Introduction
Idiopathic pulmonary fibrosis (IPF) is an invariably
fatal disease characterized by the accumulation of excess
extracellular matrix components, and it can severely compromise
lung function, which is characterized by progressive dyspnea and
coughing (1,2). Despite the progress reported by
clinical trials on IPF over the last decade, the pathogenesis and
progression of IPF have not yet been fully elucidated. Although
immunosuppressive therapy has been effective in slowing down the
progression of IPF, its deleterious side effects have led to the
reconsideration of its use in IPF (3,4).
Thus far, the currently used agents, such as immunosuppressants and
steroids, have not been found to improve the prognosis. There is an
urgent need for effective therapeutic agents for IPF.
Kisspeptin (KP), also referred to as metastin, is
encoded by the KiSS-1 metastasis suppressor (KISS-1) gene and
serves as the ligand for G protein-coupled receptor 54 (GPR54), a
Gq/11-coupled receptor also referred to as AXOR12 or hOT7T175. KP
is proteolytically cleaved into several active polypeptides,
including KP-54, KP-13 and KP-10. Tian et al (5) reported that the KISS1 gene is a
downstream target of the classic transforming growth factor-β
(TGF-β)/Smad2 signaling pathway. The KP/GPR54 signaling pathway
plays an important role in initiating the secretion of
gonadotropin-releasing hormone (GnRH) at puberty, and is involved
in the regulation of the GnRH/GnRH receptor (GnRHR) signaling
pathway (6). Previous research has
provided substantial evidence supporting the role of the
KISS-1/GPR54 signaling pathway in the regulation of the
reproductive axis, including the timing of puberty, the sexual
differentiation of the brain, the regulation of GnRH at puberty, as
well as in diabetes, adiposity, atherogenesis (7), gastrointestinal motility (8), learning and memory (9), and suppression of metastasis across a
range of cancer types (10,11).
A number of neuroendocrine factors, including
hypothalamic-pituitary-adrenal/hypothalamic-pituitary-gonadal
(HPA/HPG) axis-related hormones and GnRH, regulate the homeostasis
between cell proliferation and apoptosis (12,13).
Disruption of this homeostasis leads to inflammatory changes or
fibrosis. For example, Kyritsi et al (14) reported that inhibition of the
hepatic expression of the GnRH/GnRHR1 axis by Cetrorelix reduced
biliary duct proliferation and liver fibrosis. McMillin et
al (15) reported that
infusion of melatonin reduced cholangiocyte proliferation, hepatic
injury and fibrosis during bile duct ligation-induced liver injury.
These previous studies provided novel insight into the use of the
KISS1/GPR54/GnRH axis as a potential target for the treatment of
fibrosis or inflammation.
To the best of our knowledge, at present, no data
have been published regarding the role of KP-13 in pulmonary
fibrosis. The aim of the present study was to determine whether
KP-13 is able to attenuate bleomycin (BLM)-induced pulmonary
fibrosis in mice, and investigate the underlying mechanisms.
Materials and methods
Animals
Male C57BL/6 mice (weighing 20–22 g, 8–10 weeks-old)
were purchased from the Experimental Animal Center of Lanzhou
University. All mice were housed in cages (humidity 45–50%, sizes
20×30 cm2, bedding-wood shavings, 6 animals/cage) with
free access to tap water and food in a room, which was maintained
at 22±2°C in a 12-h light (8:00 a.m.)/dark (8:00 p.m.) cycle. All
animal protocols in the present study were approved by the Ethics
Committee of Lanzhou University (approval no. SYXK Gan
2009-0005).
Establishment of the pulmonary
fibrosis model
The animal model of pulmonary fibrosis in the
present study was established according to the protocols previously
described by Tian et al (16), Luque et al (17) and Wei et al (18). Briefly, mice were anesthetized by
an intraperitoneal (i.p.) injection of 70 mg/kg pentobarbital
sodium (Sigma-Aldrich; Merck KGaA), and were intratracheally
administered 4 mg/kg BLM (Sigma-Aldrich; Merck KGaA) to induce
fibrosis. A total of 54 mice were randomly assigned to six groups
of 9 mice each as follows: i) Control group, instilled with saline
alone; ii) BLM group, instilled with BLM alone; iii) BLM+KP-13
group, instilled with BLM and treated with KP-13 [1 mg/kg, i.p.];
iv) BLM+KP-234+KP-13 group, instilled with BLM and treated with
KP-234 (1 mg/kg, i.p.) and KP-13 (i.p.); and v)
BLM+Cetrorelix+KP-13group, instilled with BLM and treated with
Cetrorelix and KP-13 (i.p.). vi) KP-13 group were only treated with
KP-13 (i.p.). After intratracheal instillation of BLM, the mice
received all the injections daily from the third day up to 28 days.
KP-13 and KP-234 were provided by Dr Min Chang (Lanzhou
University). KP-13 and KP-234 were first dissolved in 2% DMSO and
diluted in saline immediately prior to injection, as previously
described (9). Cetrorelix
(Sigma-Aldrich; Merck KGaA) was dissolved in saline and injected
i.p. (1 mg/kg/10 ml) 30 min prior to KP-13 injection. The mice were
sacrificed on day 28 after injection of BLM. All experiments were
performed between 9:00 a.m. and 6:00 p.m. In addition, the dose
selection was based on previous studies by Jiang et al
(8,9).
Histological analysis
The histopathological assay was performed, according
to previous studies. Briefly, whole left lungs were fixed in 4%
paraformaldehyde at 4°C overnight and embedded in paraffin. The
8-µm sections were stained with hematoxylin and eosin (H&E),
Masson's trichrome stain and Picro-Sirius Red (PSR), using standard
methods. The images were performed by an ordinary optical
microscope (H&E and Masson) and polarized light microscope
(PSR; Zeiss AG).
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was conducted according to the
manufacturer's protocol (Takara Bio, Inc.). Total RNA was extracted
using TRIzol® reagent (Thermo Fisher Scientific, Inc.)
following the manufacturer's protocol and 1 µg RNA sample was
reverse transcribed into cDNA with the 5X PrimeScript RT Master Mix
(Takara Bio, Inc.), the following conditions: 37°C for 15 min, 85°C
for 5 sec, 4°C for 3 min. Amplification was conducted in a 25 µl
reaction compound composed of 12.5 µl 2X SYBR Premis Ex TaqII
(Takara Bio, Inc.), 8.5 µl ddH2O, 2 µl cDNA, 1 µl
forward primer and 1 µl reverse primer, and was carried out under
the following conditions: 95°C for 30 sec, followed by 40 cycles at
95°C for 5 sec, 60°C for 30 sec and 72°C for 30 sec. Gene
expression was evaluated using the 2−∆∆Cq method
(19) where ∆Cq=Cqtarget
gene-CqGAPDH and
∆∆Cq=CqDrug-CqControl. The primers are
presented in Table I and GAPDH was
used as the reference gene.
 | Table I.Primers used in the reverse
transcription-quantitative PCR in the present study. |
Table I.
Primers used in the reverse
transcription-quantitative PCR in the present study.
| Gene | Prime | Sequence (5′-3′) |
|---|
| IL-1β | Sense |
CAGCTTCAAATCTCGCAGCA |
|
| Anti-sense |
CTCATGTCCTCATCCTGGAAGG |
| TNF-α | Sense |
ACTCCCAGGTTCTCTTCAAGG |
|
| Anti-sense |
GGCAGAGAGGAGGTTGACTTTC |
| TGF-β | Sense |
TTGCTTCAGCTCCACAGAGA |
|
| Anti-sense |
TGGTTGTAGAGGGCAAGGAC |
| Colla1 | Sense |
GAGCGGAGAGTACTGGATCG |
|
| Anti-sense |
GCTTCTTTTCCTTGGGGTTC |
| Timp1 | Sense |
CTTCTGGCATCCTGTTGT |
|
| Anti-sense |
ACTGCAGGTAGTGATGTG |
| Acta2 | Sense |
CTGACAGAGGCACCACTGAA |
|
| Anti-sense |
CATCTCCAGAGTCCAGCACA |
| MMP2 | Sense |
TCAAGTTCCCCGGCGATG |
|
| Anti-sense |
AGTTGGCCACATCTGGGTTG |
| GAPDH | Sense |
GCCACAGACGTCACTTTCCTAC |
|
| Anti-sense |
CGGGAACACAGTCACATACCA |
Western blotting
Western blotting was performed following the
manufacturer's protocol (Bio-Rad Laboratories, Inc). Protein was
extracted with RIPA buffer containing protease inhibitor (Gibco;
Thermo Fisher Scientific, Inc.). The protein concentration was
determined using a BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). The protein samples (40 µg per lane) were
separated by SDS-PAGE on 10% gels and then transferred onto PVDF
membranes. The membranes were blocked in 5% fat-free milk in TBST
(0.1% Tween-20) at room temperature for 2 h, and incubated with
specific antibodies overnight at 4°C as follows: Anti-α-smooth
muscle actin (α-SMA; cat. no. 19245; 1:1,000; Cell Signaling
Technology, Inc.), anti-TGF-β (cat. no. 3711; 1:1,000; Cell
Signaling Technology, Inc.), anti-phosphorylated (p)-Smad2/3 (cat.
no. 8828; 1:1,000; Cell Signaling Technology, Inc.) or anti-total
(t)-Smad2/3 (cat. no. 3102; 1:1,000; Cell Signaling Technology,
Inc.), anti-Bcl-2 (cat. no. D198628-0100; 1:500; BBI Life
Sciences), anti-Bax (cat. no. D190756-0100; 1:500; BBI Life
Sciences), anti-active caspase-3 (cat. no. D195315-0100; 1:500; BBI
Life Sciences) and anti-GAPDH (cat. no. 5174; 1:2,000; Cell
Signaling Technology, Inc.). After washing three times with PBS,
the membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies (cat. no. A0208, 1:5,000; Beyotime Institute
of Biotechnology) at room temperature for 1 h. The result was
visualized with chemiluminescence reagents using an ECL kit (Thermo
Fisher Scientific, Inc.) and exposed to a film. The intensity of
the blots was quantified with densitometry (Image J 1.49v; National
Institutes of Health).
ELISA
Total protein from the lungs of mice in each group
were extracted with RIPA lysis buffer containing protease inhibitor
(Gibco; Thermo Fisher Scientific, Inc.). Total proteins were
determined using a bicinchoninic acid protein assay kit (Sangon
Biotech Co., Ltd.). Interleukin (IL)-1β, IL-6 and tumor necrosis
factor-α (TNF-α) in the lung tissue were measured using ELISA kits
(cat. no. E-EL-M0049 for TNF-α, cat.no. E-EL-M0044 for IL-6, cat.
no. E-EL-M0037 for IL-1β, Elabscience), according to the
manufacturer's protocol.
Statistical analysis
All data are presented as the mean ± SEM for two
repeats twice of each experiment. Overall survival was defined as
the time period from the first day of BLM-induced pulmonary
fibrosis to the date of succumbing, or until the 28th day of
BLM-induced pulmonary fibrosis. Comparisons of mortality were made
by analyzing Kaplan-Meier survival curves, and then log-rank tests
to assess for differences in survival. The statistical analysis was
conducted by two-way ANOVA followed by Dunnett's post-hoc test
using SPSS 19.0 (IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
KP-13 attenuates the pulmonary damage
and fibrosis induced by BLM in mice
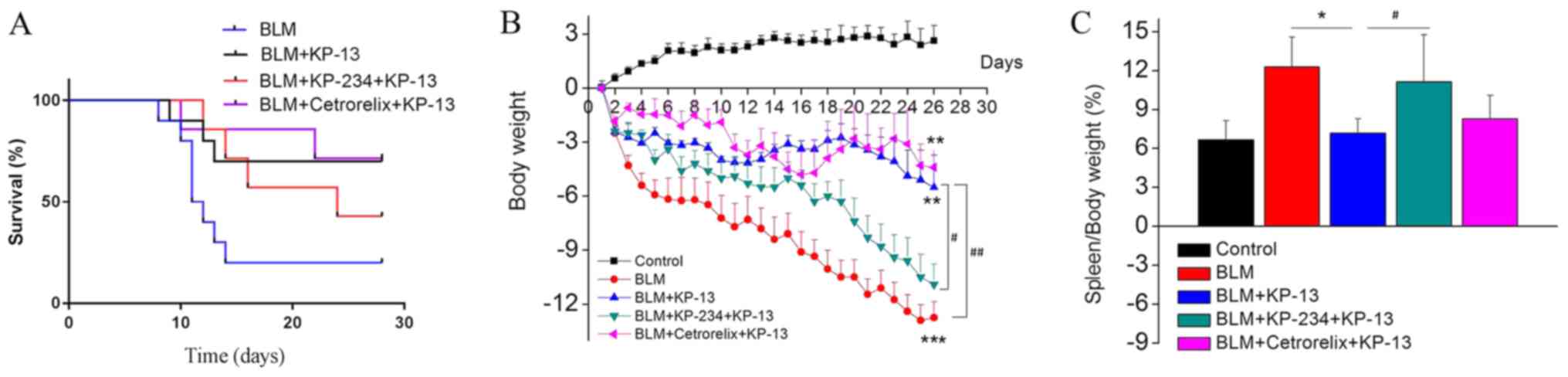
A comparison of 28-day survival curves among the
four groups of mice with pulmonary fibrosis revealed that KP-13
improved the survival of mice with BLM-induced (4 mg/kg) pulmonary
fibrosis (Fig. 1A). Additionally,
mice in the BLM group had lost an amount of body weight between
days 2 and 28 compared with the control mice, and it reached a
significant difference at 28 days (P<0.001; Fig. 1B). Treatment with KP-13markedly
inhibited these changes compared with the BLM group. The increased
spleen/body weight ratio reflected the progression of inflammation.
As shown in Fig. 1C, BLM treatment
increased this ratio, while KP-13 treatment inhibited the increase
in the ratio (P<0.05; BLM group compared with BLM+KP-13 group),
suggesting that KP-13 suppresses inflammation.
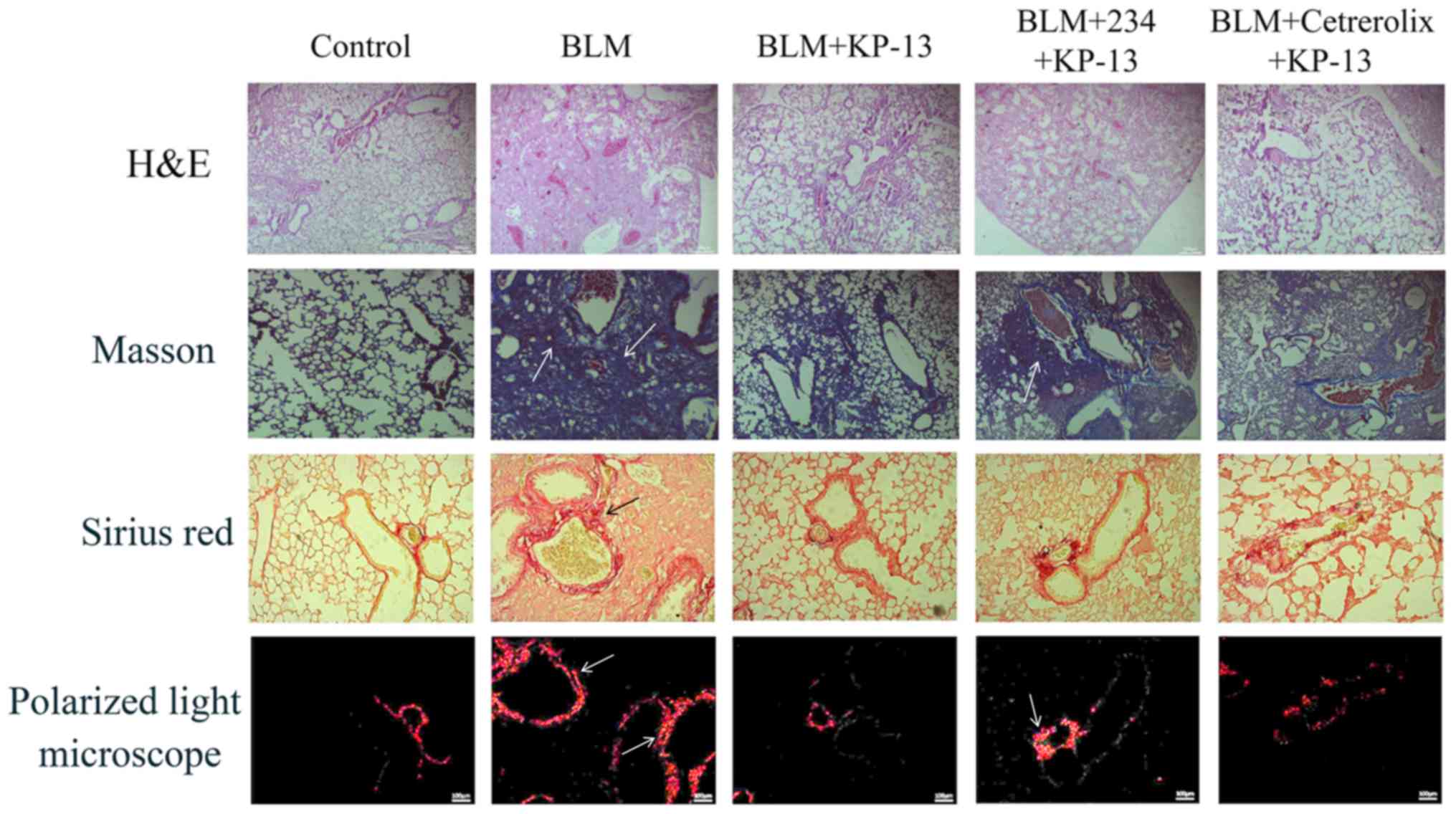
Pulmonary injury and fibrosis induced by BLM in mice
were evaluated by histopathological examination. The findings
included severe edema, alveolar collapse, alveolar membrane
thickening and inflammatory cell infiltration (Fig. 2). H&E, Masson's trichrome and
PSR staining revealed severe collagen deposition induced by BLM in
the lungs of the mice. However, treatment with KP-13 markedly
reversed these changes and alleviated collagen deposition.
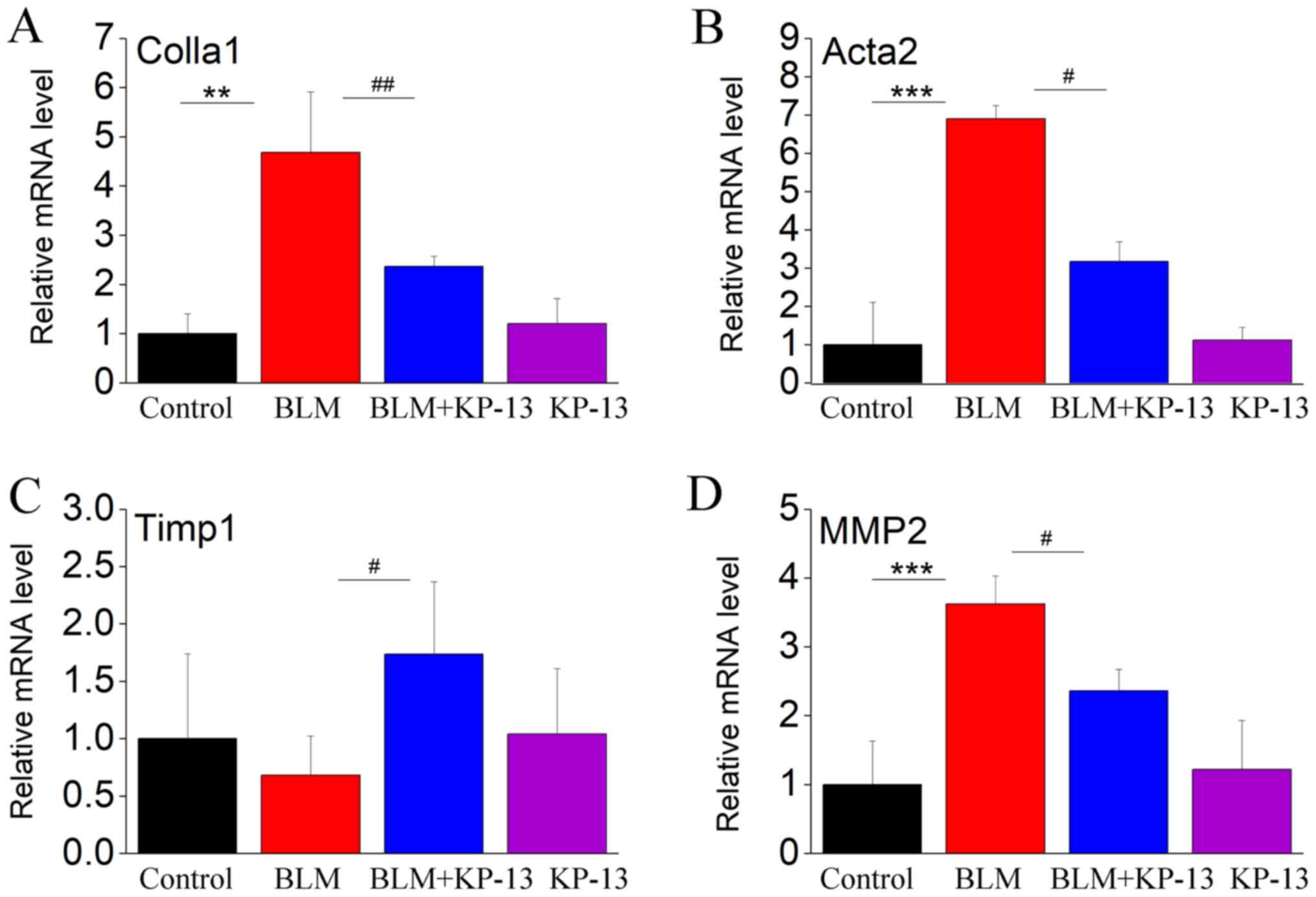
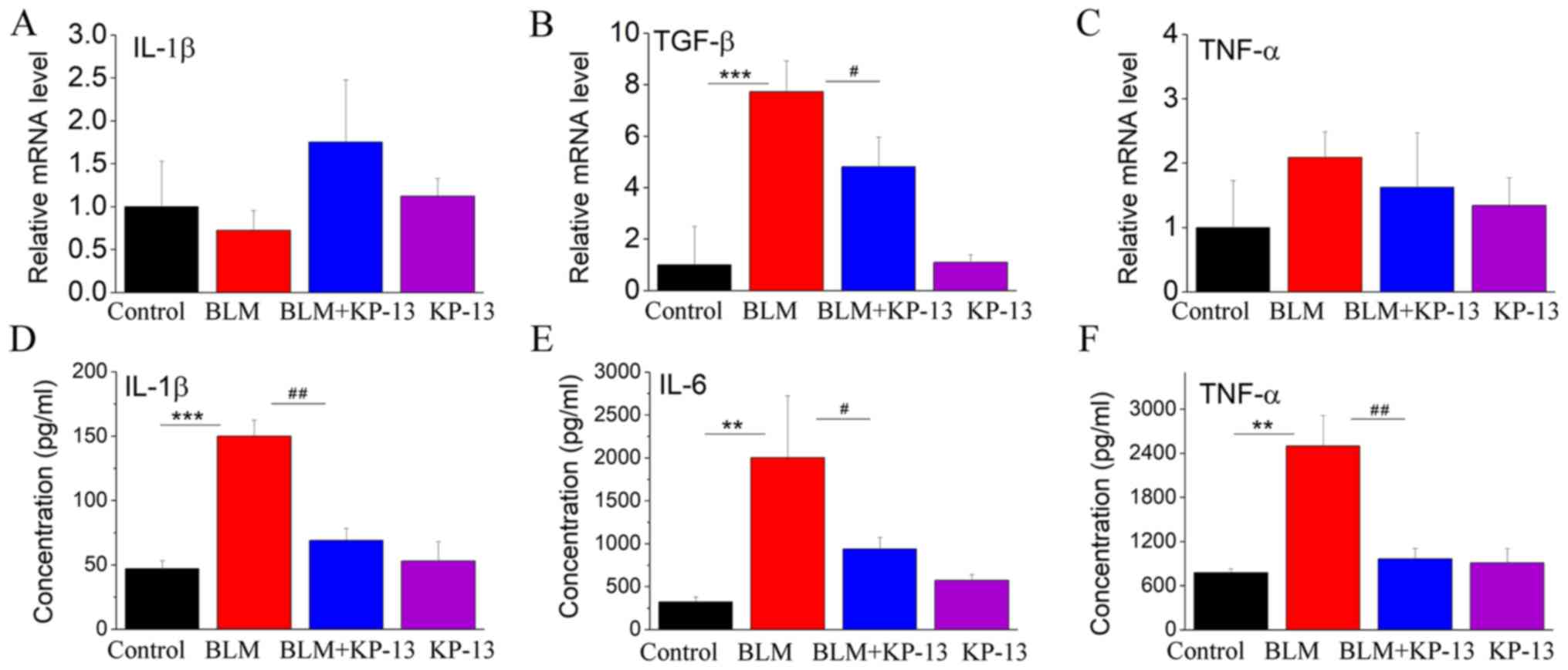
KP-13 ameliorates BLM-induced
inflammatory injury in the lungs of mice
It was recently demonstrated that BLM-induced
pulmonary fibrosis is correlated with alterations in inflammatory
cytokines (TNF-α, IL-1β and TGF-β) and fibrosis-related factors
[collagen type I α 1 (Colla1), actin α 2 (Acta2), tissue inhibitor
of metalloproteinase 1 (Timp1) and matrix metalloproteinase 2
(MMP2)] (16). In the present
study, the levels of these factors were increased in response to
intratracheal BLM instillation compared with control mice. However,
these increases were markedly inhibited by KP-13 application.
As shown in Fig. 3,
the mRNA levels of Colla1, Acta2 and MMP2 were significantly
increased following intratracheal BLM treatment (P<0.01 for
Colla1; P<0.001 for MMP2 and Acta2; Fig. 3). Administration of KP-13 to the
mice inhibited the expression of these genes induced by BLM
administration (P<0.01 for Colla1; P<0.05 for MMP2 and Acta2;
Fig. 3). TIMP1 is a collagenase
inhibitory protein in pulmonary fibrosis. Thus, its expression
level is inversely related to the level of fibrosis (20). Fig.
3C showed that BLM treated decreased the expression of TIMP1,
whereas the decrease was markedly changed by KP-13 application
(P<0.05, Fig. 3C). These
results reflect the induction of fibrosis by BLM and the beneficial
effect of KP-13 on IPF. As IPF is associated with inflammation, the
expression levels of related inflammatory cytokines were also
evaluated. The protein levels of IL-1β, TNF-α and IL-6, and the
mRNA level of TGF-β were statistically significantly increased in
the lung following intratracheal BLM treatment (P<0.001 for
IL-1β and TGF-β; P<0.01 for TNF-α and IL-6; Fig. 4), while KP-13 injection
significantly decreased the expression of those factors
(P<0.05).
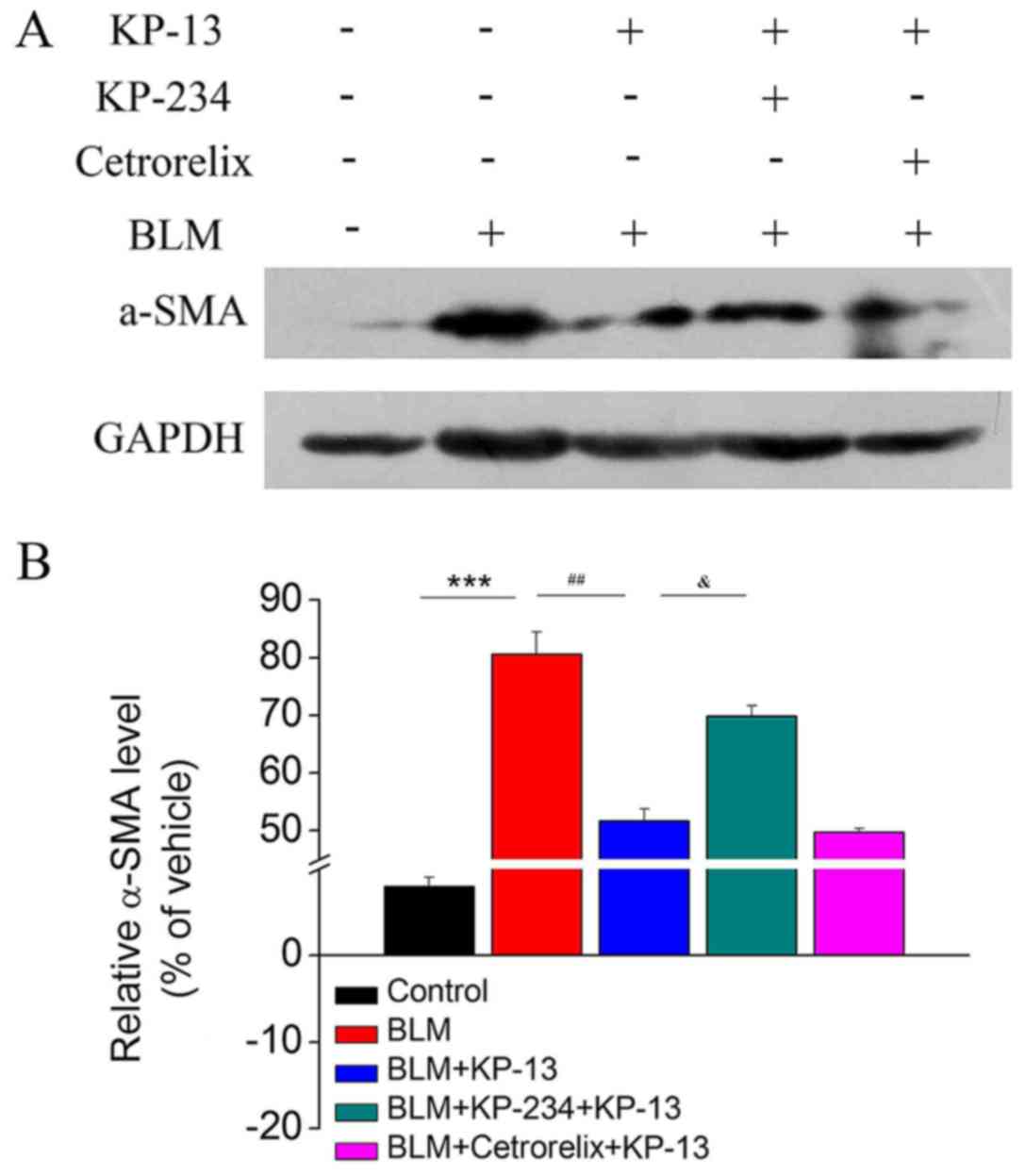
KP-13 reduces the protein expression
of α-SMA in lung tissues
A main characteristic of pulmonary fibrosis is the
over-proliferation of α-SMA-positive fibroblasts in the whole lung
(21,22). Therefore, the ability of KP-13 to
modulate the expression of α-SMA, which is a key marker of
myofibroblasts, was further evaluated. The western blotting results
demonstrated that BLM treatment upregulated the expression of α-SMA
in lung tissues compared with the control group (P<0.001;
Fig. 5), whereas the levels of
α-SMA were reduced following KP-13 treatment compared with the BLM
group (P<0.01; Fig. 5).
Mechanism underlying the inhibitory
effect of KP-13 on BLM-induced pulmonary fibrosis
A number of previous studies have demonstrated the
physiological and pathological roles of the KP/GPR54 signaling
pathway in the regulation of the reproductive system, diabetes,
adiposity, inhibition of cancer metastasis, and atherosclerotic
plaque progression and instability (11,23–25).
Recent studies have also reported that KP is a potent stimulator of
GnRH secretion, and GnRH was reported to be associated with
inflammation and fibrosis (14,15).
Therefore, KP-234, an antagonist of GPR54, and
Cetrorelix, an antagonist of GnRHR, were used to determine whether
they could block the anti-fibrotic effects of KP-13. The results
revealed that KP-234, but not Cetrorelix, significantly attenuated
the effects of KP-13 on BLM-induced pulmonary injury and fibrosis
(P<0.05 for BLM+KP-13 group and BLM+KP-234+KP-13 group; Figs. 1, 2 and 5).
KP-13 inhibits the expression of TGF-β
and phosphorylation of Smad2/3 in BLM-induced pulmonary
fibrosis
TGF-β is a key mediator of pulmonary fibrosis, as it
regulates the synthesis of extracellular matrix proteins via the
TGF-β/Smad2/3 signaling pathway (26). To elucidate the possible mechanisms
through which KP-13 ameliorates BLM-induced pulmonary injury and
fibrosis, the effects of KP-13 on the TGF-β/Smad2/3 signaling
pathway were examined. As demonstrated by the western blot analysis
results (Fig. 6A-C), the
expression of TGF-β1, as well as the phosphorylation of Smad2/3,
were significantly increased after treatment with BLM (P<0.01
for TGF-β1 and Smad2/3), which was downregulated following KP-13
application (P<0.05 for TGF-β1; P<0.001 for Smad2/3).
Furthermore, the levels of pro-apoptosis related proteins, such as
Bax and caspase-3 (P<0.05 for between Bax and caspase-3,
Fig. 6A, D-F), were increased in
the BLM group compared with the control. However, these
pro-apoptosis proteins were significantly downregulated after KP-13
application (P<0.05, Fig. 6A, E and
F). Meanwhile, anti-apoptosis related protein (Bcl-2) was
markedly decreased by BLM, whereas KP-13 upregulated its expression
level (Fig. 6A).
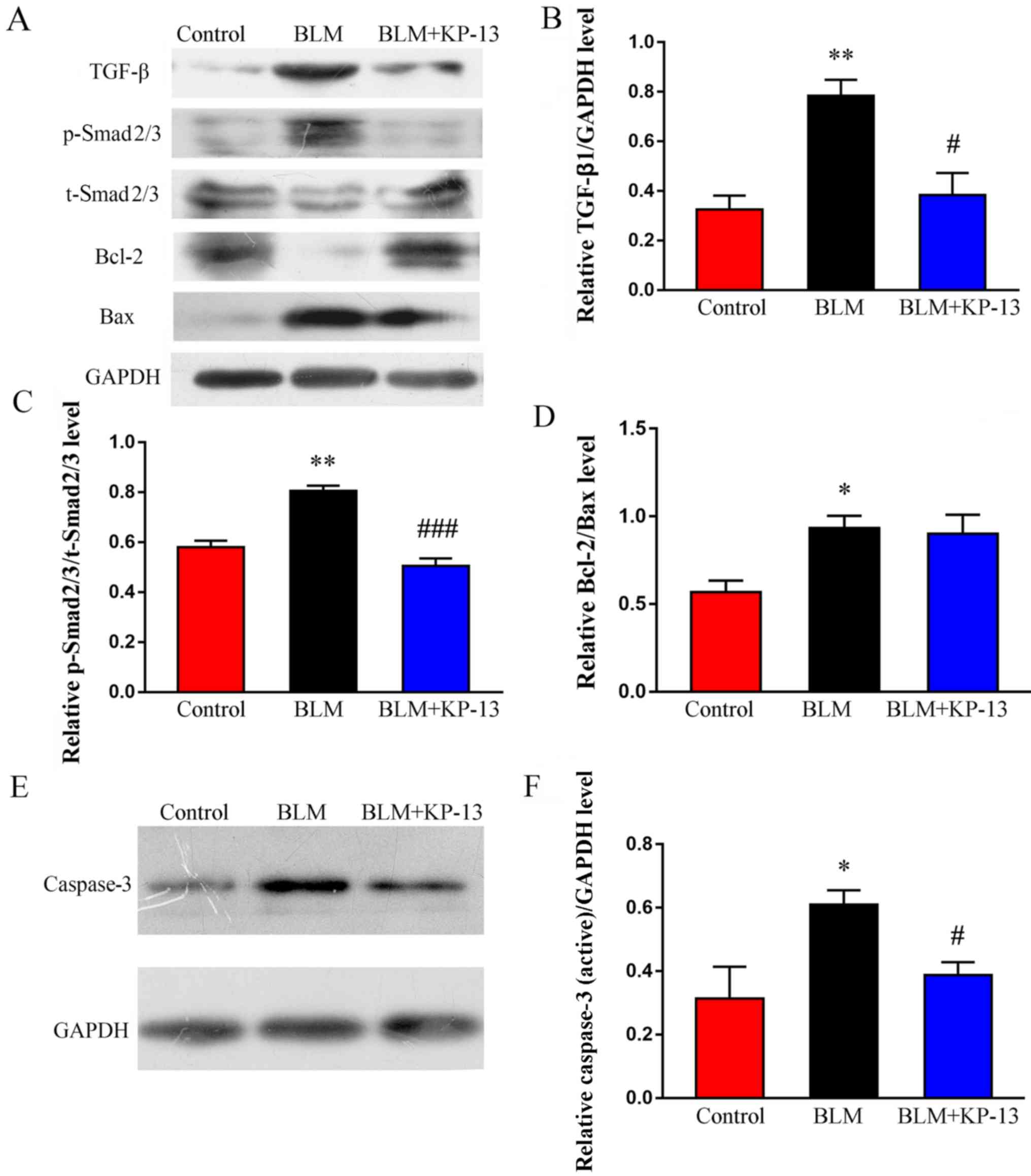 | Figure 6.Effects of KP-13 on the expression of
TGF-β, p-Smad2/3, t-Smad2/3, Bcl-2, Bax and caspase-3 were assessed
by western blot analysis in mice with pulmonary fibrosis. (A)
Western blot analysis and subsequent densitometry determined the
relative protein expression level of (B) TGF-β, (C)
p-Smad2/3/t-Smad2/3 and (D) Bcl-2/Bax. (E) Western blot analysis
and (F) subsequent densitometry determined the expression of
caspase-3, normalized to GAPDH. The data are presented as the mean
± SEM. n=5/group. *P<0.05, **P<0.01 vs. control;
#P<0.05, ###P<0.001 vs. BLM group. KP,
kisspeptin; TGF-β, transforming growth factor-β; BLM, bleomycin; p,
phosphorylated; t, total. |
Discussion
To the best of our knowledge, the present study is
the first to provide evidence demonstrating that KP-13 reduces
pulmonary injury and fibrosis induced by BLM, particularly the
inflammatory response and massive infiltration of inflammatory
cells, and the increased collagen/α-SMA deposition in the lung. In
addition, it was demonstrated that a GPR54 antagonist, but not a
GnRHR antagonist, was able to block the effects of KP-13 in an
animal model of pulmonary fibrosis.
In the present study, severe pulmonary fibrosis was
induced in mice 28 days after intratracheal instillation of BLM,
which was indicated by body weight loss, increased lung coefficient
(data not shown), decreased survival rate and exacerbated
histopathological abnormalities, with extensive collagen
deposition. By comparing the BLM-induced pulmonary fibrosis between
mice with and without KP-13 treatment, it was demonstrated that
BLM-induced pulmonary fibrosis was markedly attenuated by
KP-13.
The main characteristics of pulmonary fibrosis are
over-proliferation of α-SMA-positive fibroblasts and collagen
deposition in the whole lung; therefore, targeted inhibition of
α-SMA expression in the lung has been found to be an effective
therapeutic strategy for pulmonary fibrosis (27). Notably, KP-13 significantly
ameliorated pulmonary fibrosis by exerting an inhibitory regulatory
effect on the expression of Colla1, Acta2 and MMP2. Furthermore,
the expression levels of TNF-α and TGF-β were also markedly
decreased in the lung in response to BLM+KP-13 treatment compared
with the BLM alone-treated mice. Finally, the western blotting
results further illustrated that KP-13 downregulated the expression
of α-SMA at the protein level in lung tissues. To summarize, these
results demonstrated that KP-13 mitigated BLM-induced pulmonary
fibrosis.
A previous study reported that the KP/GPR54
signaling pathway is extensively involved in the regulation of
endothelial cells, macrophages, monocytes, cardiomyocytes, and
cells of the hypothalamus and extravillous trophoblast (17). Sato et al (28) demonstrated that the KP/GPR54
signaling cascade may serve as a potential therapeutic target for
atherosclerotic diseases. Additionally, previous experimental and
clinical studies have demonstrated the ability of GnRH agonists to
prevent postoperative adhesions, inflammation and fibrosis.
McMillin et al (15)
reported that GnRH played a key role in activated hepatic stellate
cells during cholestatic liver disease. Kyritsi et al
(14) demonstrated that targeting
the GnRH/GnRHR1 signaling pathway may be a key to the management of
hepatic fibrosis during the progression of primary sclerosing
cholangitis. Therefore, the present study investigated whether the
KP/GRP54 and GnRH/GnRHR signaling pathways were involved in the
regulation of IPF progression by KP-13 by using KP-234 and
Cetrorelix, the respective antagonists of the mentioned signaling
pathways. The findings demonstrated that KP-234, but not
Cetrorelix, was able to inhibit the anti-fibrotic effects of KP-13,
specifically in terms of body weight loss, decreased survival rate,
increased lung coefficient (data not shown), massive infiltration
by inflammatory cells and increased collagen/α-SMA deposition in
the lungs. Therefore, it was identified that the GPR54 axis, but
not the GnRH axis, was involved in the regulation of BLM-induced
IPF in mice by KP-13.
Suppression of TGF-β/Smad signaling has been
demonstrated to ameliorate experimentally induced fibrosis
(29). Previous studies have
reported that BLM-induced pulmonary fibrosis may be associated with
TGF-β/Smad2/3 signaling (26,30).
In the present study, it was observed that the expression of TGF-β
and the phosphorylation of Smad2/3 in the lung were markedly
upregulated in mice treated with BLM alone, whereas administration
of KP-13 significantly inhibited the expression of TGF-β and the
phosphorylation of Smad2/3. In addition, treatment with KP-13
markedly upregulated the expression of the anti-apoptotic factor
Bcl-2, and downregulated the expression of the pro-apoptotic factor
Bax, compared with the BLM group. These data indicated that KP-13
inhibited the enhanced TGF-β signaling in BLM-induced pulmonary
fibrosis.
To summarize, the results of the present study
indicated that KP-13 exerted antifibrotic effects via inhibition of
the inflammatory response and collagen/α-SMA deposition in the
lung. Therefore, the KP-13/GPR54 axis may represent a promising
therapeutic target for preventing the progression of pulmonary
fibrosis. However, whether the GPR54/KP systems can be used as
targets for pulmonary fibrosis in the clinic remains to be further
studied.
Acknowledgements
Not applicable.
Funding
The present study was funded by the grants from
Gansu province Natural Science Foundation of China (grant no.
1506RJZA278) and Gansu Provincial Key Laboratory Open Foundation of
China (zdsyskfkt-201707).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZL and XB conducted the experiments, performed the
analysis, and wrote the manuscript. JM analyzed the data and
provided the peptide drug KP-13. QY designed the experiments and
contributed to writing and editing the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
All of the procedures in the present study were
approved by the Ethics Committee of Lanzhou University (approval
no. SYXK Gan 2009–0005).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferrara G, Luppi F, Birring SS, Cerri S,
Caminati A, Sköld M and Kreuter M: Best supportive care for
idiopathic pulmonary fibrosis: Current gaps and future directions.
Eur Respir Rev. 27(pii): 1700762018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xie Y, Wang JJ, Li GY, Li XL and Li JS:
Acupuncture for idiopathic pulmonary fibrosis: Protocol for a
systematic review. Medicine (Baltimore). 96:e91142017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sime PJ: The antifibrogenic potential of
PPARgamma ligands in pulmonary fibrosis. J Investig Med.
56:534–538. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Selman M, King TE and Pardo A; American
Thoracic Society; European Respiratory Society; American College of
Chest Physicians, : Idiopathic pulmonary fibrosis: Prevailing and
evolving hypotheses about its pathogenesis and implications for
therapy. Ann Intern Med. 134:136–151. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tian J, Al-Odaini AA, Wang Y, Korah J, Dai
M, Xiao L, Ali S and Lebrun JJ: KiSS1 gene as a novel mediator of
TGFβ-mediated cell invasion in triple negative breast cancer. Cell
Signal. 42:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Skorupskaite K, George JT and Anderson RA:
The kisspeptin-GnRH pathway in human reproductive health and
disease. Hum Reprod Update. 20:485–500. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mead EJ, Maguire JJ, Kuc RE and Davenport
AP: Kisspeptins are novel potent vasoconstrictors in humans, with a
discrete localization of their receptor, G protein-coupled receptor
54, to atherosclerosis-prone vessels. Endocrinology. 148:140–147.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang J, Jin W, Peng Y, He Z, Wei L, Li S,
Wang X, Chang M and Wang R: In vivo and vitro characterization of
the effects of kisspeptin-13, endogenous ligands for GPR54, on
mouse gastrointestinal motility. Eur J Pharmacol. 794:216–223.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang JH, He Z, Peng YL, Jin WD, Wang Z,
Han RW, Chang M and Wang R: Kisspeptin-13 enhances memory and
mitigates memory impairment induced by Aβ1–42 in mice novel object
and object location recognition tasks. Neurobiol Learn Mem.
123:187–195. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stathaki M, Armakolas A, Dimakakos A,
Kaklamanis L, Vlachos I, Konstantoulakis MM, Zografos G and
Koutsilieris M: Kisspeptin effect on endothelial monocyte
activating polypeptide II (EMAP-II)-associated lymphocyte cell
death and metastases in colorectal cancer patients. Mol Med.
20:80–92. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kotani M, Detheux M, Vandenbogaerde A,
Communi D, Vanderwinden JM, Le Poul E, Brézillon S, Tyldesley R,
Suarez-Huerta N, Vandeput F, et al: The metastasis suppressor gene
KiSS-1 encodes kisspeptins, the natural ligands of the orphan G
protein-coupled receptor GPR54. J Biol Chem. 276:34631–34636. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meresman GF, Bilotas M, Buquet RA, Barañao
RI, Sueldo C and Tesone M: Gonadotropin-releasing hormone agonist
induces apoptosis and reduces cell proliferation in eutopic
endometrial cultures from women with endometriosis. Fertility and
sterility. 80 (Suppl 2):S702–S707. 2003. View Article : Google Scholar
|
|
13
|
Castellon E, Clementi M, Hitschfeld C,
Sánchez C, Benítez D, Sáenz L, Contreras H and Huidobro C: Effect
of leuprolide and cetrorelix on cell growth, apoptosis, and GnRH
receptor expression in primary cell cultures from human prostate
carcinoma. Cancer Invest. 24:261–268. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kyritsi K, Meng F, Zhou T, Wu N, Venter J,
Francis H, Kennedy L, Onori P, Franchitto A, Bernuzzi F, et al:
Knockdown of hepatic gonadotropin-releasing hormone by
vivo-morpholino decreases liver fibrosis in multidrug resistance
gene 2 knockout mice by down-regulation of miR-200b. Am J Pathol.
187:1551–1565. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McMillin M, DeMorrow S, Glaser S, Venter
J, Kyritsi K, Zhou T, Grant S, Giang T, Greene JF Jr, Wu N, et al:
Melatonin inhibits hypothalamic gonadotropin-releasing hormone
release and reduces biliary hyperplasia and fibrosis in cholestatic
rats. Am J Physiol Gastrointest Liver Physiol. 313:G410–G418. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tian SL, Yang Y, Liu XL and Xu QB: Emodin
attenuates bleomycin-induced pulmonary fibrosis via
anti-inflammatory and Anti-oxidative activities in rats. Med Sci
Monit. 24:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luque RM, Kineman RD and Tena-Sempere M:
Regulation of hypothalamic expression of KiSS-1 and GPR54 genes by
metabolic factors: Analyses using mouse models and a cell line.
Endocrinology. 148:4601–4611. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei YR, Qiu H, Wu Q, Du YK, Yin ZF, Chen
SS, Jin YP, Zhao MM, Wang C, Weng D and Li HP: Establishment of the
mouse model of acute exacerbation of idiopathic pulmonary fibrosis.
Exp Lung Res. 42:75–86. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong J and Ma Q: TIMP1 promotes
multi-walled carbon nanotube-induced lung fibrosis by stimulating
fibroblast activation and proliferation. Nanotoxicology. 11:41–51.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Waisberg DR, Parra ER, Barbas-Filho JV,
Fernezlian S and Capelozzi VL: Increased fibroblast telomerase
expression precedes myofibroblast α-smooth muscle actin expression
in idiopathic pulmonary fibrosis. Clinics. 67:1039–1046. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun KH, Chang Y, Reed NI and Sheppard D:
α-Smooth muscle actin is an inconsistent marker of fibroblasts
responsible for force-dependent TGFβ activation or collagen
production across multiple models of organ fibrosis. Am J Physiol
Lung Cell Mol Physiol. 310:L824–L836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cetkovic A, Miljic D, Ljubić A, Patterson
M, Ghatei M, Stamenković J, Nikolic-Djurovic M, Pekic S, Doknic M,
Glišić A, et al: Plasma kisspeptin levels in pregnancies with
diabetes and hypertensive disease as a potential marker of
placental dysfunction and adverse perinatal outcome. Endocr Res.
37:78–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hussain MA, Song WJ and Wolfe A: There is
Kisspeptin - and then there is Kisspeptin. Trends Endocrinol Metab.
26:564–572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jayasena CN, Nijher GM, Comninos AN,
Abbara A, Januszewki A, Vaal ML, Sriskandarajah L, Murphy KG,
Farzad Z, Ghatei MA, et al: The effects of kisspeptin-10 on
reproductive hormone release show sexual dimorphism in humans. J
Clin Endocrinol Metab. 96:E1963–E1972. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zank DC, Bueno M, Mora AL and Rojas M:
Idiopathic pulmonary fibrosis: Aging, mitochondrial dysfunction and
cellular bioenergetics. Front Med (Lausanne). 5:102018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sava P, Ramanathan A, Dobronyi A, Peng X,
Sun H, Ledesma-Mendoza A, Herzog EL and Gonzalez AL: Human
pericytes adopt myofibroblast properties in the microenvironment of
the IPF lung. JCI Insight. 2(pii): 963522017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sato K, Shirai R, Hontani M, Shinooka R,
Hasegawa A, Kichise T, Yamashita T, Yoshizawa H, Watanabe R,
Matsuyama TA, et al: Potent vasoconstrictor Kisspeptin-10 induces
atherosclerotic plaque progression and instability: Reversal by its
receptor GPR54 antagonist. J Am Heart Assoc. 6(pii):
e0057902017.PubMed/NCBI
|
|
29
|
Oruqaj G, Karnati S, Vijayan V, Kotarkonda
LK, Boateng E, Zhang W, Ruppert C, Günther A, Shi W and
Baumgart-Vogt E: Compromised peroxisomes in idiopathic pulmonary
fibrosis, a vicious cycle inducing a higher fibrotic response via
TGF-β signaling. Proc Natl Acad Sci USA. 112:E2048–E2057. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cutroneo KR, White SL, Phan SH and Ehrlich
HP: Therapies for bleomycin induced lung fibrosis through
regulation of TGF-beta1 induced collagen gene expression. J Cell
Physiol. 211:585–589. 2007. View Article : Google Scholar : PubMed/NCBI
|