Introduction
In 1672, English physician Thomas Willis first
described signs and symptoms of a disease that affected the muscles
(1), which is now known as the
autoimmune disease Myasthenia Gravis (‘my’ means muscle and
‘asthenia’ means illness/weakness in Greek, ‘gravis’ means severe
in Latin).
This heterogeneous disorder biologically is mainly
caused by antibodies against the muscular acetylcholine receptor
(AChR) at the neuromuscular juction and recent developments have
shown that other autoantibodies are involved in the manifestation
of MG (2). These antibodies lead to
loss of the muscle receptors, which causes a defect in
neuromuscular transmission presenting with involuntary progressive
muscle weakness. Patients usually share a variety of symptoms,
which most commonly include dyplopia (double vision), dysphagia,
drooping (ptosis) of one or both eyelids, weakness in the neck,
arms and legs (3).
The diagnosis of MG is primarily based on the
serological detection of AChR antibodies (4) and the presence of the thymus gland in
adults is taken under consideration as the thymus plays a profound
role in the pathogenesis of MG and thymectomy is one of the current
therapeutic options.
As a complex genetic disorder, MG has some shared
associations with other autoimmune disorders with the most
reproducible finding being the association of the 8.1 ancestral
haplotype of HLA-DR3 for class II and HLA-B8 and A1 for class I
(5) in Caucasians. Phenotypically,
Myasthenia Gravis presents most commonly in its generalized form
with anti-AChR antibodies as an early-onset disorder, presented
before the age of 40 years, usually in females. Other forms of the
disease can be described in both adults and juveniles, as analyzed
below.
Several polymorphisms have been identified in GWS
studies in relation with myasthenia gravis and the important role
of epigenetics is yet to be discovered. Along with genetic
predisposition, environmental factors can lead to the development
of autoimmunity as well as affect the severity of autoimmune
diseases.
The symptoms of MG can be treated very successfully
with different drugs, even leading to remission in some cases.
Harriet Edgeworth, a myasthenic herself, in 1930 discovered that
ephedrine sulfate improved her condition and muscle functionality
and a few years later, in 1934 Mary Walker was the first to
successfully administer physostigmine salicylate, and later
neostigmine bromide, to a patient with MG (1). Since then, other drugs have been added
in the fight against MG, from the anticholinesterase pyridostigmin
bromide to the latest monoclonal antibodies (mAb) drugs.
Biology of autoimmune generalized myasthenia
gravis, MG related antibodies and the thymus gland
Autoimmune myasthenia gravis (MG) is a chronic
autoimmune disease of the neuromuscular junction, characterized by
a post-synaptic blockade of the nervous transmission and presenting
clinically with varying degrees of weakness of the skeletal muscles
and generalized fatigue. MG has been identified and reported in
literature centuries ago without too much progress in regard to
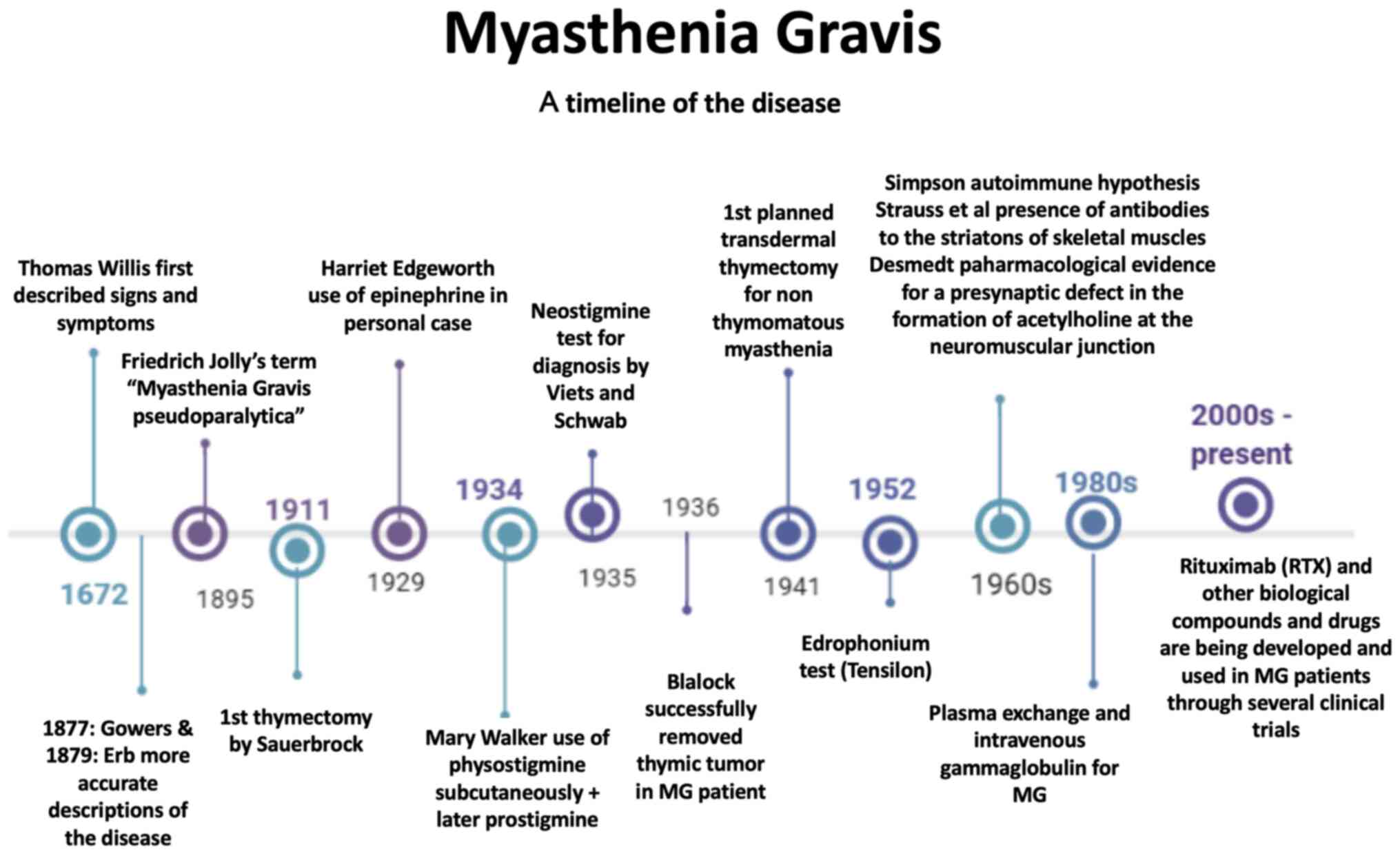
finding a cure or efficient therapy (Fig. 1). It is considered to be a rare
disease with a prevalence of 8–30 patients per 100,000 population
in Caucasians that has doubled over the last decades and continues
to increase. A 10% - 15% of all patients are estimated through
studies to be pediatric patients with the percentage going up to
50% among Asian populations.
Both pediatric and adult patients usually share a
variety of symptoms, which most commonly include dyplopia (double
vision), dysphagia, drooping of one or both eyelids (ptosis), nasal
or impaired speech, change of facial expressions, weakness in the
neck (head droop), arms and legs. It has the potential of a
life-threating disease when there is involvement of the respiratory
muscles. The symptoms may be acute or subacute and periods of
remission or relapse may occur. Despite the symptoms' pool that
most patients share, MG is considered to be a heterogeneous
disease.
As a heterogeneous disorder, myasthenia may present
as ocular or generalized in adults and juveniles along with
transient neonatal myasthenia and congenital myasthenic syndromes.
Among patients with generalized myasthenia gravis, there are
different human leukocyte antigen (HLA) associations between
early-onset and late-onset generalized MG (2).
The main characteristic of MG biologically is the
production of antibodies against the muscular acetylcholine
receptor (AChR). In ~5% of patients anti-MUSK (muscle-specific
kinase) antibodies can be present and there is a small percentage
(~10%) of seronegative patients. The need to discover and detect
new antigenic targets is significant in these patients in order to
identify them and establish new therapeutic treatments. Towards
this direction the past few years new assays have been used and it
has been possible to identify new autoantibodies in some MG
patients, like the anti-Titin antibodies or the low-density
lipoprotein receptor-related protein 4 (LRP4) as an autoantigen in
MG. Achieving identification of the autoantibodies present in each
MG patient allows not only for a correct diagnosis but also for a
more targeted therapeutic strategy with advanced antigen-specific
treatment (3).
MG with anti- AChR antibodies
An approximate 80% of patients present with
anti-AChR antibodies in their serum, which target the Ach-gated
cation channel, nicotinic α1 AChR. Acetylcholine receptors (AChRs)
are found on the surface of the muscle cells and are concentrated
between the muscle cells and the nerve cells (6). The five protein chains in adults and
neonates which compose AChRs, form a ‘bridge’ shaped as a long tube
that crosses the cell membrane and have binding sites for
acetylcholine on the external side. They contain the immunogenic
region that is recognized by anti-AChR antibodies and when
acetylcholine binds on these chains, an alteration in the
receptor's shape is caused, the channel formed by the ‘tube’ opens
and positively charged ions cross the membrane, causing the muscles
to contract. Patients with generalized anti- AChR antibodies MG
have a lower density of AChR at the neuromuscular junction, which
leads to involuntary muscle weakness, as the signal for the muscle
to work does not go through (7).
Patients in this category can be divided in those
with ocular and generalized form of the disease. The generalized
form can be further divided in groups according to the age of
onset. Early-onset myasthenia (EOMG) patients are under the age of
50 and usually present with a high number of anti- AChR antibodies
in their serum and hyperplasia of the thymus. In this category the
vast majority of patients are women, and the role of sex hormones
should be furtherly investigated. Late-onset (LOMG) patients are
over the age of 50 and usually present with a thymoma (malignant)
and are more prone to severe respiratory crises. Over the age of
60, a category of mostly male patients does not present with
thymoma and is considered to be a very late-onset form of MG. Both
forms can be also found in juvenile myasthenia gravis (JMG).
The muscle AChR composes of five subunits and each
one has an intracellular domain that is partially structured, one
highly structured extracellular domain and four transmembrane
domains. The antibodies target the extracellular domain and the
heterogeneity of the disease is that in a single patient antibody
against all subunits can be detected. Despite this, an approximate
50% of the antibodies bind to the α subunit and these appear to be
more pathogenic compared to others (8). The α subunit is directly involved in
neuro transmission due to its involvement in acetylholine binding.
Furthermore, it presents with a known functional polymorphism at
the protein level that derives from an alternative splicing of an
additional P3A exon (9). The AChR
antibodies can cause AChR loss by activating complement at the
postsynaptic membrane, as they mostly belong to the IgG1 and IgG3
subclasses (10). The signal
transduction is therefore lost and, furthermore, there is an
interference with receptor activation by acetylholine when
antibodies bind close to the ligand binding site.
The diagnosis of MG is primarily based on the
serological detection of AChR antibodies. A blood test is usually
the first step along with physical examination and an assessment of
response to acetylcholinesterase (AChE) inhibitors. The serological
testing provides a title of AChR antibodies, which does not always
correlate with the severity of the disease in some patients but can
be related to the response to therapy (11).
It is suggested by physicians to monitor the levels
of AChR antibodies in order to monitor a patient's progress and
guide the disease management by modifying, if necessary, drug
dosage. Concerning transient neonatal myasthenia, maternal antibody
titers have been found to correlate with the onset and/or severity
of the disorder in infants.
MG with anti-MuSK antibodies
The Muscle Specific Kinase (MuSK) protein has an
essential role in the neuromuscular junction and is important for
the clustering of the AChRs. In the early 21st century, anti-MuSK
antibodies were identified in about 40% of patients with negative
anti-AChR antibodies (7). These
patients often present with severe symptoms, including bulbar
dysfunction, respiratory insufficiency and atrophy of the facial
and tongue muscles.
In animal models of MuSK MG histopathological
studies have revealed that anti-MuSK antibodies cause contraction
of motor terminals, significant loss of acetylcholine receptor
(AChR) expression and a reduction in synaptic folds at the
postsynaptic membrane in the absence of complement involvement.
Failure of neuromuscular transmission at pre- and postsynaptic
membranes of the neuromuscular junctions has been observed in both
patients and animal models of MuSK MG (12). Anti-MuSK antibodies in MG patients
mainly belong to the IgG4 subclass, which has distinct properties
directly associated with the MG pathology.
Seronegative MG
Patients that are seronegative have a similar
clinical presentation of the condition with anti-AChR antibodies
patients. Although anti-AChR antibodies are not detected by the
classical assay, they appear to be present and to predominantly
belong to the IgG1 isotype that activates the complement.
MG and other antibodies
Recent developments have shown that the protein
Titin plays a role in sarcomere assembly and elasticity. The
Ryanodin receptor (RyR) is a calcium channel participating in the
muscle contraction through releasing calcium from the sarcolemma
into the cytoplasm.
Antibodies against LRP4 were recently reported in MG
patients without detectable AChR or MuSK antibodies, but that
percentage varies among populations examined and assays used from
2% to 33% (13). LRP4 is a
transmembrane protein with a central role in synaptic development
and LRP4 antibodies have been shown to activate the complement and
therefore playing a role in MG as well as in other disorders, like
ALS (amyotrophic lateral sclerosis) (14).
In more rare cases, antibodies against cytokines,
interferon-α, interferon-ω and IL-12 can be present in patients
with thymoma or LOMG, but their pathogenic role is not yet
clear.
Thymus hyperplasia
The thymus plays a profound role in the pathogenesis
of MG. A large percentage of patients have pathological changes in
their thymic tissue with >70% of them showing hyperplasia and
about 10–15% having a thymoma.
Over half the patients with AChR-MG have a
hyperplastic thymus characterized by production of antibodies by B
cells, which can be decreased by immunosuppressive drugs,
production of AChR antibodies and the presence of
anti-AChR-reactive T cells. CD4+ CD25+ Treg
cells play an important role in the control of both the autoimmune
response and the immune response and in MG patients they appear to
be defective (15). The antibodies
created exit the thymus and generate an autoimmune reaction in the
neuromuscular junction against AChRs. However, the exact mechanism
of the attack against AChRs is still unclear. A viral mechanism can
also be taken under consideration as latest data show that
poliovirus and EBV have been detected in the thymus.
Thymoma
Thymoma is observed in about 10% of adult MG
patients and is caused by the abnormal development of epithelial
cells whereas children have rarely been found to develop thymomas.
Thymomas are categorised in 5 types according to the nature of the
cortical or medullary epithelial cells involved in the tumor: Type
A, B1, B2, B3 or AB, with the most common types being B1 and B2 in
patients over 40 years of age. Autoimmune mechanisms have a strong
link with thymoma development, as over half the patients that have
a thymoma also present in MG or other autoimmune syndromes
(16).
In contrast to thymic hyperplasia, B cells and
germinal centers are not present and thymomas lack medulla. Also,
the major histocombatibility complex (MHC class II) antigens and
the autoimmunity regulator factor (AIRE) are deficient in thymoma
and that could suggest that there is a difference in the
autoimmunisation process (17). The
existing scientific data suggest that T cell selection may be
affected in different ways by a thymoma and that the lack of
CD4+ CD25+ Tregs along with a change in
expression of AChR subunits may also play a role.
Thymectomy
In early-onset Myasthenia Gravis, thymectomy is one
of the current therapeutic options. Studies show that the removal
of the thymus has a positive effect on patients with generalized
MG, (18) who usually have a
stabilization of their symptoms 3–5 years after the surgery and
often present with reduced severity or remission, especially if the
procedure is performed soon after the onset of symptoms. It is not
suggested for seronegative or late-onset patients. In children it
may have an effect for those unresponsive to treatment and may,
also, protect patients with ocular form of JMG to undergo into the
generalized form. The improvement that occurs after thymectomy may
be related to the elimination of thymic B cells but the exact
pathway is not clear yet.
Clinical Genomics and phenotypes of the
disorder
Clinical Genomics
As a complex genetic disorder, MG has some shared
associations with other autoimmune disorders. The most reproducible
finding is the association of the 8.1 ancestral haplotype of
HLA-DR3 (DRB1*03) for class II and HLA-B8 and A1 for class I with
early-onset AchR-MG and thymic hyperplasia in Caucasians (5), with a 60% frequency in patients and
the risk associated with haplotype increases about 7-times for two
copies. The Myasthenia gravis with thymus hyperplasia (MYAS1) locus
is one of the three loci of the HLA region identified and
encompasses 36 genes in a region of 1.2Mb at the boundaries of
class I on the telomeric side and class III, confirming that the B8
allele is predominant over the DR3 (19). The other two loci have effects on
serum titers of AchR antibodies: a QTL associated with elevated
titers, overlapping with MYAS1 and a locus mapped toward the class
I region, which suppresses the enhancing effect of the QTL. HLA-DR3
and DR7 may have opposing effects on the MG phenotype, with the
first associated with EOMG and the second associated with LOMG with
Titin antibodies. Using a high-resolution haplotype map as
reference, a panel of 1472 single-nucleotide polymorphisms (SNPs)
was genotyped across the classic MHC region in MG recently: The
strongest association arose from the class I region specifically in
the vicinity of the HLA complex protein 5 (HCP5) gene, while the
strongest associated HLA allele was HLA-C*0701; paucity of
significant signals was detected in the class II region. MuSK-MG
has a suggested association with LA-DR14-DQ5 while no reproducible
MHC associations have been reported in MG with thymoma (20).
The 1858T (rs2476601) functional SNP located in the
PTPN22 gene is a minor allele that may weaken T cell receptor
signaling and is represented in patients without a thymoma and with
anti-Titin antibodies, who also present a lower IL-2 expression
(7). Also, the CHRNA1 locus encodes
the α subunit of the AChR and a minor G allele of a functional SNP
is associated with myasthenia Gravis. CHRNA1 is directly involved
in acetylcholine binding and therefore in neuro transmission. It
also presents a functional polymorphism, the only one that is known
at the protein level in the AChR -subunit and that results from an
alternative splicing of the additional P3A exon. This exon codes
for 25 amino-acids in the N-terminal extra-cellular domain that
disrupt the main immunogenic region and impair the channel function
of the AChR pentamer. These features made the CHRNA1 gene that
codes for the α-subunit a good candidate for influencing the
genetic susceptibility to MG.
A number of SNPs have been associated with EOMG and
LOMG through genome-wide association and other studies (21). Rs231770, rs4263037, rs9270986 as
well as rs601006 and rs9271850 in the HLA region were analyzed in
one GWAS (21) in order to identify
their influence in AChR antibody-positive MG patients.
Apart from the HLA complex, other unlinked genetic
loci have been investigated for their involvement in autoimmune
disorders and specifically in MG. Interferon regulatory factor 5
(IRF-5), which induces IFN gene expression and upregulates
cytocines like IL-6, TNF-α, IL-12, is influenced by SNP rs10954213
in the 3′ UTR and by rs60344245 in an IRF-5 exon. The TNFα-induced
protein 3 (TNFAIP3) has an inhibitory effect on on NF-κB signaling
and the SNP rs13207033 in the 6p23 region probably affects
regulatory DNA elements (22).
Interleukin-10 (IL-10) is an anti-inflammatory cytocine that
stimulates TH2 cells and suppresses TH1 cells at the same time
(23) and has been suggested to
cause an increase of anti-AChR antibody levels in MG. Three SNPs in
the human IL-10 gene, rs45552637, rs1800872 and rs1800896 determine
the formation of three haplotypes (GCC, ACC, and ATA) with the
ACC/GCC genotype being the most frequently observed in MG patients
(19).
Several other genes were reported to be associated
(encoding β2-adrenergic receptor, IL-1β, IL-10, IFN-γ, TCRα, Ig
heavy chain, Ig κ-chain, TNF-α, TNF-β, type 2 receptors for Fc
fragment of IgG and cysteine protease cathepsin V) with various
subtypes of MG, but there is no confirmation yet.
Phenotypes of the disorder
Most commonly generalized myasthenia Gravis with
anti-AChR antibodies is an early-onset disorder, presented before
the age of 40 years, usually in females. It is rarer to appear
before the age of 20 and is usually accompanied by thymus
hyperplasia. Often patients develop within a decade of the MG onset
other autoimmune diseases, such as thyreoditis and diabetes or may
also have them prior to MG.
In children, Transient Neonatal Myasthenia (TNM),
Juvenile MG and Congenital Myasthenic Syndromes (CMS) may appear.
The CMS are a group of disorders characterized by fatigue in the
skeletal muscles with an onset shortly after or at birth with
variable severity. TMN affects newborns born to mothers with MG and
is a temporary form of the disease. It shares some similarities
with the adult and juvenile forms of MG, like affecting the
respiratory system, and occurs due to the transplacental passage of
antibodies, usually against the AChR antigen. Maternal antibodies
recognize the fetal form of the AChR and inhibit neuromuscular
transmission in the baby, leading to a transient MG at birth
(24). In a small percentage of
cases, AChR antibodies developing during pregnancy may target fetal
AChRs and result in severe arthrogryposis of the fetus even if the
mother has no clinical signs. Neonatal myasthenia is a form of the
disease presenting with AChR mutations. Maternal antibodies
recognize the fetal form of the AChR and inhibit neuromuscular
transmission in the baby, leading to a transient MG at birth
(24). In this case, the blocking
effects appear to trigger neonatal MG and are correlated with the
severity of the disease in the child.
Patients with anti-MuSK antibodies are not so common
and tend to be mainly female with no thymic pathology (thymoma is
exceptional) (25). Clinically
these patients have an oculopharyngeal onset rapidly progressing
into dysphonia, dysphagia and difficulty in chewing. Many patients
develop respiratory failure but there is rarely limb weakness in
contrast to AChR-MG. Another phenotype includes only ocular and
neck weakness, that can rapidly evolve into a myasthenic crisis
with respiratory failure.
Late-onset MG and MG with a thymoma are phenotypes
defined by the presence of anti-Titin, anti-RyR and anti-cytocine
(IL-12, IFN-α) antibodies. These phenotypes also present with
severe oropharyngeal and neck muscle involvement, along with
respiratory crisis and low response to thymectomy.
Ocular myasthenia can be characterized as the
primary onset in about half the patients that will develop
generalized MG, especially in cases with no diagnosis and no
treatment. The generalized form of the disease is developed withing
6–24 months after the ocular myasthenia and a functional SNP in the
regulatory region of the DAF gene (decay-accelerating factor)
associates with ocular myasthenia.
In certain populations familial cases of myasthenia
gravis have been observed, suggesting a role of familial factors in
the pathogenesis of the disease. In 2013 in Taiwan a
population-based study (N=23,422,955) revealed that 0.064% of
individuals had at least one first-degree relative with MG with a
familial prevalence of 0.205% in the first-degree relatives with a
family history (26). In Buenos
Aires, a case study of 190 MG patients in one public hospital was
performed, in which familial autoimmune Myasthenia Gravis
represented 3.2% of the cases (6 patients had a first-degree family
member also affected by MG) (27).
The role of Epigenetics
Alterations in genome architecture can result in
autoimmune diseases. Several polymorphisms have been identified in
GWS studies in relation with myasthenia gravis, but the
manifestation of the disease can not solely be described by these
gene abnormalities. Epigenetics play an important role and studies
in twins have shown that epigenetic deregulation contributes to the
severity and even the manifestation of this autoimmune
disorder.
Through the development of each individual, a set of
epigenetic factors can differentiate the activity of specific genes
or even whole genomic regions, including methylations in the DNA
sequence and modifications of histone proteins. These modifications
are not only the result of heritable epigenetic changes that happen
during gene expression, but environmental factors also have a great
influence. Dietary habits and stress are examples of environmental
factors that may cause the manifestation of autoimmune disorders
and their severity. In other autoimmune diseases, like Rheumatoid
Arthritis, research indicates that dietary habits may be a risk
factor and that environmental and genetic factors interact during
the pathogenesis of autoimmune diseases years before clinical onset
(28). Along with genetic
predisposition, environmental factors can lead to the development
of autoimmunity and the break of immune tolerance to self-antigens
(29). Multiple environmental
factors along diet and stress, such as air-pollution, infection and
smoking may contribute to the development of chronic illnesses
(30).
Studies of several complex human diseases have shown
differences in DNA methylation in peripheral normal blood cell
populations (CD4+ T cells, CD19+ B cells
etc.) and the analysis of these differences has led to the
understanding of the role of epigenetics in autoimmunity. Patients
with disorders like multiple sclerosis (MS) and myasthenia Gravis
(MG) that are related with the production of autoantibodies can be
benefited by fully understanding the role of the environmental
conditions, like stress. Epigenetic differences have been presented
in twin studies and these differences become clearer with age and
this can potentially explain the phenotypic differences. It is
possible that genetic factors and environmental triggers along with
epigenetic factors lead to autoimmune disorders. The environment
can lead to overexpression of silenced genes caused by subvert
epigenetic regulatory pathways. As children grow up and are exposed
to radiation, drugs, chemicals, dietary changes and stress through
their life, autoimmune diseases may become more and more common in
the coming decades.
Given the key role that B cells have in many
autoimmune disorders and the alterations described by several
studies that deal with DNA methylation (31), a need for further investigation
emerges to confirm whether and how epigenetics is involved in these
disorders. The direct or indirect role of environmental factors in
the pathogenesis of myasthenia Gravis is essential for the better
understanding of the disease and the ways to design new drugs that
will lead to full remission or even prevent MG from
manifestating.
Obesity and weight management in MG
Obesity is considered to be a chronic disease itself
caused by an imbalance between the energy spent by a person and the
energy ingested in food. Fat cells store the excess energy and that
causes their enlargement and/or increase causing a pathological
etiology for obesity (32). The
medical hazards of obesity include diabetes mellitus,
hypertriglyceridemia, changes in the levels of high and low-density
lipoprotein cholesterol, gallbladder disease, sleep apnea and
degenerative joint disease and an elevated risk for coronary heart
disease along with malignancies among the most common.
In Myasthenia gravis patients' excess weight is a
rather common problem. Some factors that lead towards patients
gaining weight are lifestyle and drug related. It is important for
MG patients to maintain a relaxed lifestyle without extreme
workouts or activities that include heavy lifting, repetitive nerve
stimulation or any muscle force that may lead to muscular skeleton
weakness and increase other symptoms like cramps, unintelligible
speech, swallowing or breathing problems and lead to generalized
fatigue. This may result in involuntary gain weight, especially if
patients do not follow a dietary plan specially designed for their
needs.
Another factor that may lead to obesity is the use
of certain drugs that are connected with alterations of the body's
appetite-regulating mechanisms resulting in excessive weight
gaining. Corticosteroids lead to improvement or remission of the
symptoms in a large percentage of patients as mentioned and
prednisone, for example, is widely used as an MG treatment as it
suppresses the immune system and helps control myasthenia gravis.
One of the main side effects of oral corticosteroids is weight gain
with fat deposits in the abdomen area and the face. This can cause
a deterioration of MG as an adverse reaction as the patient's
compliance with prescribed medication may be jeopardised and the
excess weight may itself lead to less exercise, more stress on the
body and a deterioration of the disease's progress.
A number of other drugs may change body weight as an
adverse consequence of their therapeutic use such as
antidepressants and mood stabilizers, both commonly taken among
patients with autoimmune and other chronic diseases. Studies have
shown associations that suggest that autoimmune disorders are
important etiologic factors of mood disorders, such as depression
and anxiety-related disorders, especially in patients that are
hospitalized (33). Antidepressants
such as tricyclic antidepressants and monoamine oxidase (MAO)
inhibitors are most often associated with significant weight
gain.
Overweight increases the risk of mortality as a
growing body of research tells us and a logical anticipation would
be that intentional weight loss may reduce mortality and certainly
reduce the risk of other obesity related diseases. A body mass
index (BMI kg/m2), the formula used to assess body
weight and is an indicator of obesity, is the main criteria for
evaluating normal weight. In 1995, WHO published a technical report
establishing for categories: an individual with a BMI <19.9 is
considered to be underweight, to have normal weight with the BMI
20–24.9, overweight if the BMI is between 25 - 29.9 and obese with
a BMI >30.
Intentional weight loss improves individual risk
factors, reduces the risk of diabetes mellitus and leads to changes
in blood pressure and dyslipidemia. Therefore, maintaining a normal
weight (BMI <25) is important for everyone and can be furtherly
proven important in MG patients of all age groups as it helps
stabilize the disease, ensures proper drug intake and optimal
exercise, which lead to a better and healthier lifestyle with a
more controlled form of the disease. Successful weight loss
maintenance can be achieved by dietary restrictions and with the
addition of gradually increased physical activity up to the level
of not causing MG deterioration. While weight can be a complicated
issue for many of us, it is important for MG patients to make an
effort to reach and maintain a long-term healthy weight. Further
studies evaluating the risks of MG and obesity can be proven
valuable.
Treatment options
Medical therapies are different between neonatal MG
and Juvenile and adult MG. In infants the treatment used has a much
smaller duration, varying from a few days to a few months depending
on the duration of the symptoms while in JMG and in adults the
treatment is very often a life-long commitment.
The first treatment is pyridostigmin bromide (brand
name Mestinon), an acetylcholinesterase inhibitor. Pyridostigmin as
a half-life of 20 min after an oral 60 mg dose and the dosage is
usually prescribed depending on each patient's clinical assessment
and his/her age. Mestinon timespan is an extended-release formula,
available in 180 mg tablets that has been recently developed. A
typical adult takes 30–60 mg orally every 4–6 h daily, while a
typical pediatric dose vary, from 1–7 mg/kg/day divided to up to 6
doses. There is a risk of a cholinergic crisis if the dose of AChEI
is too high, which may result in excessive saliva, extreme fatigue
and even respiratory failure, sometimes making it difficult to
differentiate from a myasthenic crisis. Neostigmine is another
acetylcholinesterase inhibitor used, but Pyridostigmine has a
longer duration of action and is, therefore, preferred. Side
effects include increased salivation, diaphoresis, muscle cramps,
broncial secretions and gastrointestinal issues.
If the patient remains symptomatic after the maximum
pyridostigmin dosage, the next step is to begin immunosuppressive
therapy, usually with prednisone, azathioprine, cyclosporine,
tacrolimus and IVIG (34).
Corticosteroids lead to improvement or remission of the symptoms in
a large percentage of patients. intravenous immunoglobulin (IVIG)
and plasma exchange (PLEX) as immunomodulating therapies are very
effective in treating MG exacerbations, myasthenic crisis and,
also, as a treatment prior to surgery and thymectomy. Azathioprine
is recommended in cases where long-term immunosuppression is
necessary, but the effect of this substance is delayed for as long
as 12 months. Cyclosporine is usually given to patients who have a
low tolerance for azathioprine and tacrolimus is an
immunosuppressive agent with a similar mechanism to cyclosporine
and appears to be a promising agent in patients with RyR
antibodies, as it increases RyR-related calcium release.
New classes of biological compounds and drugs are
currently in a clinical experimentation state, opening the
possibility of personalized medicine and specific-targeted
treatment options (35). They are
divided in three major categories: complement inhibitors, anti-B
cell therapies and nFcR (Neonatal Fc Receptor) antagonists.
Complement inhibitors include Eculizumab (ECU), a humanized
monoclonal antibody that targets complement protein C5 preventing
the effect of micro-destruction of the post-synaptic membrane and
has a good safety profile, demonstrating improvement in AChR
positive patients, especially in perceived fatigue (tiredness,
difficulty concentrating and lack of energy that differentiates
from muscle weakness) (36).
Rituximab (RTX) is a very recent development in the
treatment of MG symptoms that falls into the category of anti-B
cell therapies. RTX is a chimeric mouse/human monoclonal antibody
that targets CD20 B lymphocytes and is, at this point, prescribed
in cases with drug-resistant MG, especially with MuSK antibodies
(37). This new theurapeutic
approach has been increasingly suggested, but there are yet not
enough studies to describe the impact in the quality of life and to
establish its benefits in comparison to the other available therapy
options (37).
Finally, neonatal Fc Receptor (nFcR) antagonists are
used for the first time in MG, offering a new therapeutic option
for the disease, if their capacity to reduce circulating Igs is
proven effective. This category of specific-targeted treatment
options is subdivided into three groups, Recombinant Fc multimers,
Neonatal Fc receptor antagonists and antiFcgR antagonists. Under
current investigation through clinical trials are compounds, such
as Efgartigimod (an engineered IgG1-derived Fc fragment),
Rozanolixizumab (a humanized monoclonal antibody), Nipocalimab
(M281- a fully humanized deglycosylated monoclonal antibody to
nFcR) and RVT-1401 (a human recombinant anti-nFcR monoclonal
antibody) (35). The mechanism of
these compounds allows treatment for both AChR and MuSK- positive
MG patients.
Discussion
Pediatric myasthenia gravis can present in infants
as CMS or TNM or in adolescents as JMG which turns into adult MG,
with either ocular or generalized form. Understanding the
mechanisms involved in the pathophysiology of the autoimmune
disease generalized myasthenia gravis is crucial in order for
effective therapies to be introduced. The AChR pathogenic
autoantigen was identified in the 1970s but the genetic basis is
not clear. The number of patients has increased during the last
twenty years and it is important to explain the mechanisms
underlying MG. The study of MYAS1 could lead to a better
understanding of the HLA B8 DR3 haplotype in the pathogenesis of
this disease and perhaps many other autoimmune diseases. The
difference in the genetic basis between Early and Late Onset MG
confirms that there are more variants to be explored. The need for
better prevention, easiest diagnosis and more efficient treatment
is important in this era.
The first therapy of MG is basically a symptomatic
treatment. Other than cholinesterase inhibitors, usually sufficient
in mild cases in the beginning of the disease, corticosteroids and
variable degrees of immunosuppression are needed for the majority
of patients. Nevertheless, reducing the use of corticosteroids in
patients is a current need, as they are a risk factor for other
conditions, such as osteoporosis, metabolic, endocrine and
cardiovascular complications. A new era is beginning with the
introduction of new biological compounds directed against specific
aspects of the autoimmune process in MG.
The relationship between certain drug categories
that are forbidden or are considered cautionary drugs for
myasthenia patients and the biological pathways behind the disease,
is an area that needs to be further explored, especially in
children. Beta- blockers used for hypertension and heart disease
may worsen MG, as well as fluoroquinolones and macrolide
antibiotics (e.g., azithromycin and erythromycin), statins used to
reduce serum colesterol, botulinum toxin and other sunbstances like
quinine (used to treat malaria but also found in tonic water) and
magnesium. Other than patients knowing the side-effects and using
these drugs with caution (if at all), the biochemical information
connecting these drugs with a neuromuscular disease like MG can
prove even life-saving in some cases.
Myasthenia Gravis has lately been associated in 3
cases with severe acute respiratory syndrome coronavirus 2
(SARS-CoV-2) and observers propose that a viral infection of nerve
cells may cause MG symptoms (38).
More investigation in this direction for other viruses should also
be planned in the future as new emerging pathogens could create new
threats for MG. Novel drug design pipelines should be considered
for further studies especially on ssRNA viruses under the prism of
MG (39–41). Research in this direction could be
proven valuable at all levels of investigation, starting from in
silico and computational work, to in vitro and in
vivo and eventually reaching clinical trials.
The study of epigenetic mechanisms, like DNA
methylation and histone modifications, and discovering microRNAs
that are MG-specific along with comprehending environmental factors
that might play a role in myasthenia, may lead to valuable
information about the genetic and biological profile of myasthenia
and could, furthermore, lead to the discovery of new and more
effective therapeutic treatments.
Acknowledgements
Not applicable.
Funding
The current study was supported by the
‘Competitiveness, Entrepreneurship & Innovation, EPAnEK 2nd
Cycle’ Operational Program (grant no. Τ2ΕΔΚ-02222, MIS: 5074548),
which was co-funded by Greece and the European Union.
Availability of data and materials
Not applicable.
Authors' contributions
RG and DV conceived the current study. RG, LP, AE,
FB, GPC, EE and DV wrote, drafted, revised, edited and reviewed the
manuscript All authors have read and approved the final manuscript.
Data sharing is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Herrmann C Jr: Myasthenia gravis and the
myasthenic syndrome. Calif Med. 113:27–36. 1970.PubMed/NCBI
|
|
2
|
Vandiedonck C, Giraud M and Garchon HJ:
Genetics of autoimmune myasthenia gravis: The multifaceted
contribution of the HLA complex. J Autoimmun. 25:6–11. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lazaridis K and Tzartos SJ: Autoantibody
specificities in Myasthenia Gravis; implications for improved
diagnostics and therapeutics. Front Immunol. 11:2122020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Frykman H, Kumar P and Oger J:
Immunopathology of Autoimmune Myasthenia Gravis: Implications for
Improved Testing Algorithms and Treatment Strategies. Front Neurol.
11:5966212020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garchon HJ: Genetics of autoimmune
myasthenia gravis, a model for antibody-mediated autoimmunity in
man. J Autoimmun. 21:105–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pal J, Rozsa C, Komoly S and Illes Z:
Clinical and biological heterogeneity of autoimmune myasthenia
gravis. J Neuroimmunol. 231:43–54. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mori S and Shigemoto K: Mechanisms
associated with the pathogenicity of antibodies against
muscle-specific kinase in myasthenia gravis. Autoimmun Rev.
12:912–917. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kordas G, Lagoumintzis G, Sideris S,
Poulas K and Tzartos SJ: Direct proof of the in vivo pathogenic
role of the AChR autoantibodies from myasthenia gravis patients.
PLoS One. 9:e1083272014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beeson D, Morris A, Vincent A and
Newsom-Davis J: The human muscle nicotinic acetylcholine receptor
alpha-subunit exist as two isoforms: A novel exon. EMBO J.
9:2101–2106. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rødgaard A, Nielsen FC, Djurup R, Somnier
F and Gammeltoft S: Acetylcholine receptor antibody in myasthenia
gravis: Predominance of IgG subclasses 1 and 3. Clin Exp Immunol.
67:82–88. 1987.
|
|
11
|
Oosterhuis HJ, Limburg PC, Hummel-Tappel E
and The TH: Anti-acetylcholine receptor antibodies in myasthenia
gravis. Part 2. Clinical and serological follow-up of individual
patients. J Neurol Sci. 58:371–385. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Berrih-Aknin S and Le Panse R: Myasthenia
gravis: A comprehensive review of immune dysregulation and
etiological mechanisms. J Autoimmun. 52:90–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zisimopoulou P, Evangelakou P, Tzartos J,
Lazaridis K, Zouvelou V, Mantegazza R, Antozzi C, Andreetta F,
Evoli A, Deymeer F, et al: A comprehensive analysis of the
epidemiology and clinical characteristics of anti-LRP4 in
myasthenia gravis. J Autoimmun. 52:139–145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tzartos JS, Zisimopoulou P, Rentzos M,
Karandreas N, Zouvelou V, Evangelakou P, Tsonis A, Thomaidis T,
Lauria G, Andreetta F, et al: LRP4 antibodies in serum and CSF from
amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol.
1:80–87. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhong H, Zhao C and Luo S: HLA in
myasthenia gravis: From superficial correlation to underlying
mechanism. Autoimmun Rev. 18:1023492019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
NCBI, MYAS1 Myasthenia gravis with thymus
hyperplasia [homo sapiens (human)], . Gene ID: 246750, updated on
16 Aug 2019. Updated Aug 16 2019.
|
|
17
|
Thomann HK and Pandya S: Myasthenia
gravis: pathophysiology, diagnosis, differential diagnosis and
management. Clin Eye Vis Care. 7:3–13. 1995. View Article : Google Scholar
|
|
18
|
Wolfe GI, Kaminski HJ, Aban IB, Minisman
G, Kuo HC, Marx A, Ströbel P, Mazia C, Oger J, et al: Randomized
Trial of Thymectomy in Myasthenia Gravis. N Engl J Med Aug.
11:375:511–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zagoriti Z, Georgitsi M, Giannakopoulou O,
Ntellos F, Tzartos SJ, Patrinos GP and Poulas K: Genetics of
myasthenia gravis: A case-control association study in the Hellenic
population. Clin Dev Immunol. 2012:4849192012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Price P, Witt C, Allcock R, Sayer D,
Garlepp M, Kok CC, French M, Mallal S and Christiansen F: The
genetic basis for the association of the 8.1 ancestral haplotype
(A1, B8, DR3) with multiple immunopathological diseases. Immunol
Rev. 167:257–274. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Seldin MF, Alkhairy OK, Lee AT, Lamb JA,
Sussman J, Pirskanen-Matell R, Piehl F, Verschuuren JJGM,
Kostera-Pruszczyk A, Szczudlik P, et al: Genome-wide association
study of late-onset Myasthenia Gravis: Confirmation of TNFRSF11A
and identification of ZBTB10 and three distinct HLA associations.
Mol Med. 21:769–781. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vereecke L, Beyaert R and van Loo G: The
ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of
immunopathology. Trends Immunol. 30:383–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moore KW, de Waal Malefyt R, Coffman RL
and O'Garra A: Interleukin-10 and the interleukin-10 receptor. Annu
Rev Immunol. 19:683–765. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vecchio D, Ramdas S, Munot P, Pitt M,
Beeson D, Knight R, Rodríguez Cruz P, Vincent A, Jayawant S, DeVile
C, et al: Paediatric myasthenia gravis: Prognostic factors for drug
free remission. Neuromuscul Disord. 30:120–127. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tournier-Lasserve E and Bach JF: The
immunogenetics of myasthenia gravis, multiple sclerosis and their
animal models. J Neuroimmunol. 47:103–114. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu FC, Kuo CF, See LC, Tsai HI and Yu HP:
Familial aggregation of myasthenia gravis in affected families: A
population based study. Clin Epidemiol. 9:527–535. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aguirre F and Villa AM: Myasthenia gravis.
Register of 190 cases in a single center. Medicina (B Aires).
80:10–16. 2020.(In Spanish). PubMed/NCBI
|
|
28
|
Gioia C, Lucchino B, Tarsitano MG,
Iannuccelli C and Di Franco M: Dietary habits and nutrition in
rheumatoid arthritis: Can diet influence disease development and
clinical manifestations? Nutrients. 12:14562020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shi J, Knevel R, Suwannalai P, van der
Linden MP, Janssen GMC, van Veelen PA, Levarht NEW, van der
Helm-van Mil AH, Cerami A, Huizinga TWJ, et al: Autoantibodies
recognizing carbamylated proteins are present in sera of patients
with rheumatoid arthritis and predict joint damage. Proc Natl Acad
Sci USA. 108:17372–17377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lucchino B, Spinelli FR, Iannuccelli C,
Guzzo MP, Conti F and Di Franco M: Mucosa-environment interactions
in the pathogenesis of rheumatoid arthritis. Cells. 8:7002019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zouali M: DNA methylation signatures of
autoimmune diseases in human B lymphocytes. Clin Immunol.
222:1086222021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bray GA: Medical consequences of obesity.
J Clin Endocrinol Metab. 89:2583–2589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Benros ME, Waltoft BL, Nordentoft M,
Ostergaard SD, Eaton WW, Krogh J and Mortensen PB: Autoimmune
diseases and severe infections as risk factors for mood disorders:
A nationwide study. JAMA Psychiatry. 70:812–820. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Katz N and Barohn JR: The history of
acetylcholinesterase inhibitors in the treatment of myasthenia
gravis. Neuropharmacology. 182:1083032021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mantegazza R and Antozzi C: From
traditional to targeted immunotherapy in Myasthenia Gravis:
Prospects for research. Front Neurol. 11:9812020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Andersen H, Mantegazza R, Wang JJ, O'Brien
F, Patra K and Howard JF Jr; REGAIN Study Group, : Eculizumab
improves fatigue in refractory generalized myasthenia gravis. Qual
Life Res. 28:2247–2254. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Peres J, Martins R, Alves JD and Valverde
A: Rituximab in generalized myasthenia gravis: Clinical, quality of
life and cost-utility analysis. Porto Biomed J. 2:81–85. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Restivo DA, Centonze D, Alesina A and
Marchese-Ragona R: Myasthenia Gravis associated with SARS CoV 2
infection. Ann Intern Med. 173:1027–1028. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Papageorgiou L, Loukatou S, Sofia K,
Maroulis D and Vlachakis D: An updated evolutionary study of
Flaviviridae NS3 helicase and NS5 RNA-dependent RNA polymerase
reveals novel invariable motifs as potential pharmacological
targets. Mol Biosyst. 12:2080–2093. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vlachakis D, Fakourelis P, Megalooikonomou
V, Makris C and Kossida S: DrugOn: A fully integrated pharmacophore
modeling and structure optimization toolkit. PeerJ. 3:e7252015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vlachakis D and Kossida S: Molecular
modeling and pharmacophore elucidation study of the Classical Swine
Fever virus helicase as a promising pharmacological target. PeerJ.
1:e852013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hughes T: The early history of myasthenia
gravis. Neuromuscul Disord. 15:878–886. 2005. View Article : Google Scholar : PubMed/NCBI
|