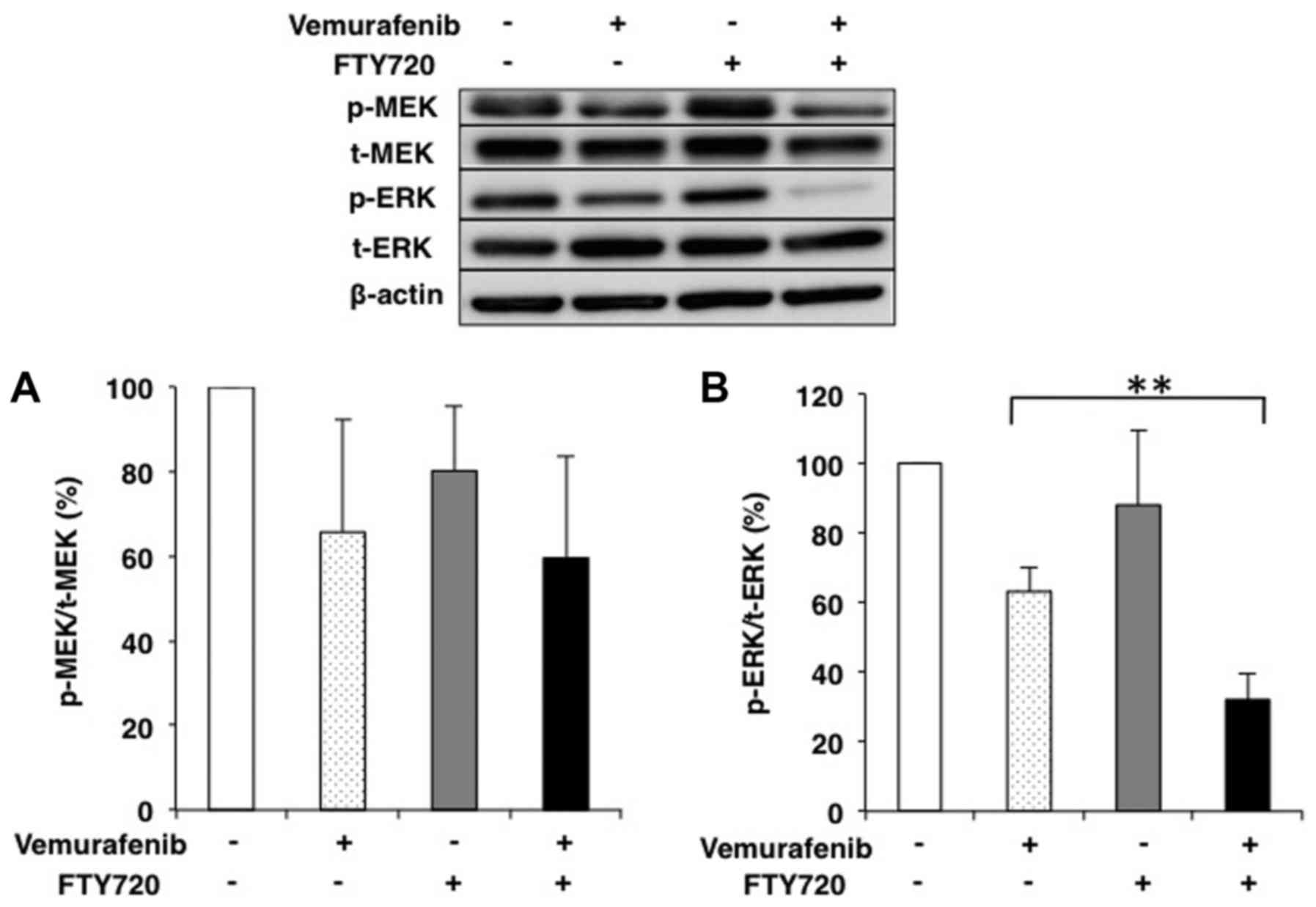
The effects of combination treatment with 2 µM
vemurafenib and 3 µM FTY720 on MEK and ERK phosphorylation in
WM-115 were evaluated by western blot analysis. The ratio of
phosphorylated to total MEK (p-MEK/t-MEK) was decreased by
combination treatment, but the difference was not significant
(Fig. 3A). In contrast, the ratio
of phosphorylated to total ERK (p-ERK/t-ERK) was remarkably
decreased by combination treatment compared with vemurafenib alone
(P<0.01; Fig. 3B). We
investigated the changes in the ratio of phosphorylated to total
Akt (p-Akt/t-Akt) after the treatment with 2 µM vemurafenib, 3 µM
FTY720 or both agents in WM-115 cells.
The treatment with 2 µM vemurafenib or 3 µM FTY720
alone induced no significant changes in Akt protein expression
(Fig. 4A). Conversely, the
treatment with combined FTY720 and vemurafenib caused a synergistic
decrease in t-Akt protein expression (Fig. 4A), whereas there were no
significant changes in p-Akt/t-Akt (Fig. 4B).
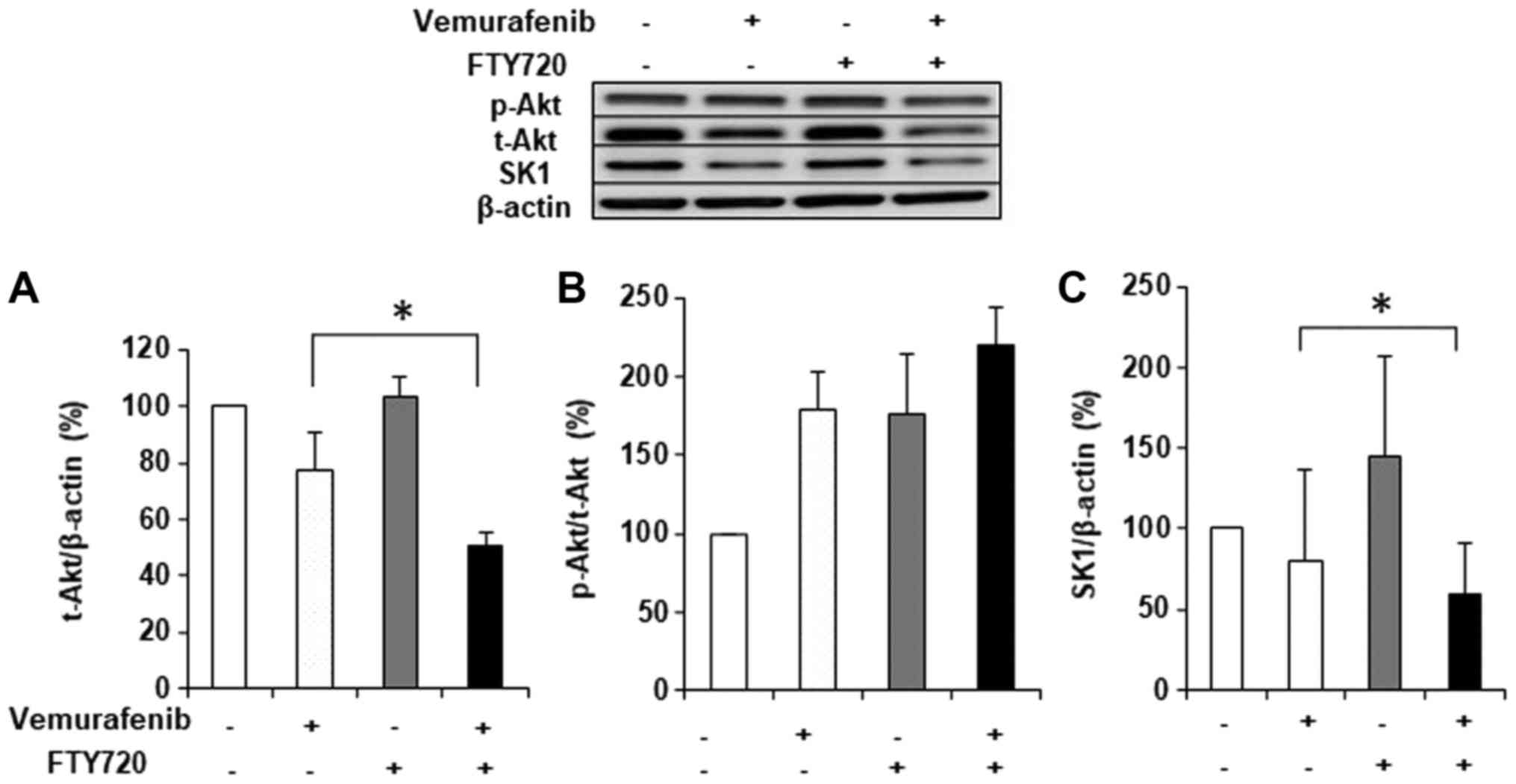 | Figure 4.Effects of FTY720 and vemurafenib on
the protein expression of (A and B) Akt and (C) SK1 in WM-115
cells. WM-115 cells were treated with 3 µM of FTY720 and 2 µM
vemurafenib, alone or in combination, for 48 h, and cell lysates
were subjected to western blot analysis with antibodies against the
total forms of (A) Akt and (C) SK1, and against the total (t) and
phosphorylated (p) forms of (B) Akt. β-actin was used as the
loading control. The results are expressed as ratios of treated to
untreated cells. Each condition was examined in triplicate, and
data are expressed as the means ± standard deviation. *P<0.05,
as indicated. FTY720, fingolimod; Akt, protein kinase B; SK1,
sphingosine kinase 1; p-, phosphorylated; t-, total. |
The effects of the combination treatment on SK1 with
2 µM vemurafenib and 3 µM FTY720 was evaluated by western blotting.
The expression of SK1 was significantly decreased with combination
treatment compared with vemurafenib treatment alone (Fig. 4C).
Establishing vemurafenib-resistant
R-SK-Mel cells and evaluating changes in the expression of cell
signaling proteins in these cells
To confirm the role of signalling proteins in the
sensitivity to vemurafenib and FTY720, we attempted preparation of
the vemurafenib-resistant cell line by treating the original
vemurafenib-sensitive SK-Mel-28 cells with a high concentration of
vemurafenib as described in the Materials and Methods.
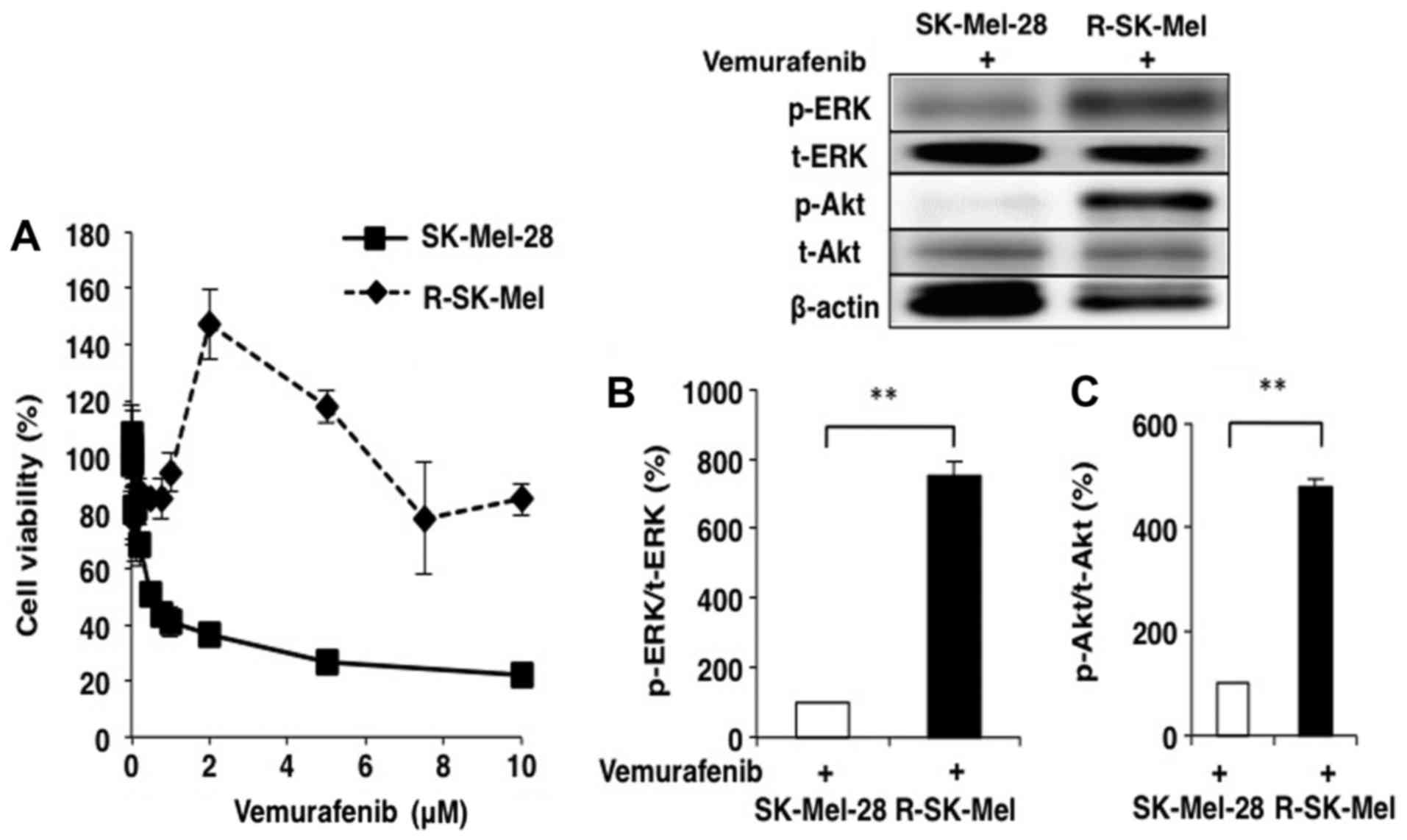
Consequently, we succeeded in establishing the
vemurafenib-resistant cell line R-SK-Mel, which can proliferate
even in the presence of 10 µM vemurafenib. As shown in Fig. 5A, the IC50 of
vemurafenib in R-SK-Mel cells was 50-fold higher than that in the
parent cell line. Changes in the expression of cell signaling
proteins was examined between the parent and resistant cells. The
expression levels of p-ERK/t-ERK and p-Akt/t-Akt were remarkably
increased in R-SK-Mel cells compared with those in parent SK-Mel-28
cells treated with 2 µM vemurafenib (Fig. 5B and C).
Synergistic effects of FTY 720 and/or
vemurafenib combination treatment on cell viability and apoptosis
in R-SK-Mel cells
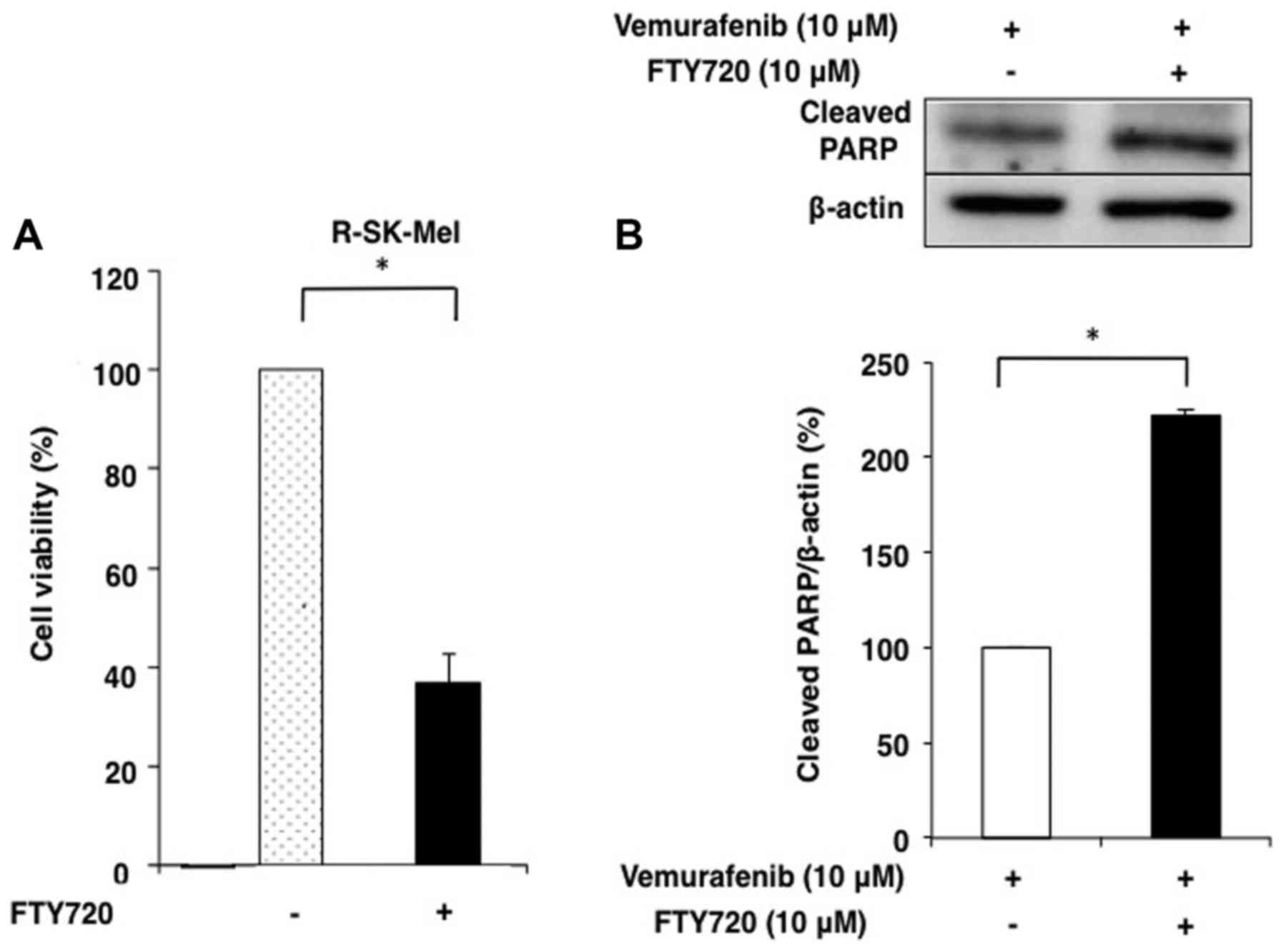
We used MTT assays to compare the cell viability of
SK-Mel-28 and R-SK-Mel cells treated with FTY720. The
IC50 of vemurafenib in R-SK-Mel cells was remarkably
reduced by treatment with FTY720 compared with that in SK-Mel-28
cells (Fig. 6A). The effects of
FTY720 and vemurafenib combination treatment on R-SK-Mel cell
apoptosis were evaluated using western blotting to measure the
expression of cleaved PARP. In R-SK-Mel cells, only combination
treatment with 10 µM vemurafenib and 10 µM FTY720 induced a
synergistic increase in PARP degradation (Fig. 6B).
Changes in the expression of
p-ERK/t-ERK and p-Akt/t-Akt by combination treatment with
vemurafenib and FTY720 in R-SK-Mel cells
Changes in the expression of proteins in the MAPK
and PI3K-Akt signalling pathways upon combination treatment with 10
µM vemurafenib and 10 µM FTY720 in R-SK-Mel cells were
investigated. The expression levels of p-ERK/t-ERK and p-Akt/t-Akt
were remarkably decreased by combination treatment (Fig. 7A and B). However, p-MEK/t-MEK
expression did not significantly change with this combination
treatment (Fig. 7C).
 | Figure 7.Effects of FTY720 on the expression
of (A) ERK, (B) Akt and (C) MEK in vemurafenib-resistant R-SK-Mel
cells. Vemurafenib-resistant R-SK-Mel cells were treated with 10 µM
vemurafenib with or without 10 µM FTY720 for 48 h. Cell lysates
were subjected to western blot analysis with antibodies against
total (t) and phosphorylated (p) forms of (A) ERK, (B) Akt and (C)
MEK. The ratios of phosphorylated to total ERK, Akt and MEK were
calculated. β-actin was used as the loading control. The results
are expressed as a percentage relative to the treatment without
FTY720. The data are expressed as the mean ± standard deviation.
*P<0.05 and **P<0.01, as indicated. FTY720, fingolimod;
R-SK-Mel, SK-Mel-28 resistant; MEK, mitogen-activated protein
kinase kinase; ERK, extracellular signal-related kinase; Akt,
protein kinase B; p-, phosphorylated; t-, total. |
Discussion
The MAPK and PI3K-Akt pathways are major pathways
underlying cancer development and progression (36), and the latter pathway is
particularly important for cell survival in melanoma (37). Several studies have previously
described the mechanism of resistance to BRAF inhibitors, and noted
that acquisition of the activated NRAS mutation (38) leads to reactivation of the MAPK
pathway. Furthermore, there have been many reports on the
mechanisms underlying resistance to BRAF inhibitors such as
activation of receptor tyrosine kinase (39), increased expression of mutated BRAF
kinase, increased expression of Cancer Osaka Thyroid (40), acquisition of MAP2K1
mutations (41) and loss of
NF1 (42).
In this study, we demonstrated that the melanoma
cell line WM-115 was most resistant to the BRAF inhibitor
(vemurafenib), and exhibited much higher expression levels of p-ERK
compared with vemurafenib-sensitive cells (data not shown).
Furthermore, when changes in the expression of cell signaling
molecules in R-SK-Mel cells were compared with the parent cells, we
found that the levels of p-ERK and p-AKT were remarkably increased
in R-SK-Mel cells, suggesting that both the MAPK and PI3K-Akt
pathways were enhanced in the vemurafenib resistant-melanoma
cells.
We also examined the effects of FTY720 and
vemurafenib combination treatment on the resistant cells. This
treatment strongly reduced cell viability, and also induced a
synergistic increase in cleaved PARP in the vemurafenib-resistant
WM-115 and R-SK-Mel cells. These results suggested that the
addition of FTY720 to vemurafenib was effective in enhancing
apoptosis compared with vemurafenib treatment alone. Furthermore,
we also examined the change in protein expression in cell signaling
pathway components by this combination treatment in the resistant
cells. In the MAPK pathway, p-ERK/t-ERK, and t-Akt and SK1 were
remarkably decreased by this treatment, whereas there was no
significant change in p-MEK/t-MEK. Moreover, this combination
treatment induced remarkable decreases in the levels of p-ERK/t-ERK
and also p-Akt/t-Akt in R-SK-Mel cells.
We previously reported that the combination
treatment with cisplatin and FTY720 had synergistic effects on
apoptosis induction in cisplatin-resistant melanoma cells by SK1
degradation, possibly due to downregulation of the PI3K/Akt/mTOR
pathway via the S1P receptor and reduced EGFR expression (34). In addition, FTY720 mediates many
anticancer effects through inactivation of the PI3K/Akt pathway
mediated via a variety of mechanisms including the inhibition of
PI3K, increased PTEN expression, activation of protein phosphatase
2A activity and SK1 inhibition (43). In accordance with these studies,
our results strongly suggest that the combination treatment with
FTY720 and vemurafenib induces apoptosis by downregulating both
PI3K/Akt and MAPK pathways in melanoma cells.
Recently, several cases of malignant melanoma have
been documented in patients with multiple sclerosis who were being
treated with FTY720 (44–46), and a relationship between treatment
by FTY720 and occurrence of melanoma has been suggested. However,
since the exact mechanism of the relationship has not been
elucidated, we believe that further studies are necessary.
Furthermore, we strongly suggest that FTY720 is most effective as
combination treatment with anticancer drugs rather than as
treatment by itself in clinical applications.
This combination treatment in vemurafenib-resistant
melanoma cells did not induce significant changes in p-MEK/t-MEK
expression. A recent study demonstrated that another pathway is
activated by BRAF mutation, which directly activates ERK through
Abl and Arg activation, but not through MEK (47). The authors also indicated that
Abl/Arg cooperates with a parallel, compensatory signalling pathway
(PTEN loss/Akt activation) to promote melanoma growth and survival.
Based on this report, FTY720 likely plays a major role in
inhibiting Akt activity, which consequently enhances cell
apoptosis. The latest therapy for patients affected by BRAF mutated
melanoma is a combination therapy of BRAF inhibitor and MEK
inhibitor. Our data suggest that more effective pharmacodynamic
actions can be obtained by using FTY720, which may block signalling
pathways via ERK, but not via MEK, as well as the Akt pathway.
The results of this study suggest that FTY720 may be
an effective agent for enhancing antineoplastic effects and
apoptosis, and thus, decreasing resistance to vemurafenib in
patients being treated with this agent.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TT, HK, YB and MS conceived and designed the study.
TT, NA and YB performed the experiments. TT, YB and MS wrote the
paper. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Davies H, Bignell GR, Cox C, Stephens P,
Edkins S, Clegg S, Teaque J, Woffendin H, Garnett MJ, Bottomley W,
et al: Mutations of the BRAF gene in human cancer. Nature.
417:949–954. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lovly CM, Dahlman KB, Fohn LE, Su Z,
Dias-Santagata D, Hicks DJ, Hucks D, Berry E, Terry C, Duke M, et
al: Routine multiplex mutational profiling of melanomas enables
enrollment in genotype-driven therapeutic trials. PLoS One.
7:e353092012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jakob JA, Bassett RL Jr, Ng CS, Curry JL,
Joseph RW, Alvarado GC, Rohlfs ML, Richard J, Gershenwald JE, Kim
KB, et al: NRAS mutation status is an independent prognostic factor
in metastatic melanoma. Cancer. 118:4014–4023. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gray-Schopfer VC, Karasarides M, Hayward R
and Marais R: Tumor necrosis factor-α blockes apoptosis in melanoma
cells when BRAF signaling is inhibited. Cancer Res. 67:122–129.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Testori A,
et al: Improved survival with vemurafenib in melanoma with BRAF
V600E mutation. N Eng J Med. 364:2507–2516. 2011. View Article : Google Scholar
|
|
6
|
Hauschild A, Grob JJ, Demidov LV, Jouary
T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr,
Kaempgen E, et al: Dabrafenib in BRAF-mutated metastatic melanoma:
A multicentre, open-label, phase 3 randomised controlled trial.
Lancet. 380:358–365. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lito P, Rosen N and Solit DB: Tumor
adaptation and resistance to RAF inhibitors. Nat Med. 21:1401–1409.
2013. View
Article : Google Scholar
|
|
8
|
Falchook GS, Lewis KD, Infante JR, Gordon
MS, Vogelzang NJ, DeMarini DJ, Sun P, Moy C, Szabo SA, Roadcap LT,
et al: Activity of the oral MEK inhibitor trametinib in patients
with advanced melanoma: A phase 1 dose-escalation trial. Lancet
Oncol. 13:782–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimizu T, Tolcher AW, Papadopoulos KP,
Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B,
et al: The clinical effect of the dual-targeting strategy involving
PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced
cancer. Clin Cancer Res. 18:2316–2325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taha TA, Hannun YA and Obeid LM:
Sphingosine kinase: Biochemical and cellular regulation and role in
disease. J Biochem Mol Biol. 39:113–131. 2006.PubMed/NCBI
|
|
11
|
Ogretmen B and Hannun YA: Biologically
active sphingolipids in cancer pathogenesis and treatment. Nat Rev
Cancer. 4:604–616. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Olivera A, Kohama T, Edsall L, Nava V,
Cuvillier O, Poulton S and Spiegel S: Sphingosine kinase expression
increases intracellular sphingosine-1-phosphate and promotes cell
growth and survival. J Cell Biol. 147:545–558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia P, Wang L, Gamble JR and Vadas MA:
Activation of sphingosine kinase by tumor necrosis factor-alpha
inhibits apoptosis in human endothelial cells. J Biol Chem.
274:34499–34505. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spiegel S and Milstien S:
Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol
Cell Biol. 4:397–407. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xia P, Gamble JR, Wang L, Pitson SM,
Moretti PA, Wattenberg BW, D'Andrea RJ and Vedas MA: An oncogenic
role of sphingosine kinase. Curr Biol. 10:1527–1530. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ho JW, Man K, Sun CK, Lee TK, Poon RT and
Fan ST: Effects of a novel immunomodulating agent, FTY720, on tumor
growth and angiogenesis in hepatocellular carcinoma. Mol Cancer
Ther. 4:1430–1438. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pchejetski D, Doumerc N, Golzio M, Naymark
M, Teissié J, Kohama T, Waxman J, Malavaud B and Cuvillier O:
Chemosensitizing effects of sphingosine kinase-1 inhibition in
prostate cancer cell and animal models. Mol Cancer Ther.
7:1836–1845. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
French KJ, Schrecengost RS, Lee BD, Zhuang
Y, Smith SN, Eberly JL, Yun JK and Smith CD: Discovery and
evaluation of inhibitors of human sphingosine kinase. Cancer Res.
63:5962–5969. 2003.PubMed/NCBI
|
|
19
|
Shida D, Takabe K, Kapitonov D, Milstien S
and Spiegel S: Targeting SphK1 as a new strategy against cancer.
Curr Drug Targets. 9:662–673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vadas M, Xia P, McCaughan G and Gamble J:
The role of sphingosine kinase 1 in cancer: Oncogene or
non-oncogene addiction? Biochim Biophys Acta. 1781:442–447. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Azuma H, Takahara S, Horie S, Muto S,
Otsuki Y and Katsuoka Y: Induction of apoptosis in human bladder
cancer cells in vitro and in vivo caused by FTY720 treatment. J
Urol. 169:2372–2377. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ubai T, Azuma H, Kotake Y, Inamoto T,
Takahara K, Ito Y, Kiyama S, Sakamoto T, Horie S, Muto S, et al:
FTY720 induced Bcl-associated and Fas-independent apoptosis in
human renal cancer cells in vitro and significantly reduced in vivo
tumor growth in mouse xenograft. Anticancer Res. 27:75–88.
2007.PubMed/NCBI
|
|
23
|
Billich A, Bornancin F, Dévay P,
Mechtcheriakova D, Urtz N and Baumruker T: Phosphorylation of the
immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem.
278:47408–47415. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paugh SW, Payne SG, Barbour SE, Milstien S
and Spiegel S: The immunosuppressant FTY720 is phosphorylated by
sphingosine kinase type2. FEBS Lett. 554:189–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang JD, Takahara S, Nonomura N, Ichimaru
N, Toki K, Azuma H, Matsumiya K, Okuyama A and Suzuki S: Early
induction of apoptosis in androgen-independent prostate cancer cell
line by FTY720 requires caspase-3 activation. Prostate. 40:50–55.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matloubian M, Lo CG, Cinamon G, Lesneski
MJ, Xu Y, Brinkmann V, Allende ML, Proia RL and Cyster JG:
Lymphocyte egress from thymus and peripheral lymphoid organs is
dependent on S1P receptor 1. Nature. 427:355–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pereira FV, Arruda DC, Figueiredo CR,
Massaoka MH, Matsuo AL, Bueno V and Rodrigues EG: FTY720 induces
apoptosis in B16F10-NEX2 murine melanoma cells, limits metastatic
development in vivo, and modulates the immune system. Clinics (Sao
Paulo). 68:1018–1027. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
LaMontagne K, Littlewood-Evans A, Schnell
C, O'Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau
G, et al: Antagonism of sphingosine-1-phosphate receptors by FTY720
inhibits angiogenesis and tumor vascularization. Cancer Res.
66:221–231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tonelli F, Lim KG, Loveridge C, Long J,
Pitson SM, Tigyi G, Bittman R, Pyne S and Pyne NJ: FTY720 and
(S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and
promote its proteasomal degradation in human pulmonary artery
smooth muscle, breast cancer and androgen-independent prostate
cancer cells. Cell Signal. 22:1536–1542. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kihara Y, Maceyka M, Spiegel S and Chun J:
Lysophospholipid receptor nomenclature review: IUPHAR review 8. Br
J Pharmacol. 171:3575–3594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang N, Qi Y, Wadham C, Wang L, Warren A,
Di W and Xia P: FTY720 induces necrotic cell death and autophagy in
ovarian cancer cells: A protective role of autophagy. Autophagy.
6:1157–1167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Azuma H, Takahara S, Ichimaru N, Wang JD,
Itoh Y, Otsuki Y, Morimoto J, Fukui R, Hoshiga M, Ishihara T, et
al: Marked prevention of tumor growth and metastasis by a novel
immunosuppressive agent, FTY720, in mouse breast cancer models.
Cancer Res. 62:1410–1419. 2002.PubMed/NCBI
|
|
33
|
Chua CW, Lee DT, Ling MT, Zhou C, Man K,
Ho J, Chan FL, Wang X and Wong YC: FTY720, a fungus metabolite,
inhibits in vivo growth of andorogen-independent prostate cancer.
Int J Cancer. 117:1039–1048. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ishitsuka A, Fujine E, Mizutani Y, Tawada
C, Kanoh H, Banno Y and Seishima M: FTY720 and cisplatin
synergistically induce cell death of cisplatin-resistant melanoma
cells through downregulation of PI3K pathway and decrease in
epidermal growth factor receptor expression. Int J Mol Med.
34:1169–1174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stordal BK, Davey MW and Davey RA:
Oxaliplatin induces drug resistance more rapidly than cisplatin in
H69 small cell lung cancer cells. Cancer Chemother Pharmacol.
58:256–265. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meier F, Schittek B, Busch S, Garbe C,
Smalley K, Satyamoorthy K, Li G and Herlyn M: The RAS/RAF/MEK/ERK
and PI3K/AKT signaling pathways present molecular targets for the
effective treatment of advanced melanoma. Front Biosci.
10:2986–3001. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sinnberg T, Lasithiotakis K, Niessner H,
Schittek B, Flaherty KT, Kulms D, Maczey E, Campos M, Gogel J,
Garbe C and Meier F: Inhibition of PI3K-AKT-mTOR signaling
sensitizes melanoma cells to cisplatin and temozolomide. J Invest
Dematol. 129:1500–1515. 2009. View Article : Google Scholar
|
|
38
|
Nazarian R, Shi H, Wang Q, Kong X, Koya
RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al: Melanoma
acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS
upregulation. Nature. 468:973–977. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Villanueva J, Vultur A, Lee JT,
Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu
X, Gimotty PA, Kee D, et al: Acquired resistance to BRAF inhibitors
mediated by a RAF kinase switch in melanoma can be overcome by
cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 18:683–695. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Johannessen CM, Boehm JS, Kim SY, Thomas
SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP,
Barretina J, et al: COT drives resistance to RAF inhibition through
MAP kinase pathway reactivation. Nature. 468:968–972. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Emery CM, Vijayendran KG, Zipser MC,
Sawyer AM, Niu L, Kim JJ, Hatton C, Chapra R, Oberholzer PA,
Karpova MB, et al: MEK1 mutations confer resistance to MEK and
B-RAF inhibition. Proc Natl Acad Sci USA. 106:20411–20416. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hodis E, Watson IR, Kryukov GV, Arold ST,
Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C,
et al: A landscape of driver mutations in melanoma. Cell.
150:251–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Patmanathan SN, Yap LF, Murray PG and
Paterson IC: The antineoplastic properties of FTY720: Evidence for
the repurposing of fingolimod. J Cell Mol Med. 19:2329–2340. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Filoni A, Lospalluti L, Giudice G,
Bonamonte D and Vestita M: Fingolimod and melanoma risk: Is there
sufficient evidence? Clin Exp Dermatol. 42:427–428. 2017.
View Article : Google Scholar
|
|
45
|
Haebich G, Mughal A and Tofazzal N:
Superficial spreading malignant melanoma in a patiet on fingolimod
therapy for multiple sclerosis. Clin Exp Dermatol. 41:433–434.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Conzett KB, Kolm I, Jelcic I, Kamarachev
J, Dummer R, Braun R, French LE, Linnebank M and Hofbauer GF:
Melanoma occurring during treatment with fingolimod for multiple
sclerosis: a case report. Arch Dermatol. 147:991–992. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jain A, Tripathi R, Turpin CP, Wanq C and
Pattner R: Abl kinase regulation by BRAF/ERK and cooporation with
Akt in melanoma. Oncogene. 36:4585–4596. 2017. View Article : Google Scholar : PubMed/NCBI
|