Introduction
Sepsis, the most common cause of death among
patients in the intensive care unit, is defined as life-threatening
organ dysfunction caused by a dysregulated host response to
infection (1). It is recognized as
a global public health problem due to its high mortality and
morbidity, as well as its substantial economic burden (2). Rudd et al (3) recently reported that an estimated 48.9
million cases of sepsis and 11.0 million sepsis-related deaths were
recorded worldwide in 2017, representing 19.7% of all global
deaths. Septic shock is a subset of sepsis that can lead to
multi-organ dysfunction and rapid death (1). Myocardial dysfunction is recognized as
a common complication of septic shock and contributes to adverse
outcomes (4). Endotoxins, such as
lipopolysaccharide (LPS), which is an important structural
component of the gram-negative bacterial outer membrane, are
important factors responsible for septic myocardial dysfunction
(5). Previous studies have
demonstrated that LPS can mimic the myocardial dysfunction of
septic shock (6,7). A number of underlying
pathophysiological mechanisms, including genetic, molecular,
metabolic and structural mechanisms, have been proposed to be
involved in sepsis-induced myocardial dysfunction (8). However, the specific mechanisms have
not yet been elucidated.
Toll-like receptors (TLRs) are the first line of
defense in the mammalian immune system and recognize
pathogen-associated molecular patterns, such as lipoproteins, LPS,
flagellin and nucleic acids of bacterial origin (9). Among these receptors, TLR4 has been
identified as the only receptor for LPS and plays a key role in
LPS-mediated inflammatory responses and myocardial dysfunction
(6). The intracellular signaling
pathways activated after LPS-TLR4 binding have been classified as
the myeloid differentiation primary response protein 88
(MyD88)-independent and TIR-containing adapter inducing
interferon-β (TRIF)-dependent pathway (10). The MyD88-independent pathway induces
the activation of nuclear factor-κB and mitogen-activated protein
kinases (MAPKs), thus leading to the release of pro-inflammatory
cytokines and regulation of cardiac function during sepsis
(11). MAPKs, including p38,
extracellular signal-regulated kinase (ERK)1/2, c-Jun N-terminal
kinase (JNK) and ERK5 (12),
transduce a variety of extracellular signals that regulate the
cellular response implicated in proliferation, differentiation,
apoptosis, stress and inflammatory response (13,14)
and play an important role in regulating sepsis-induced cardiac
dysfunction (15). JNK, also known
as stress-activated protein kinase, is a member of the MAPK family.
However, the role of the JNK signaling pathway in tumor necrosis
factor-α (TNF-α) expression and myocardial dysfunction induced by
sepsis has not yet been clearly defined. The majority of previous
studies (16,17) have reported that the activation of
the JNK signaling pathway promotes the production of
pro-inflammatory cytokines and the development of LPS-induced
cardiac dysfunction. Research from Peng et al (18) revealed that LPS activates JNK1, thus
leading to the inhibition of TNF-α expression and improvement of
the myocardial function in sepsis. In addition, only a few studies
have investigated whether TLR4 mediates LPS-induced myocardial
dysfunction by modulating the JNK signaling pathway. Thus, the aim
of the present study was to examine the changes in cardiac
function, myocardial histopathology and TNF-α expression during LPS
stimulation. Furthermore, this study also attempted to explore the
regulatory role of the TLR4/JNK signaling pathway in LPS-induced
TNF-α expression and myocardial dysfunction. The results
demonstrated that the production of TNF-α increased significantly,
cardiac function decreased, and the activities of TLR4 and JNK in
the myocardium were upregulated during LPS stimulation. Whereas,
inhibition of TLR4 activation downregulated JNK activation, reduced
TNF-α expression and improved cardiac function in response to LPS.
The present results suggested that TLR4 mediated myocardial
dysfunction by regulating the JNK signaling pathway during
sepsis.
Materials and methods
Animals
A total of 54 male wild-type C57BL/6 mice (8–12
weeks old and weighing 22–28 g), were purchased from the Laboratory
Animal Center, Academy of Military Medical Sciences, [certificate
no. SCXK (Jing) 2014-0013; Beijing, China]. The mice were housed in
a specific pathogen-free environment at a constant temperature
(22±1°C) with 50% humidity and 12-h light/dark cycles. All mice had
free access to food and water. All the experimental procedures were
approved by the Animal Experiments Ethics Committee of Nankai
University (Tianjin, China; approval no. 10011).
Experimental protocol
To induce sepsis, mice were injected
intraperitoneally (i.p.) with 12 mg/kg LPS (from Escherichia
coli, a phenol extract of serotype 011:B4; Sigma-Aldrich; Merck
KGaA), a dose that was sufficient to induce cardiac dysfunction as
determined in our preliminary experiment (19). Mice were randomly assigned to three
groups: Sham group (n=18), LPS group (n=18) and TAK-242 group
(n=18). Mice in the LPS and TAK-242 groups were i.p. treated with
saline or a TLR4 inhibitor (TAK-242; 2 mg/kg), respectively,
followed by LPS (12 mg/kg, i.p.) 1 h later. Mice in the sham group
were injected with equal 0.9% saline. For time course experiments,
mice were sacrificed at 3, 12 and 24 h after LPS or saline
injection (n=6 for each time point). Left ventricular (LV) function
was assessed at 3, 12 and 24 h after LPS injection during
anesthesia. After measurement of LV function, mice were euthanized
by excessive inhalation of isoflurane, and then the plasma and
ventricular myocardium were collected and stored at −80°C.
Echocardiographic assessment of LV
function
A Vevo 2100 ultrahigh resolution small animal
ultra-sound imaging system (VisualSonics, Inc.) with a MS-400
ultrasound scanning transducer was used to evaluate LV function
(20–22). Briefly, mice were anesthetized with
1–1.25% isoflurane via a mask, and their chest was shaved before
the animals were placed in the supine position on a 37°C pad. Warm
electrode gel was applied to the limb leads, allowing for the
electrocardiogram and respiration rate to be recorded during
ultrasound imaging. Briefly, an even layer of ultrasound coupling
agent was spread on the mouse thoracic, and the console was used at
a downward sloping angle of 30–45°. Two-dimensional cine loops and
guided M-mode frames were recorded from the parasternal long axis.
The following parameters were measured as indicators of function:
LV end diastolic diameter (LVEDD), LV end systole diameter (LVESD),
LV end diastolic volume (LVEDV), LV end systolic volume (LVESV), LV
ejection fraction percentage (LVEF%), and LV fractional shortening
percentage (LVFS%). All data were analyzed off-line at the end of
the study with the software in-built in the ultrasound system
(20).
Histopathological examination of
myocardial tissues
Immediately after the sacrifice of the mice, their
hearts were removed, fixed in a 10% paraformaldehyde solution for
24 h at room temperature and embedded in paraffin. Serial sections
(4 µm) of the ventricle were affixed to slides, deparaffinized, and
stained with hematoxylin and eosin (H&E) for 5 min at room
temperature. Pathological changes of the myocardial tissues were
observed under a light microscope (magnification, ×400).
Transmission electron microscopy
Transmission electron microscopy was performed as
described in a previous study by Wang et al (23). After pretreatment, the samples were
cut into thin sections and visualized at 120 kV with a H7650
transmission electron microscope (Hitachi, Ltd.). A total of 10–15
micrographs per sample were obtained using a Philips CM12 (Philips
Medical Systems, Inc.) by random sampling.
Measurement of cardiac troponin I
(cTnI) and TNF-α protein levels
cTnI and TNF-α protein levels in the plasma were
determined using a mouse cTnI ELISA kit (Biotopped Life Sciences;
cat. no. TOPEL02104) and a TNF-α ELISA kit (BioLegend; cat. no.
430907) according to the manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the myocardial tissues
using TRIzol® reagent (Ambion; Thermo Fisher Scientific,
Inc.), following the manufacturer's instructions. RT of the
purified RNA (4 µg) was performed using random primers and the
RevertAid First Strand cDNA Synthesis kit (Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions. TB
Green Premix EX Taq II (Tli RNase H Pus) (Takara Bio, Inc.; cat.
no. RR820A) in a CFX96 system (Bio-Rad Laboratories, Inc.) was used
to determine the mRNA expression of TLR4 and JNK. All reactions
were performed in triplicate. The sequences of the sense and
antisense primers used for amplification are listed in Table I. The relative expression of the
target genes was determined by calculating the values of the Δ
cycle quantification (ΔCq) by normalizing the average Cq value to
that of the endogenous control (GAPDH), and then calculating the
2−∆∆Cq values (24).
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Gene | Primer sequences
(5→3) |
|---|
| TLR4 | F:
GCTAAGTGCCGAGTCTGAGTGTAA |
|
| R
TGCAGCCTTTCAGAAACACATT |
| JNK | F:
GCTCTCAGCATCCATCGTCTTC |
|
| R:
AGGTCCAGCTGATGCTTCTAGACT |
| GAPDH | F:
CTCTGCTCCTCCCTGTTCCA |
|
| R:
ATACGGCCAAATCCGTTCAC |
Western blot analysis
The Whole Protein Extraction kit (Applygen
Technologies, Inc.) was used to extract the whole protein of
myocardial tissue, according to manufacturer's instructions. The
concentration of whole protein was determined with a Qubit™ Protein
Assay kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions, and the mean values of concentration
were determined. The samples were stored at −80°C until use.
Following quantification, equal amounts of protein samples (11.25
mg) from the homogenized total tissues were separated using 5%
stacking and 10% separating gels, and subsequently transferred to a
PVDF membrane. After blocking at room temperature for 2 h with 5%
non-fat milk and Tris-buffered saline with 0.05% Tween-20 (TBST) or
5% bovine serum albumin (cat. no. A8010; Beijing Solarbio Science
& Technology Co., Ltd.) and washing with TBST, the membranes
were incubated overnight at 4°C with primary antibodies against
TLR4 (1:100; Santa Cruz Biotechnology, Inc.; cat. no. sc-293072),
JNK (1:500; Abcam; cat. no. ab112501), phosphorylated (p)-JNK
(1:500; Abcam; cat. no. ab4821) or GAPDH (1:10,000; ProteinTech
Group, Inc.; cat. no. 60004-1-lg). Subsequently, the membranes were
rinsed and incubated with the corresponding secondary antibody,
either horseradish peroxidase-conjugated goat anti-rabbit or goat
anti-mouse IgG (1:5,000; Boster Biological Technology; cat. nos.
BA1055 and BA1051), for 2 h at room temperature. Immunoblots were
visualized with an enhanced chemiluminescence reagent (Bio-Rad
Laboratories, Inc.), and developed and analyzed using Quantity One
software (version 4.6; Bio-Rad Laboratories, Inc.). The protein
signals were quantified by gray scale values. GAPDH was used as an
internal control. The total protein level was normalized to the
GAPDH protein level, and the level of p-JNK to total JNK were
presented.
Statistical analysis
All experiments were repeated ≥3 times. All data are
presented as the mean ± SD. Statistical analysis was performed
using SPSS 22.0 statistical software (IBM Corp.) and graphs were
generated using Prism 6.0 software (GraphPad Software, Inc.).
Comparisons among multiple groups were analyzed using one-way ANOVA
followed by the SNK post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Changes in cardiac function
The beneficial effect of TAK-242 pretreatment on
LPS-induced cardiac dysfunction was confirmed by quantitative
analysis of echocardiograms. Representative echocardiograms in the
different groups are presented in Fig.
1. The results of echocardiography analysis in various groups
are summarized in Fig. 2. Compared
with the values in the sham groups, the LPS groups showed a
significant decrease in LVEDD at 24 h, and in LVEDV at 12 and 24 h
(P<0.05), as well as a significant increase in LVESD and LVESV
at 3 and 12 h (P<0.05), and a significant decrease in LVEF% and
LVFS% at all time points after LPS injection (P<0.05). However,
compared with the values in the LPS groups, pretreatment with
TAK-242 induced a significant increase in LVEDD and LVEDV at 12 and
24 h (P<0.05), alongside a significant reduction in LVESD and
LVESV at 3 h (P<0.05), and a significant increase in LVEF% and
LVFS% at 3 h following LPS injection (P<0.05).
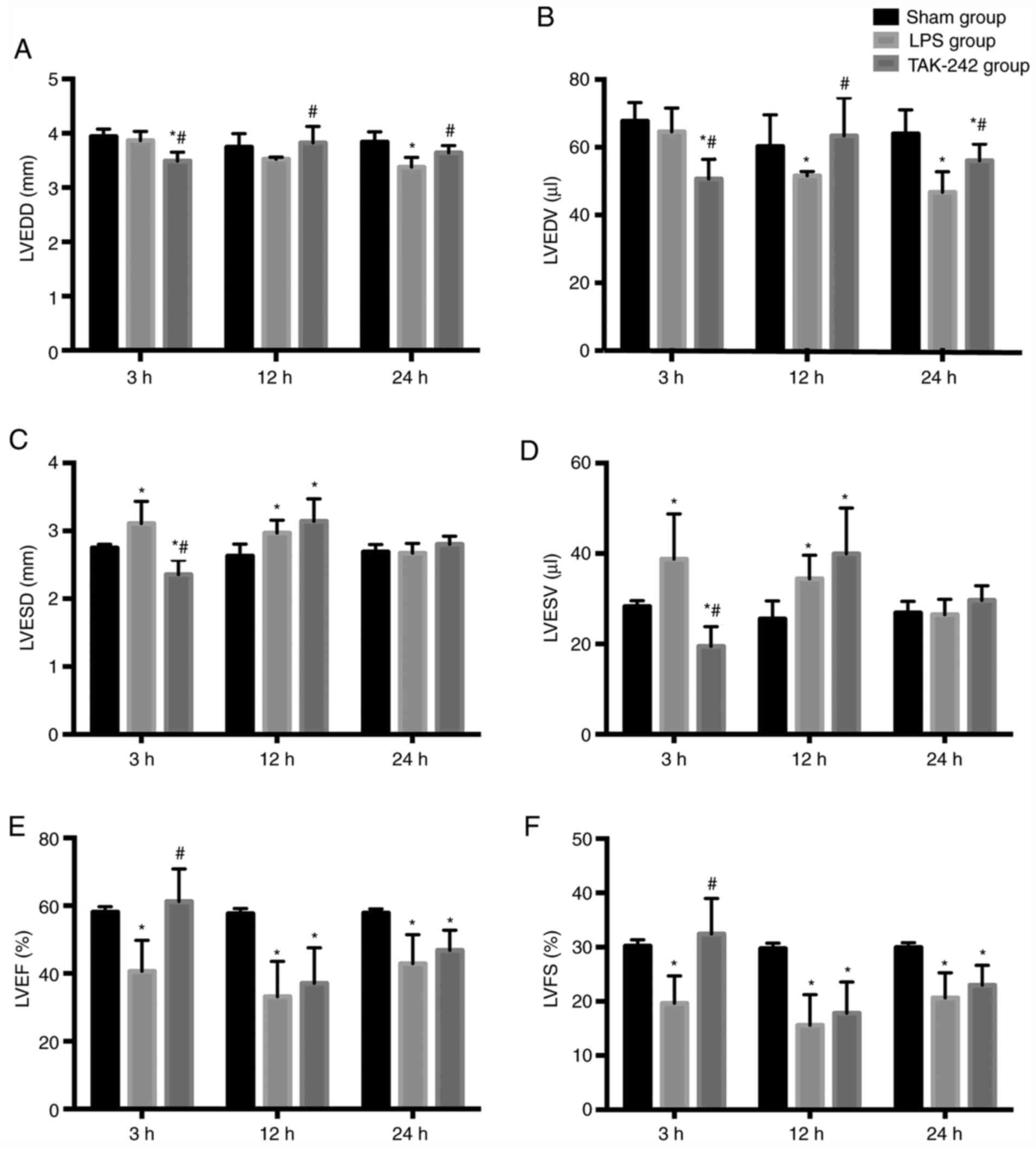 | Figure 2.Echocardiographic characterization of
cardiac function in different groups. Quantitative assessment of
dilation and systolic function of heart based on (A) LVEDD, (B)
LVEDV, (C) LVESD, (D) LVESV, (E) LVEF and (F) LVFS. Data are
expressed as the mean ± SD from six animals in each group.
*P<0.05 vs. sham group; #P<0.05 vs. LPS group.
LVEDD, left ventricular end diastolic diameter; LVESD, left
ventricular end systole diameter; LVEDV, left ventricular end
diastolic volume; LVESV, left ventricular end systolic volume;
LVEF, left ventricle ejection fraction; LVFS, left ventricle
fractional shortening; LPS, lipopolysaccharide. |
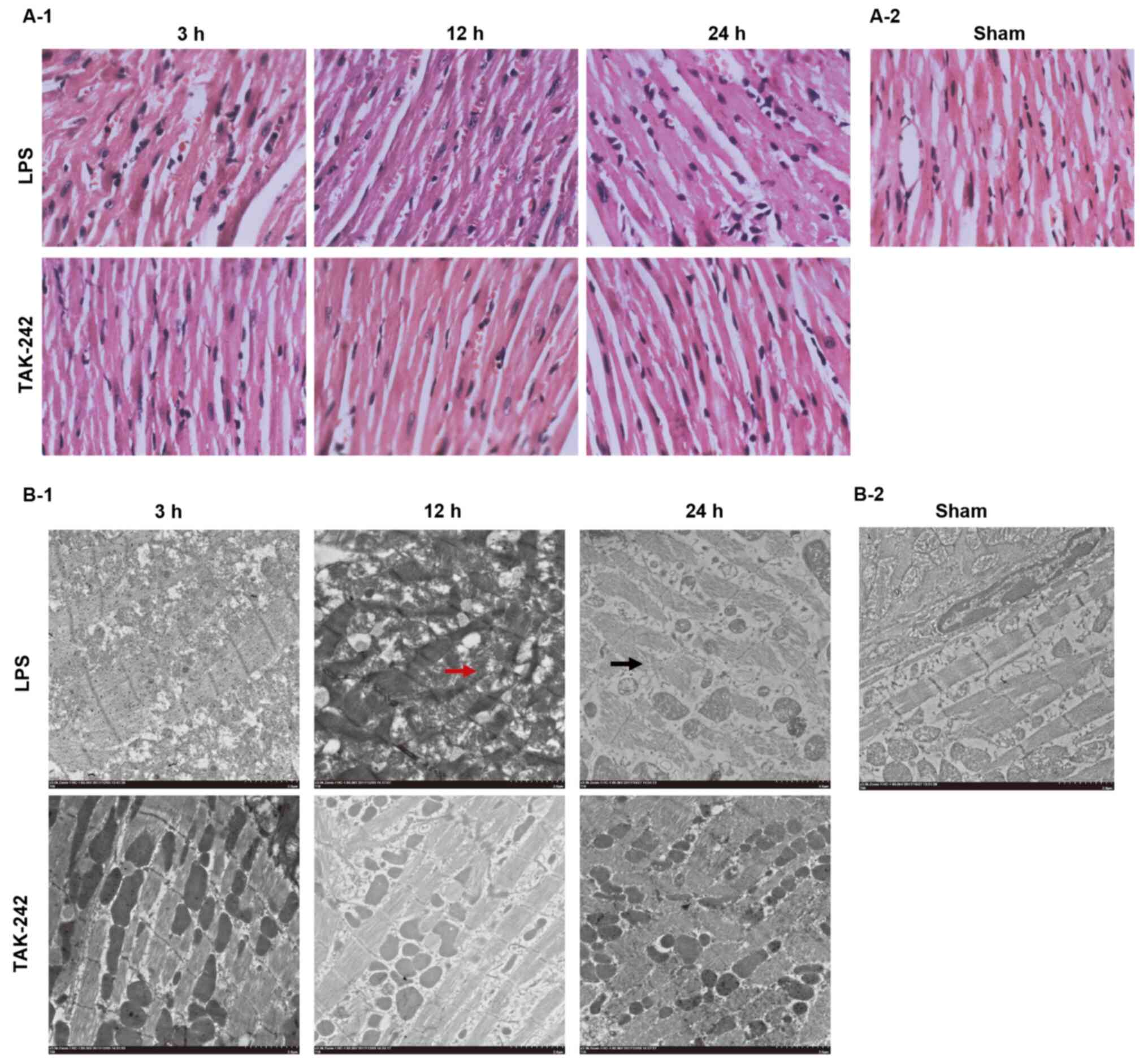
Histological changes in the
myocardium
To directly observe the effect of TAK-242
pretreatment on the LPS-induced alterations in myocardial
structure, the histology of the LV myocardial tissues was examined
(Fig. 3). H&E staining showed
that distinct myocardial injury occurred in the myocardial tissues
of the LPS groups, including myocardial interstitial edema,
prominent hemorrhaging, rupture of myocardial fibers, myocardial
cell swelling, degeneration, loss of transverse striations and
infiltration of leukocytes. Under the transmission electron
microscope, the myocardial ultrastructure in the LPS groups was
damaged, which was characterized by myofibrillar disarray, cellular
disorganization, disarrangement of sarcomere and/or disruption,
mitochondrial swelling and cracking, disappearance of mitochondrial
crista, as well as autophagic vacuoles. As time increased, the
degree of myocardial damage in the LPS groups gradually worsened.
However, the LPS-induced myocardial injuries were ameliorated by
pretreatment with TAK-242.
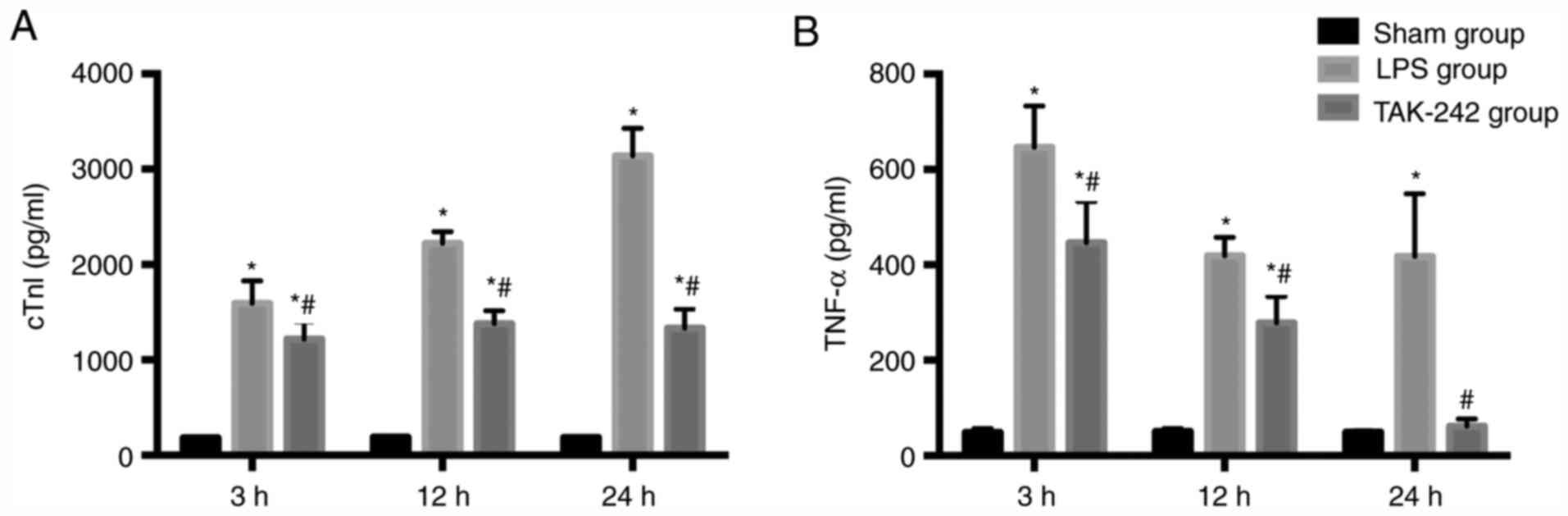
Serum levels of cTnI and TNF-α
Significant increases in the enzyme cTnI, a
myocardial injury marker, were observed in the LPS groups compared
with the values of the sham groups, and the level of cTnI increased
gradually with time (P<0.05). Pretreatment with TAK-242
significantly reduced the level of cTnI compared with that of the
LPS group (P<0.05). Compared with the levels of the sham groups,
the serum TNF-α level increased significantly in the LPS groups
(P<0.05) and reached the maximal level at 3 h following LPS
injection. Pretreatment with TAK-242 induced a significant decrease
in the serum TNF-α level (P<0.05), compared with that of the LPS
groups (Fig. 4).
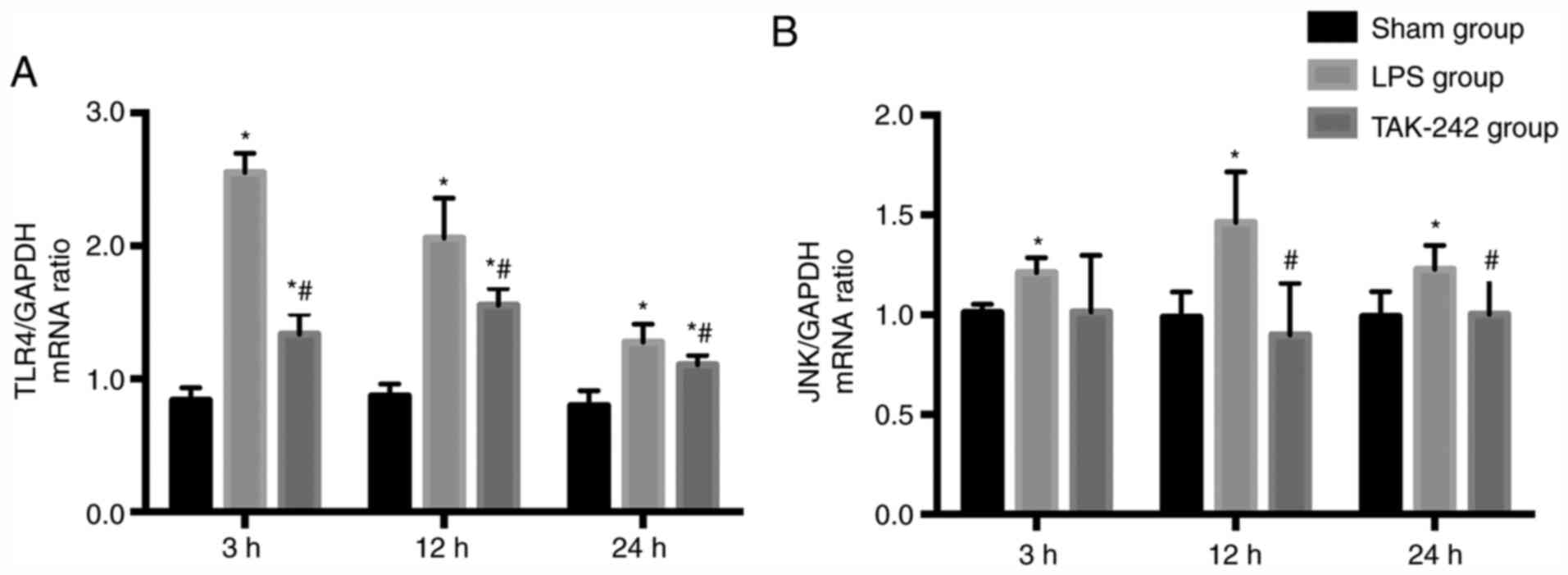
TLR4 and JNK gene expression in the
myocardium
As shown in Fig. 5,
the relative TLR4 and JNK mRNA expression levels were significantly
increased in response to LPS (P<0.05) and reached the maximal
levels at 3 and 12 h after LPS injection, respectively. Compared
with that of the LPS groups, pretreatment with TAK-242
significantly downregulated the expression levels of TLR4 and JNK
mRNA at 3 and 12 h after LPS injection, respectively
(P<0.05).
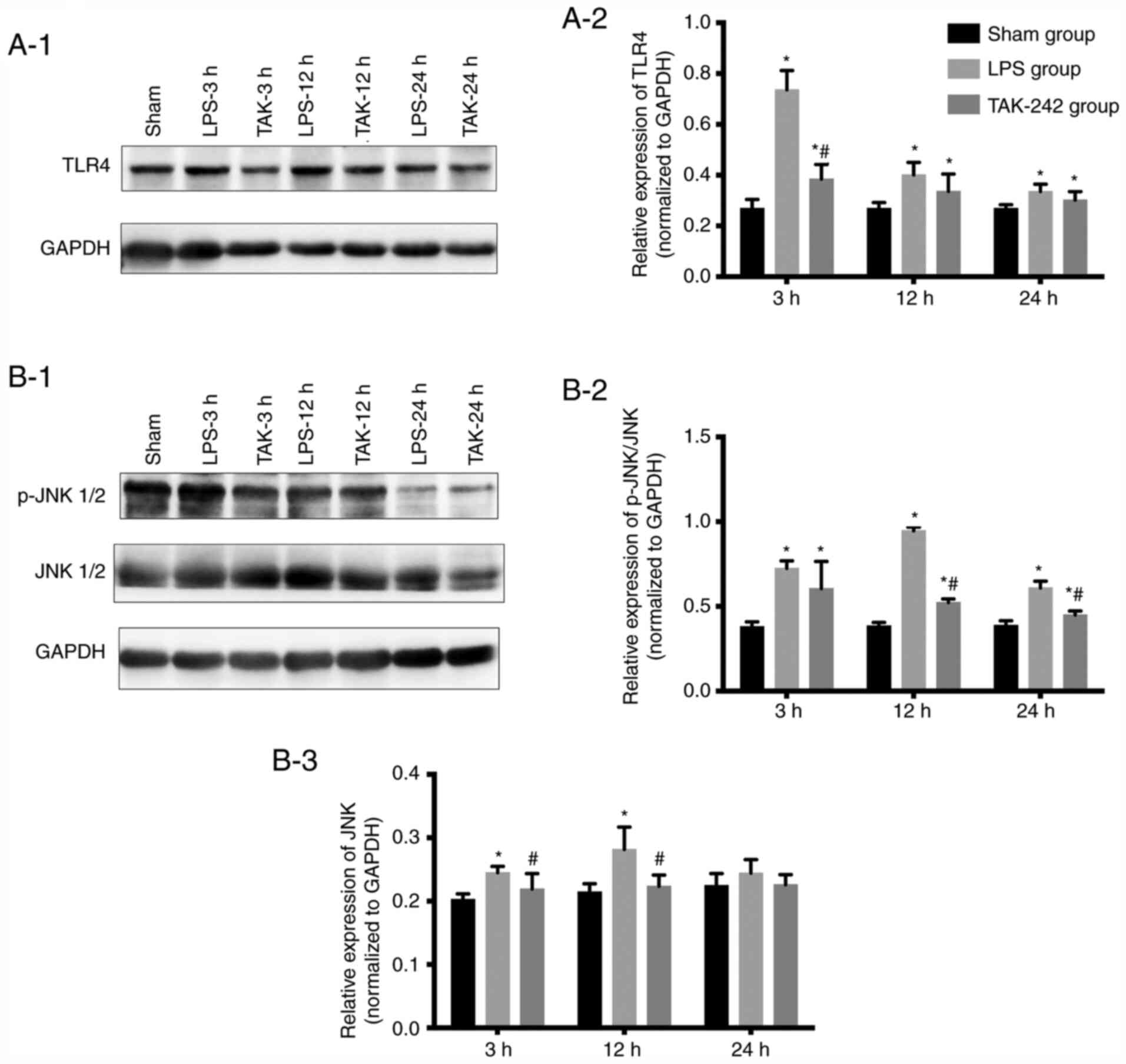
TLR4, JNK and p-JNK protein expression
in the myocardium
As revealed by western blot analysis (Fig. 6), compared with that of the sham
groups, the relative protein expression of TLR4 and p-JNK was
significantly increased in response to LPS (P<0.05), and reached
the peak ~3 and 12 h following LPS injection, respectively.
Pretreatment with TAK-242 significantly reduced the protein levels
of TLR4 and p-JNK (P<0.05). LPS induced a significant increase
in the protein level of the total JNK, compared with the level of
the sham groups (P<0.05). Although TAK-242 significantly
decreased the protein expression of total JNK compared with the LPS
groups at 3 and 12 h (P<0.05), there was no significant
difference at 24 h (P>0.05).
Discussion
The present study demonstrated that, in response to
LPS, the activation of TLR4 and JNK in the myocardium was
upregulated, and the serum levels of TNF-α and cTnI were increased.
Pathological myocardial damage was observed using H&E staining
and transmission electron microscopy, and cardiac function was
shown to be reduced. Inhibition of TLR4 activation led to the
reduction of JNK activation and protein expression of TNF-α in the
plasma, and alleviated histopathological myocardial injury and
improved cardiac function during sepsis in mice. The present data
supported the notion that the TLR4/JNK signaling pathway plays an
important role in myocardial dysfunction induced by sepsis.
Sepsis is a complicated syndrome that begins with a
systemic immune response to an infection and can progress to septic
shock leading to multiple organ failure and death (25). Although the identification of
clinical biomarkers in patients to facilitate earlier diagnosis and
rapid therapeutic intervention is likely to have the greatest
impact on improving prognosis, understanding the underlying
mechanisms of the disease and evaluating the therapeutic efficacy
of novel interventions using animal models are also of critical
importance. Animal models of sepsis are broadly divided into three
categories: Toxemia models, bacterial infection models and
host-barrier disruption models (26). The advantage of toxemia models is
that there is a rapid onset of pathological changes, and they can
have relatively low inter-animal variability because the exact dose
and route of administration can be standardized (26). In addition, toxemia models are often
used to study the basic biology of septic shock, and in particular,
they are employed in mechanistic studies on the role of TLR
signaling (26). Considering the
aforementioned reasons, in the present study, the sepsis models
were established by i.p. injection of 12 mg/kg LPS, a dose
demonstrated to ensure cardiac dysfunction in our preliminary
experiment (19).
TLR4 is a type I transmembrane protein and has a
modular structure composed of an extracellular domain formed by
17–31 leucine-rich repeats and an intracellular domain that is
known as the toll-IL-1 receptor domain, which is responsible for
signaling transmission (27). Since
the identification of TLR4 as the LPS receptor, it has been assumed
that this molecule triggers all the responses to LPS (28). The TLR4 signaling pathway plays an
important role in initiating the innate immune response and
inflammation in sepsis. Typically, activation of TLR4 is preceded
by binding of LPS to CD14 protein, and then CD14 transfers the LPS
to the TLR4/myeloid differentiation factor 2 (MD-2) complex, which
dimerizes and triggers MyD88- and TRIF-dependent production of
pro-inflammatory cytokines (29).
The signaling pathway associated with MAPKs is an important signal
transduction pathway that mediates sepsis-induced myocardial
dysfunction. It has been reported that the activation of ERK, JNK
and p38 is increased in response to LPS stimulation in
cardiomyocytes (30), which is
consistent with the results of the current study that found LPS
administration increased the phosphorylation levels of JNK. A
previous study demonstrated that TLR4/MyD88 triggers a signaling
cascade that leads to early-phase activation of MAPKs (29). In the present study, to investigate
whether TLR4 plays a role in the activation of JNK in response to
LPS, TAK-242 was employed to block TLR4 signaling. TAK-242 is a
small-molecule compound that selectively inhibits the TLR4
signaling pathway by binding directly to the intracellular domain
of TLR4 (27). Pretreatment with
TAK-242 inhibited the activation of JNK induced by LPS, indicating
that JNK is located downstream of TLR4 signaling.
Myocardial dysfunction is regarded as a
well-recognized clinical manifestation of sepsis and septic shock,
and is characterized by biventricular dilation, reduced ejection
fraction (EF), and a recovery period of 7–10 days in patients with
sepsis (31). Echocardiographic
techniques have revealed that either systolic or diastolic
dysfunction is commonly present in sepsis (32). Currently, two-dimensional
echocardiography is the most common diagnostic tool for assessing
myocardial dysfunction in sepsis (33), and the M-mode-derived fractional
shortening, EF and ventricular dimensions are regarded as common
parameters to evaluate cardiac function (20). Among these parameters, EF is most
commonly employed to evaluate LV systolic function (34). Diastolic dysfunction occurs in ~50%
patients with sepsis and is associated with high mortality
(35). LV dilation can be assessed
by estimating the LVEDV, a simple measure of ventricular dilation
that is associated with worse clinical outcomes (34). In the present study, suppressed
cardiac function was confirmed by reduced EF% and FS% in response
to LPS, and was improved by pretreatment with TAK-242 at 3 h
following LPS injection, which suggested that TLR4/JNK signaling
plays an important role in LPS-induced systolic dysfunction.
However, the present study showed that TAK-242 failed to reverse
the inhibitory effect of LPS on LVEF% and LVFS% at 12 and 24 h. The
reasons for this finding may be because TAK-242 has been
demonstrated to act early in the process of TLR4 signaling
(36) or it may be related to the
half-life of TAK-242. Further studies will explore whether TAK-242
can reverse the inhibitory effect of LPS on LVEF% and LVFS% at
different time points through continuous and intermittent
administration of TAK-242. Contrary to previous studies (37), the current data showed that the
decreases in LVEDD and LVEDV were induced by LPS stimulation, which
may be related to decompensation of ventricular diastolic function
and further studies are necessary to explore the reasons.
However, there were some limitations of the present
study. The results showed that LPS had no significant effect on
LVEDD, but in the TAK-242 group, there was either a decrease or an
increase in LVEDD at 3 and 12 h, respectively. These results
suggested that the effect of TAK-242 was independent of LPS
exposure. TLR4 is an important pathway and is most commonly known
for inducing inflammation in response to LPS, which is the most
understood TLR4 agonist. However, it has also been established that
TLR4, as a single protein, has a remarkably low affinity for LPS.
Triggering the TLR4 pathway by LPS requires at least four
co-receptors: MD-2, CD14 and CD11b bound to CD18 (38). In response to LPS, TLR4-expressing
normal cells significantly increase the production of inflammatory
factors due to the combined upregulation of TLR4, its intracellular
adapters and co-receptors, which enhances cooperation among the
pathway's components (38). TAK-242
is a small-molecule compound that selectively inhibits the TLR4
signaling pathway by binding directly to Cys747 in the
intracellular TLR4 domain (27).
Therefore, theoretically, the effect of TAK-242 cannot be
independent of LPS exposure. In order to examine the effect of
TAK-242 on various parameters, an additional group of sham animals
treated with TAK-242 is needed, so further relevant experiments
will be performed in the future.
Previous evidence suggests that structural
abnormalities may play a critical role in the pathophysiology of
sepsis-induced cardiac dysfunction (39). In previous years, cardiac biomarkers
have been used to detect myocardial injury (40). Troponin-I, a sensitive and specific
marker of myocardial injury, is the subunit of the troponin
complex. Previous studies have demonstrated that cardiac troponin
release in sepsis is related to LV dysfunction and poor outcomes
(41,42). In the present study, consistent with
previous findings (40,43), LPS-induced myocardial injury was
confirmed by the loss of integrity of myocardial membranes on
histological examination (Figs. 3
and 4) and elevation of the cTnI
enzyme. Furthermore, myocardial injury and elevation of cTnI could
be attenuated by TAK-242 administration. According to the current
results, it could be suggested that the TLR4/JNK signaling pathway
plays a critical role in myocardial injury induced by LPS.
Systemic inflammatory response syndrome (SIRS) is a
key characteristic in the development of organ injury during sepsis
and overactivated inflammatory response plays a critical role in
sepsis (40). Although there is no
definitive cause-and-effect association between systemic cytokine
levels and survival outcomes in sepsis, numerous studies have
reported that inhibition of the cardiac inflammatory processes is
beneficial in sepsis-induced cardiac dysfunction (43,44).
The production of pro-inflammatory cytokines stimulated by LPS has
been demonstrated to be one of the primary underlying mechanisms of
cardiac dysfunction (44). As a
trigger of inflammation, TNF-α participates in the production of
IL-1β, and together with IL-1β, it induces the formation of
secondary inflammatory factors, such as IL-6, resulting in an
inflammatory cascade (45). In
addition, cytokines, in particular TNF-α, can also induce the
release of additional inflammatory factors, such as inducible
nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) and reactive
oxygen species (ROS), which eventually induce myocardial
dysfunction (46,47). Administration of TNF-α directly
suppresses myocardial function in animal and human cardiomyocytes
(48,49), and treatment with anti-TNF-α
protects cardiac function in sepsis animal models and patients with
sepsis (50,51). In the present study, LPS increased
the production of TNF-α in mouse serum, which is supported by a
previous finding that inflammatory cytokines such as TNF-α were
induced by LPS in mice (30).
Inhibition of TLR4 by pretreatment with TAK-242 significantly
reduced the protein levels of TNF-α induced by LPS. These results
indicated that TLR4/JNK signaling was an important pathway leading
to TNF-α expression in response to LPS stimulation. However, the
present study failed to include assays that demonstrated the
differential expression of inflammatory-related proteins, such as
iNOS, COX-2 and IL-6, and accumulation of ROS. Relevant experiments
to determine the expression of inflammatory-related proteins will
be performed in the future.
In conclusion, the present study demonstrated that
the TLR4/JNK signaling pathway appeared to be of critical
importance in sepsis, as its inhibition attenuated cardiac
dysfunction and the overactivated inflammatory response. Therefore,
this study indicated that the TLR4/JNK signaling pathway plays an
important role in regulating myocardial dysfunction in sepsis.
Acknowledgements
Not applicable.
Funding
This study was supported by The Health and Family
Planning Commission of Tianjin of China (grant no. 2014KY32).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
PL and CC conceived, designed and coordinated the
present study. LH performed the majority of experiments, analyzed
the data and drafted the manuscript. SS, YS, SL and JW helped
peform the experiments. All authors reviewed the results, and read
and approved the final manuscript.
Ethics approval and consent to
participate
All the experimental procedures were approved by the
Animal Experiments Ethics Committee of Nankai University (Tianjin,
China; approval no. 10011).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Markwart R, Saito H, Harder T, Tomczyk S,
Cassini A, Fleischmann-Struzek C, Reichert F, Eckmanns T and
Allegranzi B: Epidemiology and burden of sepsis acquired in
hospitals and intensive care units: A systematic review and
meta-analysis. Intensive Care Med. 46:1536–1551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the Global Burden of Disease
Study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Antonucci E, Fiaccadori E, Donadello K,
Taccone FS, Franchi F and Scolletta S: Myocardial depression in
sepsis: From pathogenesis to clinical manifestations and treatment.
J Crit Care. 29:500–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Buras JA, Holzmann B and Sitkovsky M:
Animal models of sepsis: Setting the stage. Nat Rev Drug Discov.
4:854–865. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fallach R, Shainberg A, Avlas O, Fainblut
M, Chepurko Y, Porat E and Hochhauser E: Cardiomyocyte Toll-like
receptor 4 is involved in heart dysfunction following septic shock
or myocardial ischemia. J Mol Cell Cardiol. 48:1236–1244. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suffredini AF, Fromm RE, Parker MM,
Brenner M, Kovacs JA, Wesley RA and Parrillo JE: The cardiovascular
response of normal humans to the administration of endotoxin. N
Engl J Med. 321:280–287. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fenton KE and Parker MM: Cardiac function
and dysfunction in sepsis. Clin Chest Med. 37:289–298. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gay NJ, Symmons MF, Gangloff M and Bryant
CE: Assembly and localization of Toll-like receptor signalling
complexes. Nat Rev Immunol. 14:546–558. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Conejeros I, Gibson AJ, Werling D,
Muñoz-Caro T, Hermosilla C, Taubert A and Burgos RA: Effect of the
synthetic Toll-like receptor ligands LPS, Pam3CSK4, HKLM and FSL-1
in the function of bovine polymorphonuclear neutrophils. Dev Comp
Immunol. 52:215–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Y, Liu X, Yue H, Ji J, Dou H and Hou
Y: Anti-inflammatory effects of benzenediamine derivate FC-98 on
sepsis injury in mice via suppression of JNK, NF-κB and IRF3
signaling pathways. Mol Immunol. 67:183–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang C, Zhang C, Zhao X, Kuang X, Tang H
and Xiao X: Differential regulation of mitogen-activated protein
kinase signaling pathways in human with different types of mitral
valvular disease. J Surg Res. 181:49–59. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sabio G and Davis RJ: TNF and MAP kinase
signalling pathways. Semin Immunol. 26:237–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Plotnikov A, Zehorai E, Procaccia S and
Seger R: The MAPK cascades: Signaling components, nuclear roles and
mechanisms of nuclear translocation. Biochim Biophys Acta.
1813:1619–1633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng F, Qi Y, Dong C and Yang C: PVT1
regulates inflammation and cardiac function via the MAPK/NF-κB
pathway in a sepsis model. Exp Ther Med. 16:4471–4478.
2018.PubMed/NCBI
|
|
16
|
Lee SM, Suk K and Lee WH: Myristoylated
alanine-rich C kinase substrate (MARCKS) regulates the expression
of proinflammatory cytokines in macrophages through activation of
p38/JNK MAPK and NF-κB. Cell Immunol. 296:115–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Z, Wu Q, Nie X, Guo J and Yang C:
Infusion of esmolol attenuates lipopolysaccharide-induced
myocardial dysfunction. J Surg Res. 200:283–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng T, Zhang T, Lu X and Feng Q:
JNK1/c-fos inhibits cardiomyocyte TNF-alpha expression via a
negative crosstalk with ERK and p38 MAPK in endotoxaemia.
Cardiovasc Res. 81:733–741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hu L, Li P, Chang C, Liu S, Song Y, Zhao F
and Liu T: Establishment and evaluation of mouse models of septic
myocardial injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue.
30:342–345. 2018.(In Chinese). PubMed/NCBI
|
|
20
|
Chen J, Wei J, Wang L, Zhu Y, Li L, Akinyi
M, Gao X and Fan G: Cardioprotection against ischemia/reperfusion
injury by QiShenYiQi Pill® via ameliorate of multiple
mitochondrial dysfunctions. Drug Des Devel Ther. 9:3051–3066. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Täng MS, Redfors B, Shao Y and Omerovic E:
Velocity vector imaging fails to quantify regional myocardial
dysfunction in a mouse model of isoprenaline-induced
cardiotoxicity. Echocardiography. 29:818–826. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teng B, Tilley SL, Ledent C and Mustafa
SJ: In vivo assessment of coronary flow and cardiac function after
bolus adenosine injection in adenosine receptor knockout mice.
Physiol Rep. 4:42016. View Article : Google Scholar
|
|
23
|
Wang GQ, Tang T, Wang ZS, Liu YY, Wang L,
Luo PF and Xia ZF: Overexpression of hypo-phosphorylated iκBβ at
Ser313 protects the heart against sepsis. PLoS One.
11:e01608602016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kenneth J: Livak, Thomas D. Schmittgen;
Analysis of relative gene expression data using real-time
quantitative PCR and the 2−ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iskander KN, Osuchowski MF,
Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C and Remick
DG: Sepsis: Multiple abnormalities, heterogeneous responses, and
evolving understanding. Physiol Rev. 93:1247–1288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lilley E, Armstrong R, Clark N, Gray P,
Hawkins P, Mason K, López-Salesansky N, Stark AK, Jackson SK,
Thiemermann C, et al: Refinement of animal models of sepsis and
septic shock. Shock. 43:304–316. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuzmich NN, Sivak KV, Chubarev VN, Porozov
YB, Savateeva-Lyubimova TN and Peri F: TLR4 signaling pathway
modulators as potential therapeutics in inflammation and sepsis.
Vaccines (Basel). 5:52017.
|
|
28
|
Beutler B, Du X and Poltorak A:
Identification of Toll-like receptor 4 (Tlr4) as the sole conduit
for LPS signal transduction: Genetic and evolutionary studies. J
Endotoxin Res. 7:277–280. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Płóciennikowska A, Hromada-Judycka A,
Borzęcka K and Kwiatkowska K: Co-operation of TLR4 and raft
proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life
Sci. 72:557–581. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li P, Chen XR, Xu F, Liu C, Li C, Liu H,
Wang H, Sun W, Sheng YH and Kong XQ: Alamandine attenuates
sepsis-associated cardiac dysfunction via inhibiting MAPKs
signaling pathways. Life Sci. 206:106–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jeong HS, Lee TH, Bang CH, Kim JH and Hong
SJ: Risk factors and outcomes of sepsis-induced myocardial
dysfunction and stress-induced cardiomyopathy in sepsis or septic
shock: A comparative retrospective study. Medicine (Baltimore).
97:e02632018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sevilla Berrios RA, O'Horo JC, Velagapudi
V and Pulido JN: Correlation of left ventricular systolic
dysfunction determined by low ejection fraction and 30-day
mortality in patients with severe sepsis and septic shock: A
systematic review and meta-analysis. J Crit Care. 29:495–499. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haileselassie B, Su E, Pozios I, Niño DF,
Liu H, Lu DY, Ventoulis I, Fulton WB, Sodhi CP, Hackam D, et al:
Myocardial oxidative stress correlates with left ventricular
dysfunction on strain echocardiography in a rodent model of sepsis.
Intensive Care Med Exp. 5:212017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vallabhajosyula S, Pruthi S, Shah S, Wiley
BM, Mankad SV and Jentzer JC: Basic and advanced echocardiographic
evaluation of myocardial dysfunction in sepsis and septic shock.
Anaesth Intensive Care. 46:13–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hoffman M, Kyriazis ID, Lucchese AM, de
Lucia C, Piedepalumbo M, Bauer M, Schulze PC, Bonios MJ, Koch WJ
and Drosatos K: Myocardial strain and cardiac output are preferable
measurements for cardiac dysfunction and can predict mortality in
septic mice. J Am Heart Assoc. 8:e0122602019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takashima K, Matsunaga N, Yoshimatsu M,
Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y and Ii M:
Analysis of binding site for the novel small-molecule TLR4 signal
transduction inhibitor TAK-242 and its therapeutic effect on mouse
sepsis model. Br J Pharmacol. 157:1250–1262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sanfilippo F, Corredor C, Fletcher N,
Landesberg G, Benedetto U, Foex P and Cecconi M: Diastolic
dysfunction and mortality in septic patients: A systematic review
and meta-analysis. Intensive Care Med. 41:1004–1013. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ran S, Bhattarai N, Patel R and
Volk-Draper L: TLR4-induced inflammation is a key promoter of tumor
growth, vascularization, and metastasis. Translational Studies on
Inflammation. 2020. View Article : Google Scholar
|
|
39
|
Lin Y, Xu Y and Zhang Z: Sepsis-induced
myocardial dysfunction (SIMD): The pathophysiological mechanisms
and therapeutic strategies targeting mitochondria. Inflammation.
43:1184–1200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fan TT, Feng XY, Yang YZ, Gao F and Liu Q:
Downregulation of PI3K-γ in a mouse model of sepsis-induced
myocardial dysfunction. Cytokine. 96:208–216. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cheng H, Fan WZ, Wang SC, Liu ZH, Zang HL,
Wang LZ, Liu HJ, Shen XH and Liang SQ: N-terminal pro-brain
natriuretic peptide and cardiac troponin I for the prognostic
utility in elderly patients with severe sepsis or septic shock in
intensive care unit: A retrospective study. J Crit Care.
30:654.e9–654.e14. 2015. View Article : Google Scholar
|
|
42
|
Klouche K, Pommet S, Amigues L, Bargnoux
AS, Dupuy AM, Machado S, Serveaux-Delous M, Morena M, Jonquet O and
Cristol JP: Plasma brain natriuretic peptide and troponin levels in
severe sepsis and septic shock: Relationships with systolic
myocardial dysfunction and intensive care unit mortality. J
Intensive Care Med. 29:229–237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang T, Yan T, Du J, Wang S and Yang H:
Apigenin attenuates heart injury in lipopolysaccharide-induced
endotoxemic model by suppressing sphingosine kinase 1/sphingosine
1-phosphate signaling pathway. Chem Biol Interact. 233:46–55. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu X, Jia B, Wang F, Lv X, Peng X, Wang Y,
Li H, Wang Y, Lu D and Wang H: α1 adrenoceptor
activation by norepinephrine inhibits LPS-induced cardiomyocyte
TNF-α production via modulating ERK1/2 and NF-κB pathway. J Cell
Mol Med. 18:263–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Z, Bu L, Yang P, Feng S and Xu F:
Alleviation of sepsis induced cardiac dysfunction by overexpression
of Sestrin2 is associated with inhibition of p S6K and activation
of the pAMPK pathway. Mol Med Rep. 20:2511–2518. 2019.PubMed/NCBI
|
|
46
|
Suzuki T, Suzuki Y, Okuda J, Kurazumi T,
Suhara T, Ueda T, Nagata H and Morisaki H: Sepsis-induced cardiac
dysfunction and β-adrenergic blockade therapy for sepsis. J
Intensive Care. 5:222017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Li M, Wang H and Astragaloside
IV: Astragaloside IV alleviates the myocardial damage induced by
lipopolysaccharide via the Toll-like receptor 4 (TLR4)/nuclear
factor kappa B (NF-κB)/proliferator-activated receptor α (PPARα)
signaling pathway. Med Sci Monit. 25:7158–7168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Natanson C, Eichenholz PW, Danner RL,
Eichacker PQ, Hoffman WD, Kuo GC, Banks SM, MacVittie TJ and
Parrillo JE: Endotoxin and tumor necrosis factor challenges in dogs
simulate the cardiovascular profile of human septic shock. J Exp
Med. 169:823–832. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cain BS, Meldrum DR, Dinarello CA, Meng X,
Joo KS, Banerjee A and Harken AH: Tumor necrosis factor-alpha and
interleukin-1beta synergistically depress human myocardial
function. Crit Care Med. 27:1309–1318. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Boillot A, Capellier G, Racadot E,
Wijdenes J, Herve P and Barale F: Pilot clinical trial of an
anti-TNF alpha monoclonal antibody for the treatment of septic
shock. Clin Intensive Care. 6:52–56. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Herbertson MJ, Werner HA, Goddard CM,
Russell JA, Wheeler A, Coxon R and Walley KR: Anti-tumor necrosis
factor-alpha prevents decreased ventricular contractility in
endotoxemic pigs. Am J Respir Crit Care Med. 152:480–488. 1995.
View Article : Google Scholar : PubMed/NCBI
|