Introduction
Parkinson's disease (PD) is a multifactorial
neurodegenerative disease (1). It
is the second most frequently diagnosed type of neurodegenerative
disease (2) and its incidence
ranges from 5/100,000 to over 35/100,000 new cases each year,
according to worldwide data, mainly obtained from Europe and the
USA (3), with this value
significantly increasing from the sixth to ninth decade of life
(4). The prevalence has grown with
the aging population (4), and the
number of individuals suffering from PD worldwide is projected to
exceed 12 million by 2040 (5).
The main brain area affected by neurodegeneration in
PD is the substantia nigra pars compacta (SNpc) in the midbrain,
which demonstrates selective loss of dopaminergic neurons. However,
during PD development, extensive involvement of other structures in
either the central (e.g. in the basal ganglia, cerebellum or
thalamus) (6) or peripheral (e.g.
in the sensory nerves) (7) nervous
system can be observed (8).
The clinical manifestation of PD is primarily based
on motor symptoms like bradykinesia, muscle rigidity, resting
tremor, posture and gait abnormalities (9). As the disease progresses, more
advanced signs of neurodegeneration such as dysarthria and
dysphagia can also occur (9).
Furthermore, PD is associated with numerous non-motor symptoms,
including hyposmia, sleep disturbances, bowel and urinary
dysfunction and depression (10).
One important clinical characteristic of PD is continuous cognitive
decline, which increases with the progression of neurodegenerative
processes (11).
In terms of PD development, several groups of risk
factors can be distinguished. The first group comprises numerous
genetic causes that have been identified in recent years. In most
populations, 3–5% of PD cases could be explained by monogenic
mutations (12) in genes such as
synuclein α (SNCA; encoding α-synuclein), VPS35
(involved in endosomal trafficking), parkinsonism associated
deglycase and PTEN induced kinase 1 (mitochondrial genes), leucine
rich repeat kinase 2 (serving a role in autophagy and microtubule
stability) and GBA (encoding a key enzyme for the proper
functioning of lysosomes) (13).
In contrast, the hereditary risk of developing non-monogenic PD
varies from 16–36% and at least 90 genetic risk variants have been
reported overall (12).
Sporadic/idiopathic PD is strongly age-related (14) and associated with a heterogenous
group of environmental risk factors including pesticides,
low-frequency magnetic fields (15) or previous head injury (16).
The development of neurodegeneration at the cellular
level has frequently been attributed to the accumulation of
misfolded proteins; more specifically, the underlying central
pathogenetic mechanism of PD is associated with the aggregation of
misfolded α-synuclein (αS) protein. Following this, αS fibrils
further accumulate to eventually form proteinaceous intracellular
inclusion bodies in neuronal somas or neurites, called Lewy bodies
or Lewy neurites, respectively (17). Furthermore, αS aggregates interact
with the substrates of the outer mitochondrial membrane, which
initiates mitochondrial dysfunction; this phenomenon occurs in both
genetic and sporadic PD (18), and
results in increased production of reactive oxygen species (ROS),
which generate oxidative stress conditions and thus potentiate the
neurodegeneration process in dopaminergic neurons (19). Moreover, pathological αS activity
can stem from the malfunction of degradation pathways like the
autophagy-lysosome system, which are responsible for toxicity
control and cell death prevention by timely removal of long-lived
proteins and impaired organelles (20).
As a consequence of these events, the accumulation
of αS aggregates and the disruption of protein clearance trigger
endoplasmic reticulum (ER) stress conditions within neural cells
(21). The ER is an important
eukaryotic organelle, which serves a vital role in the protein
quality control system. It is involved in protein folding and
regulates intracellular calcium levels (22). ER-related proteostasis is
maintained by numerous specific chaperones, such as
glucose-regulated protein 78 (GRP78) (23). As the misfolded proteins accumulate
in the ER lumen, proteostasis becomes disrupted, which induces ER
stress conditions. Subsequently, the unfolded protein response
(UPR) signaling pathway is activated as an adaptive mechanism
(24). Initially, the UPR may
involve neuroprotective mechanisms and alleviate protein overload
in the ER, but in the case of persistent ER stress, the UPR
executes pro-apoptotic cascades that aggravate neurodegeneration
(21). The UPR is controlled by
three specific transmembrane proteins in the ER, which act as
stress sensors: Activating transcription factor (ATF6),
inositol-requiring enzyme 1 (IRE1) and protein kinase RNA-like ER
kinase (PERK) (25). Under evoked
ER stress conditions, activated PERK undergoes oligomerization and
autophosphorylation. The phosphorylated form of PERK (p-PERK) in
turn phosphorylates the a subunit of eukaryotic initiation factor
2α (eIF2α), which is involved in the repression of global protein
synthesis by inhibition of the 80S ribosome assembly. Despite this,
the activating transcription factor 4 (ATF4) mRNA undergoes
preferential translation as a result of eIF2α activation, due to
the presence of open reading frames in its 5′-untranslated region
(26,27). One of the roles of ATF4 is to
induce the transcription of the CCAAT enhancer binding protein
homologous protein (CHOP) (28),
which strongly promotes apoptotic cell death in neural cells
(29). CHOP is encoded by the DNA
damage-inducible transcript 3 (DDIT3) gene (30). Apoptosis induced by CHOP
overexpression is associated with activation and mitochondrial
translocation of protein (31). It
has been reported that CHOP regulates the expression of certain
proteins from the Bcl-2 family, including pro-apoptotic proteins
(such as Bcl-2-like protein 11, p53 upregulated modulator of
apoptosis and phorbol-12-myristate-13-acetate-induced protein 1),
and upregulates the expression of proteins, such as DNA
damage-inducible 34 (GADD34), Tribbles-related protein 3 and
endoplasmic reticulum oxidoreductin 1α (32).
Previous studies have reported that ER dysfunction,
ER stress conditions and UPR activation are key events in the
pathogenesis of PD. For instance, post mortem examination of
the brain tissues of PD patients has revealed elevated levels of
the p-PERK and p-eIF2α (33).
Furthermore, in an in vivo model of PD, the overexpression
of ATF4, a crucial member of the PERK-dependent signaling pathway,
resulted in severe dopaminergic neurodegeneration in the substantia
nigra region (34). Moreover, it
has been reported that aggregates of misfolded aS interact directly
with the GRP78 chaperone, which results in UPR activation (17,35).
A previous study reported that the interaction between αS
aggregates and ER calcium pump, sarco/endoplasmic reticulum
Ca2+-ATPase (SERCA) induces cell sensitization to ROS
production and apoptosis (36). It
is important to notice that certain genetic mutations directly
connected with the familial type of PD are also linked to ER
dysfunction and UPR activation (25).
Contemporary therapeutic strategies for PD still
only treat the symptoms and are mainly based on pharmacotherapy and
non-pharmacological supporting methods (such as surgery or
physiotherapy). Current pharmacological treatment of PD is focused
on increasing the level of the neurotransmitter dopamine in the
brain (12). This can be achieved
using drugs which act on several pharmacological targets associated
with dopamine metabolism, such as the dopamine precursor Levodopa,
passing through blood-brain barrier (BBB), or dopamine degradation
inhibitors (e.g. monoamine oxidase B inhibitors or
catechol-o-methyltransferase inhibitors) (37). To the best of our knowledge, there
is currently no available and approved therapy that can slow or
halt the progression of the disease, and hence, prevent or reverse
the ongoing neurodegeneration process (38).
As evidence from previous studies indicates that the
ER stress and the PERK-dependent branch of the UPR may be strongly
associated with PD pathogenesis at the molecular level, the present
study evaluated the efficacy of the small-molecule PERK inhibitor
LDN-87357 in an in vitro model of PD. Recent results
indicated that targeting the individual components of the UPR
signaling branches may lead to the development of innovative,
disease-modifying therapeutic options for PD (25).
Materials and methods
Identification of the small-molecule
PERK inhibitor LDN-87357
The investigated small-molecule PERK inhibitor
LDN-87357 was screened, characterized and provided for further
analysis courtesy of the Department of Biochemistry and Molecular
Biology, Hollings Cancer Center (Medical University of South
Carolina, USA). The selection was performed according to the
protocol described previously by Pytel et al (39). High-throughput assay screening
(inhibitor selection) was performed using the Laboratory for Drug
Discovery in Neurodegeneration (LDDN) compound library; the LDDN is
part of the Department of Neurology at Brigham and Women's Hospital
and the Harvard Medical School (Boston, MA, USA) (40). The library consists of 150,000
compounds, of which 80,000 were selected for further analysis based
on certain computational filters that have previously been
described (39). The calculations
of desirability filters, such as polar surface area or Lipinski's
‘rule of five’ were performed to select the compounds with an
increased probability of good oral bioavailability and ability to
cross the BBB.
Cell culture
All experiments were performed using an in
vitro model of PD with a commercially available neuroblastoma
SH-SY5Y cell line, purchased from the American Type Culture
Collection (ATCC; cat. no. CRL-2266TM), which is derived from human
neuroblastoma tissues and widely used in PD research (41). The culture was maintained according
to the supplier's protocol, under standard conditions, which were
37°C, 5% CO2 and 95% humidity. The complete culture
medium for SH-SY5Y cells was composed of: ATCC-formulated Eagle's
Minimum Essential Medium (EMEM) (cat. no. 30-2003TM; ATCC) and F12
Medium (cat. no. 11765-054; Thermo Fisher Scientific, Inc.), mixed
in a 1:1 ratio. The medium was supplemented with 10% fetal bovine
serum (cat. no. 30-2020; ATCC) and 100 U/ml penicillin with 100
µg/ml streptomycin solution (cat. no. 15140-122; Gibco; Thermo
Fisher Scientific, Inc.). Cell passage was performed every 3–4 days
(when cells reached 90–95% confluence). The cells were dissociated
using 0.25% trypsin and 0.53 mM EDTA solution.
Analysis of the mRNA expression levels
of the pro-apoptotic ER stress-related genes
To assess the mRNA expression levels of specific
pro-apoptotic ER stress-related genes, total RNA was isolated from
SH-SY5Y cells using a PureLink RNA Mini Kit (Thermo Fisher
Scientific Inc.). The RNA obtained was reverse transcribed into
complementary (c)DNA using GoScript TM Reverse Transcriptase
(Promega Corporation) at a final concentration of 100 ng. All steps
were performed in accordance with the manufacturers' protocols.
Following this, TaqMan Gene Expression Assays were performed for
the analysis of the expression of pro-apoptotic, ER stress-related
genes, including DDIT3, BAX, ATF4, eIF2α, Bcl-2, GADD34.
ACTB was used as a reference gene (details of the assays
used were presented in Table I).
The mixture for the qPCR analysis (with a total volume of 10 µl),
consisted of the following reagents: cDNA (1 µl), 5× HOT
FIREPol® Probe quantitative (q)PCR Mix (2 µl; Solis
BioDyne OÜ), primers (1 µl) and nuclease free water (6 µl). The
thermocycling conditions for the qPCR analysis were as follows:
enzyme activation (15 min at 95°C), DNA denaturation (40 cycles of
10 sec at 95°C) and annealing/extension (40 cycles of 60 sec at
60°C), according to the manufacturer's protocol. Gene expression
was determined by the 2−ΔΔCq quantification method
(42) using a Bio-Rad CFX96
(Bio-Rad Laboratories, Inc.) system.
 | Table I.Assays used for assessment of mRNA
expression levels, using the TaqMan gene expression assay. |
Table I.
Assays used for assessment of mRNA
expression levels, using the TaqMan gene expression assay.
| Gene | Assay ID | Chromosome
location |
|---|
| DDIT3 | Hs01090850_m1 | Chr.12:
57516588-57520517 |
| BAX | Hs00180269_m1 | Chr.19:
48954825-48961798 |
| ATF4 | Hs00909569_g1 | Chr.22:
39519709-39522686 |
| eIF2α | Hs00230684_m1 | Chr.3:
150546678-150586016 |
| Bcl-2 | Hs00608023_m1 | Chr.18:
63123346-63319778 |
| GADD34 | Hs00169585_m1 | Chr.19:
48872392-48876062 |
| ACTB | Hs99999903_m1 | Chr.7:
5527148-5530601 |
Analysis of the cytotoxicity and
pharmacological efficacy of the LDN-87357
The cytotoxicity of the investigated PERK inhibitor,
LDN-87357, was evaluated using the colorimetric
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
(XTT) assay (Thermo Fisher Scientific, Inc.). The assay assessed
cell viability as a function of the cellular redox potential.
Actively-respiring cells transform XTT into an orange-colored
formazan product, both of which are soluble in water. All
experiments were repeated three times, with similar results.
Briefly, SH-SY5Y cells were cultured in 96-well plates
(5×103 cells/well) for 24 h at 37°C in 100 µl complete
growth medium. Cells were then exposed at 37°C to 100 µl of
complete culture medium containing LDN-87357, at the following
concentrations: 0.75, 3, 6, 12, 25, 50, 75 or 100 µM, or 50 mM or
0.1% DMSO (MilliporeSigma), which was the solvent used for
LDN-87357. Cells untreated with the inhibitor and cultured in
complete medium were used as a negative control, whereas cells
treated with 100% DMSO served as the positive control.
The present study also evaluated the effectiveness
of LDN-87357 after the induction of ER stress conditions in SH-SY5Y
cells. The cells were seeded in 96-well plates (5×103
cells/well) and cultured for 24 h in 100 µl of complete culture
medium. After incubation, the cells were exposed to 100 µl complete
culture medium containing LDN-87357 at the aforementioned
concentration range for 1 h, and then treated with 500 nM
thapsigargin (Th) to invoke ER stress. Certain cells were treated
only with 500 nM Th. Cells cultured in complete medium only were
used as a negative control, and 100% DMSO-treated cells were used
as a positive control. All samples were incubated for 16, 24 or 48
h, and then 25 µl XTT/PMS mixture was added to each well and
incubated for 2 h at 37°C in a 5% CO2 incubator. The
absorbance was quantified using a Synergy HT spectrophotometer
(Agilent Technologies, Inc.) at 450 nm.
Assessment of apoptosis using a
caspase-3 activity assay
The activity of caspase-3, one of the major
pro-apoptotic proteins, was assessed using the Caspase-3 Assay Kit
(Colorimetric) (Abcam). Activated caspase-3 cleaved the labeled
DEVD-p-NA substrate, and levels of the obtained product,
chromophore p-nitroaniline (p-NA), were assessed using a
spectrophotometer. All tests were performed in triplicate with
similar results. Briefly, SH-SY5Y cells were cultured in 6-well
plates (5×105 cells/well) in a complete culture medium
for 24 h at 37°C. The cells were then incubated for another 24 h at
37°C with LDN-87357 (0.75, 3, 6, 12, 25, 50 and 100 µM) or with the
solvent, 0.1% DMSO (MilliporeSigma). One set of cells was exposed
to 1 µM staurosporine (cat. no. S4400-1MG; MilliporeSigma) for 16 h
at 37°C as a positive control, and another set of cells were
cultured for 24 h in complete culture medium alone as a negative
control. To assess the effectiveness LDN-87357 in SH-SY5Y cells
exposed to ER stress, the cells were transferred to 6-well plates
(5×105 cells/well) and the culture was maintained in
complete medium for 24 h at 37°C. The cells were then exposed to
complete culture medium including LDN-87357 at the aforementioned
concentration range for 1 h at 37°C, and then Th was added at 500
nM for 24 h at 37°C. An additional group of cells was treated only
with 500 nM Th (cat. no. 58-600-51MG; MilliporeSigma) for 24 h at
37°C. The positive control consisted of SH-SY5Y cells treated with
1 µM staurosporine for 16 h at 37°C and the negative control
consisted of cells incubated for 24 h at 37°C in complete growth
medium alone. After removal of complete medium, the SH-SY5Y cells
were rinsed with 1X Dulbecco's phosphate-buffered saline (DPBS;
MilliporeSigma). The cells were then passaged with 0.25% trypsin
and 0.53 mM EDTA solution (ATCC) for 5 min at 37°C. The obtained
cell suspension was then centrifuged at 140 × g for 5 min at room
temperature. After removal of the supernatant, the cell pellet was
resuspended in complete culture medium), counted using a TC20
Automated Cell Counter (Bio-Rad Laboratories, Inc.), centrifuged at
140 × g for 5 min at room temperature, and the final pellet
(~1×106 cells) was then resuspended in 50 µl of cold
Cell Lysis Buffer (Abcam). The cell suspension was incubated for 10
min on ice, and then centrifuged at 10,000 × g for 1 min at room
temperature. The supernatant was then transferred to fresh 2 ml
tubes. Protein concentration was assessed using a standard Bradford
assay, with BSA used as a protein standard; each assay used cell
lysate containing 100 µg of protein. Following this, each sample
was supplemented with 2X Reaction Buffer (Abcam)(including DTT at
10 mM) and DEVD-pNA (4 mM) substrate (Abcam) at a final
concentration of 200 µM. The cells were incubated for two h at
37°C, and the p-NA level was assessed spectrophotometrically at 405
nm using a Synergy HT spectrophotometer (Agilent Technologies,
Inc.).
Analysis of cell cycle distribution
and progression
Cell cycle distribution was determined by flow
cytometry using propidium iodide (PI) staining. Due to the
inability of PI to penetrate living cells, prior treatment with
ethanol was required. The method provided assessment of the cell
cycle based on the differences in fluorescence intensity and the
increased fluorescence present in cells preparing for division due
to increased DNA levels. The cell cycle was interpreted based on
the following phases: sub G0/G1 (representation of apoptotic cell
subpopulation), G0/G1 (regulation of the entry of the quiescent
cell into the cycle), S (DNA replication) and G2/M (checkpoint
responsible for the prevention of cells with damaged DNA from
undergoing mitosis).
All experiments were performed in triplicate, with
similar results. SH-SY5Y cells were cultured in 96-well plates
(5×105 cells/well) and incubated for 24 h in complete
culture medium. After cell adhesion, LDN-87357 (0.75, 3, 6, 12, 25,
50 and 100 µM) was added to each well for 24 h; some wells received
only the solvent, 0.1% DMSO (MilliporeSigma). One group of cells
were exposed to 1 µM nocodazole (cat. no. M1404; MilliporeSigma)
for 16 h as a positive control, and another group were cultured in
complete culture medium for 24 h at 37°C as a negative control. To
evaluate the influence of the inhibitor LDN-87357 on SH-SY5Y cells
in ER stress conditions, cells were transferred to 6-well plates
(5×105 cells/well) and the culture was maintained in
complete culture medium for 24 h at 37°C.
The cultures were then pretreated with complete
culture medium including LDN-87357 inhibitor at the aforementioned
concentrations for 1 h at 37°C, and then treated with 500 nM Th for
24 h at 37°C. An additional set of cells were incubated only with
500 nM Th for 24 h at 37°C. The SH-SY5Y cells treated with 1 µM
nocodazole for 16 h at 37°C were used as a positive control, and
cells incubated for 24 h at 37°C with complete culture were used as
a negative control.
SH-SY5Y cells were then collected and rinsed twice
with cold 1X DPBS (MilliporeSigma). Aliquots of 1×106
cells/ml were then placed in ice-cold 70% ethanol for 20 min at
−20°C. The ethanol-suspended cells were then centrifuged for 5 min
at 3,630 × g at 4°C. The remaining cell pellets were then
resuspended in 250 µl of 1X DPBS, and treated with 10 mg/ml RNase A
solution (cat. no. EZ0002; Canvax Reagents S.L.) and incubated for
1 h at 37°C before they were stained using 10 µg/ml PI solution
(MilliporeSigma) at 4°C for 30 min. Finally, the samples were
assessed using a CytoFLEX flow cytometer (Beckman Coulter, Inc.).
The percentage of cells in each cell cycle phase (sub G0/G1, G0/G1,
S and G2/M), was determined depending on the DNA content, using
Kaluza Analysis Software (version 1.5A, Beckman Coulter, Inc.) and
analyzed using Cyflogic™ software (version 1.2.1; CyFlo Ltd.).
Statistical analysis
All obtained results were subjected to statistical
analysis using SigmaPlot (version 11.0; Systat Software, Inc.). In
all the experiments performed in the present study, the normality
of data distribution was determined using the Shapiro-Wilk test. As
all the data were characterized by a normal distribution, further
statistical analysis and comparison among multiple groups was
performed using ANOVA with Dunnett's post hoc test. Correlations
among changes in ER stress markers, cell survival and cell cycle
data were analyzed using Pearson's correlation coefficients. Three
independent tests were performed for the statistical analyses in
all individual experiments. P<0.05 was considered to indicate a
statistically significant difference among the groups.
Results
Evaluation of the cytotoxic and
pharmacological effect of the inhibitor LDN-87357
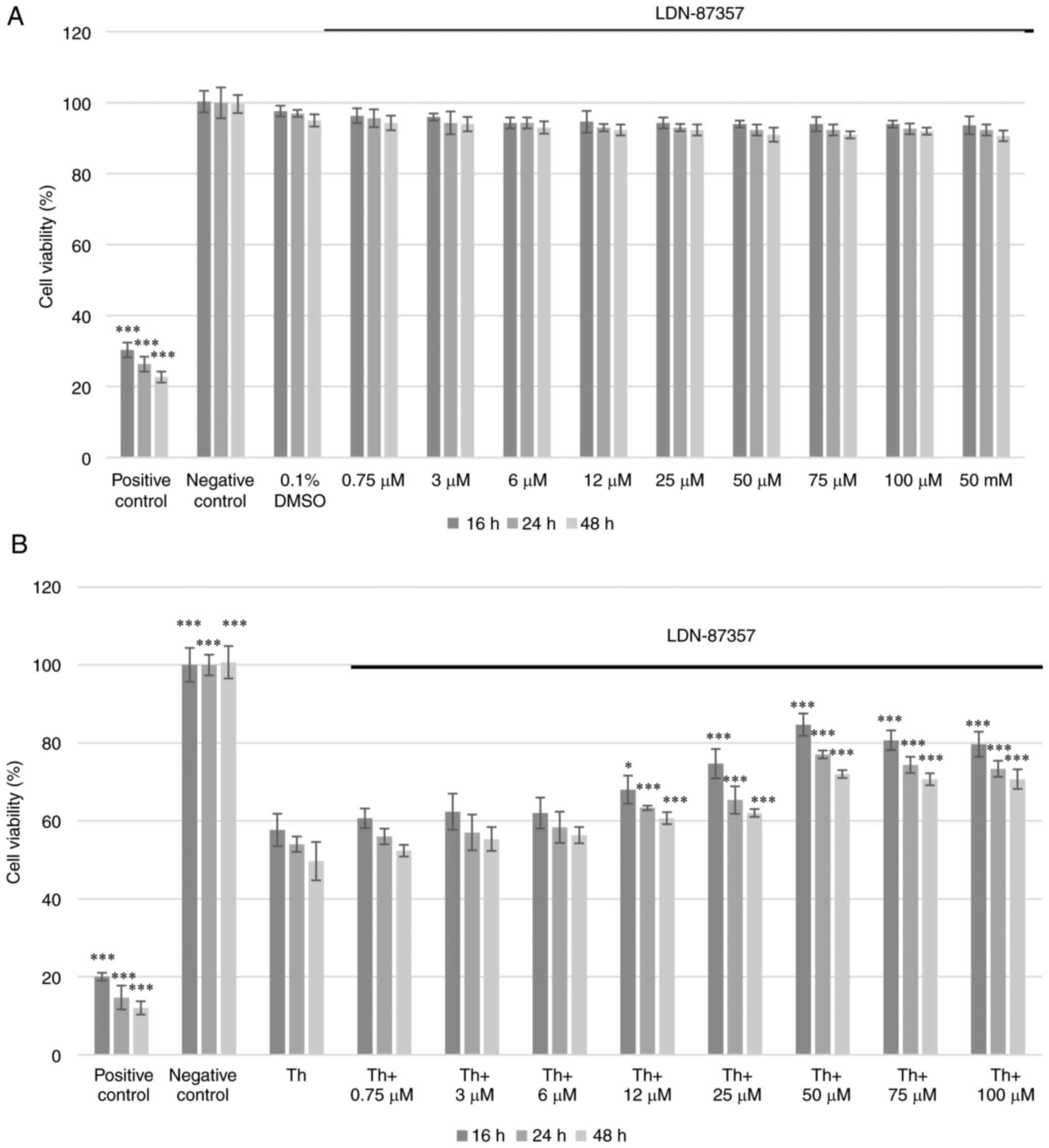
The cytotoxic effect of LDN-87357 on SH-SY5Y cells
was assessed using the XTT assay. No significant toxicity was
demonstrated at any of the applied concentrations after 16, 24 or
48 h. Moreover, the solvent (0.1% DMSO), did not induce significant
cytotoxicity either (Fig. 1A). The
effect of LDN-87357 (0.75–100 µM), on cell viability was also
assessed under ER stress conditions induced by Th. A significant
decrease in the percentage of viable SH-SY5Y cells was demonstrated
after 16, 24 and 48 h treatment with Th, compared with negative
controls. However, treatment with LDN-87357 (≥50 µM) resulted in a
significant increase in cell viability compared with Th treatment
alone, for all incubation times (Fig.
1B).
Assessment of the level of apoptosis
in SH-SY5Y cells by the colorimetric caspase-3 assay
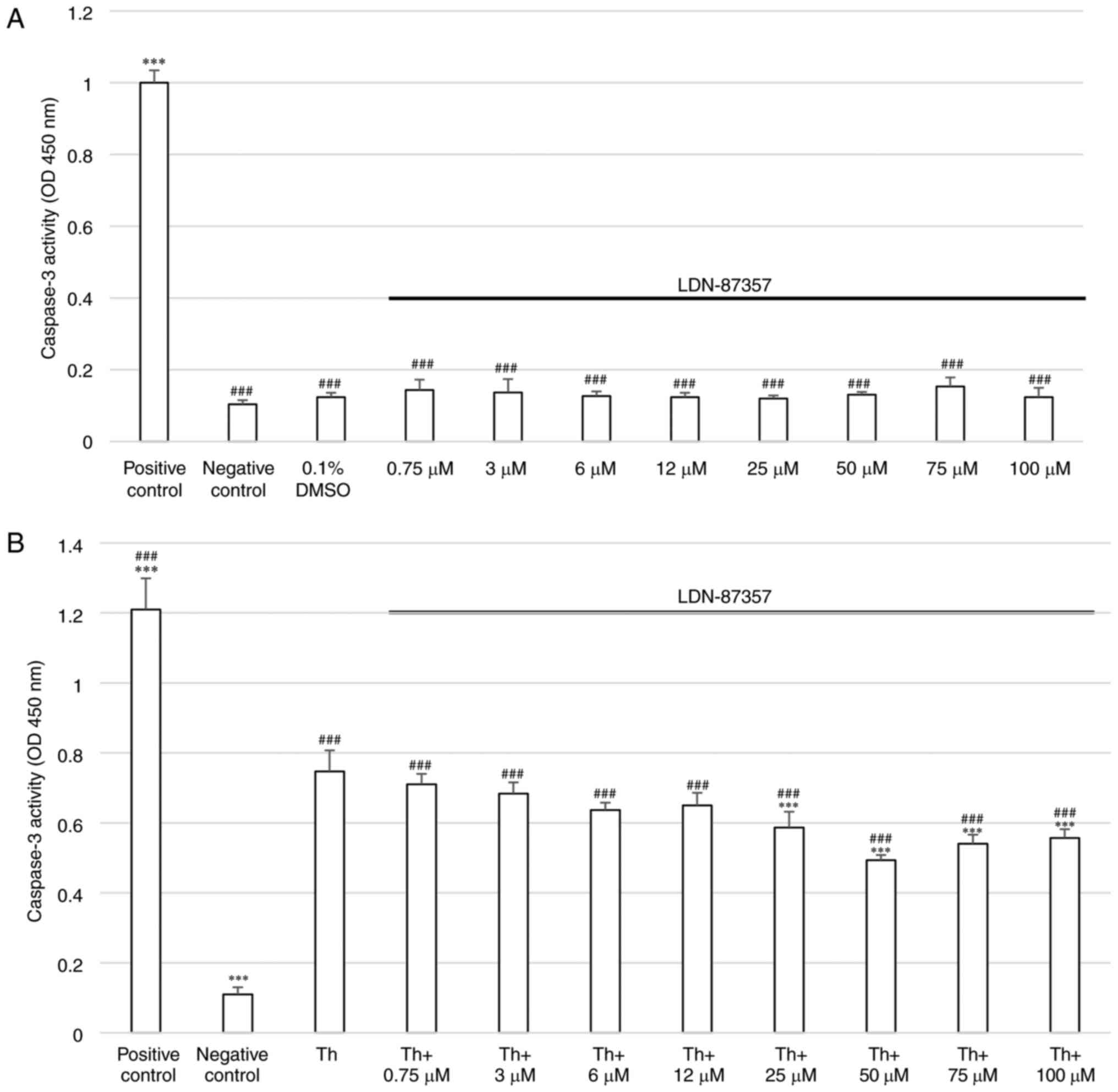
To evaluate the caspase-3 activity in SH-SY5Y cells
after treatment with LDN-87357, a colorimetric caspase-3 assay was
performed. The results demonstrated that SH-SY5Y cells incubated
with 1 µM staurosporine for 16 h were characterized by a
significant increase in the level of caspase-3 activity compared
with the negative control. No significant elevation of caspase-3
activity compared with the control was demonstrated in cell
cultures incubated for 24 h with LDN-87357 at any concentration.
Moreover, 24 h incubation with 0.1% DMSO (the solvent used for
LDN-87357) did not significantly induce caspase-3-mediated
apoptosis (Fig. 2A).
Cells pretreated with Th for 16 h and then exposed
to LDN-87357 (0,75–100 µM) demonstrated a significant increase in
caspase 3-activity compared with the negative control cells, which
were incubated with the dedicated culture medium for 24 h. However,
a significant decline in caspase-3 activity was demonstrated in
cells treated for 25 h with ≥25 µM LDN-87357 and Th, compared with
cells treated with Th alone (Fig.
2B).
Evaluation of the effect of LDN-87357
on cell cycle distribution and progression
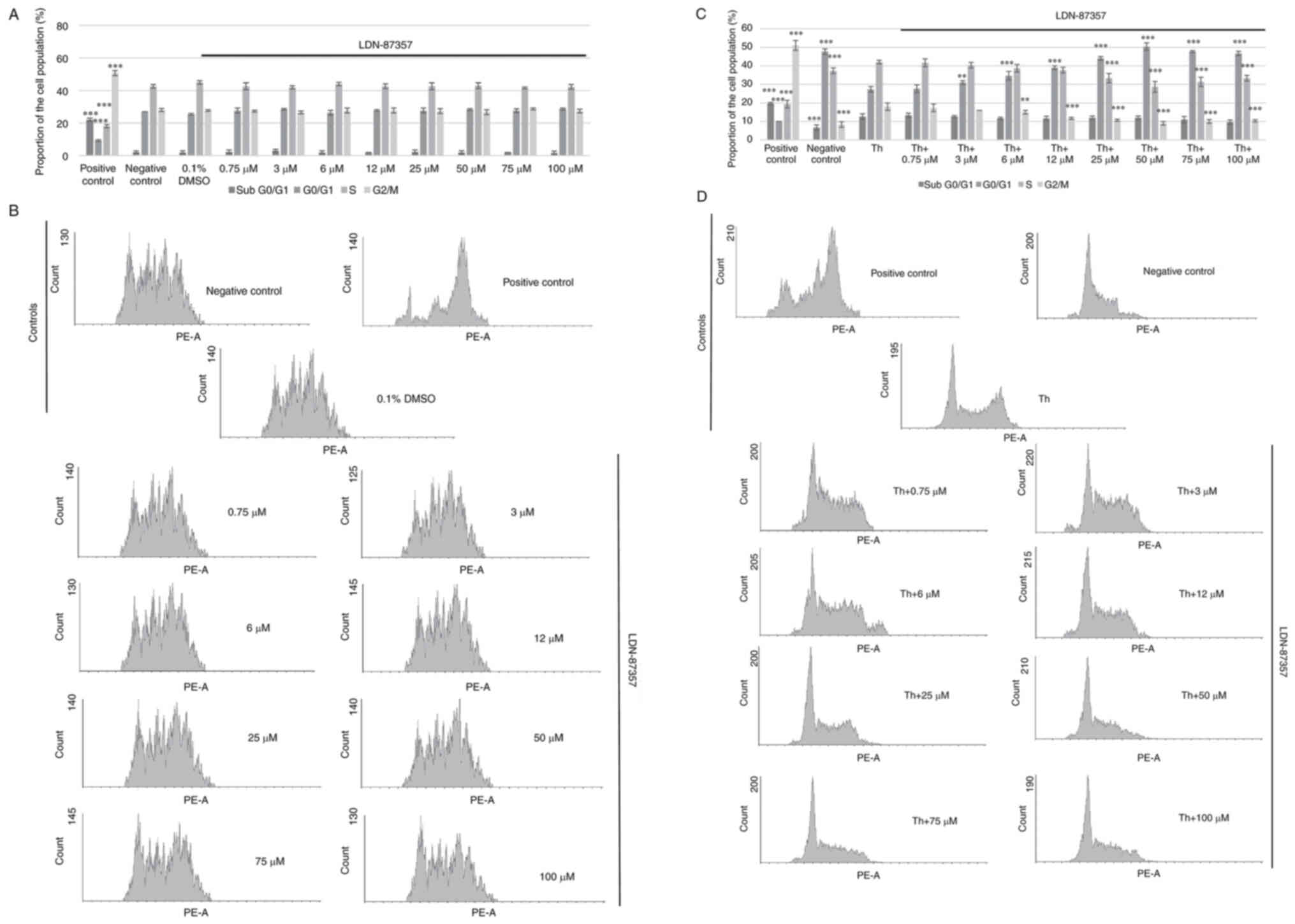
Treatment with LDN-87357 did not significantly
affect the course of cell cycle distribution in SH-SY5Y cells. G2/M
phase cell cycle arrest was only demonstrated in the positive
control sample, where SH-SY5Y cells were exposed to 1 µM nocodazole
for 16 h. No significant differences in the proportion of control
(cultured in complete medium) and experimental SH-SY5Y cells,
exposed to LDN-87357 for 24 h were demonstrated and in each case,
no cell cycle arrest was observed. Furthermore, the inhibitor
solvent (0.1% DMSO) demonstrated no effect on the cell cycle course
in SH-SY5Y cells after 24 h (Fig. 3A
and B).
Significant cell cycle arrest at G2/M phase was
demonstrated in the SH-SY5Y cells treated with nocodazole for 16 h
(positive control) in comparison with the cells untreated with any
compound (negative control). Furthermore, a significantly higher
proportion of SH-SY5Y cells treated only with Th were found in the
G2/M phase compared with the negative control. However, SH-SY5Y
cells treated with Th and then with LDN-87357 (≥6 µM) for 24 h
demonstrated a significant reduction in the proportion at G2/M and
a significant increase in the proportion in G0/1, compared with
samples incubated with Th alone (Fig.
3C and D).
The effect of LDN-87357 on the mRNA
expression level of pro-apoptotic ER stress-related genes
The mRNA expression levels of DDIT3 (encoding
CHOP), BAX, ATF4, eIF2α, Bcl-2, GADD34 in SH-SY5Y cells was
quantified under Th-induced ER stress conditions, after the
treatment of SH-SY5Y cells with the small-molecule PERK inhibitor
LDN-87357 alone as well as after treatment with both Th and 0.75 or
50 µM LDN-87357. The pro-apoptotic genes, DDIT3, Bax, ATF4
and GADD34, demonstrated a significant decrease in their
mRNA expression level when treated with Th and 50 µM LDN-87357 when
compared with SH-SY5Y cells treated with Th alone. However, a
significant increase in the mRNA expression levels of
anti-apoptotic gene Bcl-2 were demonstrated compared with
SH-SY5Y cells treated only with Th (Fig. 4).
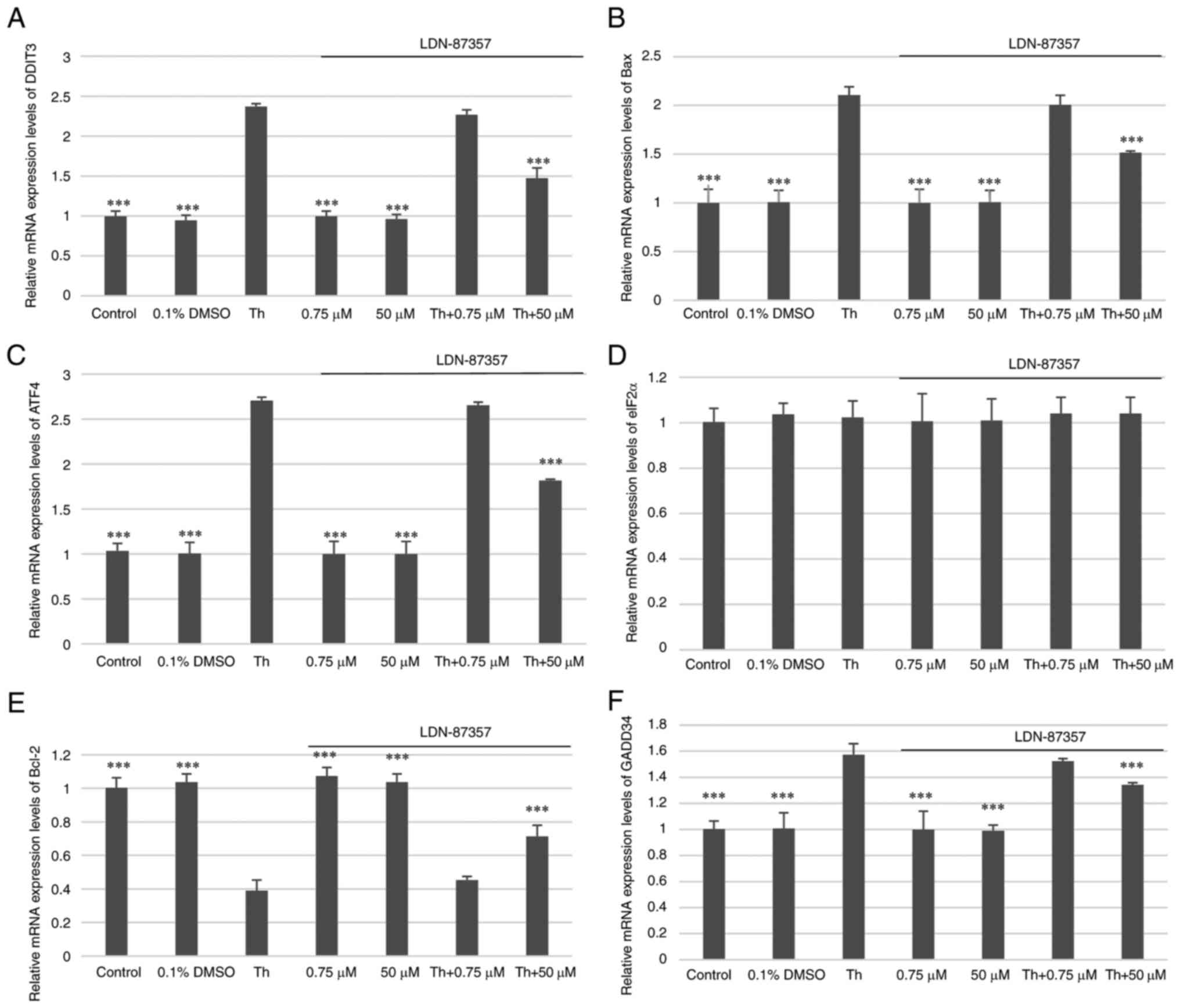 | Figure 4.Analysis of the mRNA expression
levels of pro-apoptotic ER stress-related genes. The relative mRNA
expression levels of (A) DDIT3, (B) Bax, (C)
ATF4, (D) eIF2α, (E) Bcl-2 and (F)
GADD34 in SH-SY5Y cells with Th-induced ER stress
conditions, after treatment with the small-molecule PERK inhibitor
LDN-87357 alone or with both LDN-87357 and Th. The analysis was
performed using the TaqMan gene expression assay. Three independent
tests were performed for each statistical analysis in all
experiments. The data are presented as mean ± SE. ***P<0.001 vs.
Th. Untreated SH-SY5Y cells were used as the control. DMSO,
dimethyl sulfoxide; Th, thapsigargin; DDIT3, DNA
damage-inducible transcript 3; ATF4, activating
transcription factor 4; GADD34, DNA damage-inducible
34. |
Pearson's correlation coefficient
The correlation between cell viability, cell cycle
arrest in the G2/M phase, and the expression levels of
the pro-apoptotic (DDIT3, BAX, ATF4, eIF2α, GADD34) and
anti-apoptotic (Bcl-2) genes in SH-SY5Y cells were evaluated
using Pearson's correlation coefficient. Cell viability was
negatively correlated with the expression levels of DDIT3,
BAX and ATF4, and positively correlated with
Bcl-2 gene expression. Moreover, a positive correlation was
detected between cell cycle arrest in the G2/M phase and
the expression levels of DDIT3 and ATF4. Furthermore,
cell viability and cell cycle arrest in the G2/M phase
were negatively correlated. Notably, a positive correlation was
observed between the expression levels of the following genes:
DDIT3 and ATF4, BAX and DDIT3, BAX and
GADD34. By contrast, the gene expression levels of
Bcl-2 and ATF4, Bcl-2 and GADD34 were
negatively correlated (Table
II).
| Table II.Pearson's correlation coefficients
between cell viability, cell cycle arrest and gene expression. |
Table II.
Pearson's correlation coefficients
between cell viability, cell cycle arrest and gene expression.
| Parameter | Cell viability | DDIT3
expression | BAX expression | ATF4
expression | eIF2α
expression | Bcl-2
expression | GADD34
expression | Population of cells
in G2/M phase |
|---|
| Cell viability |
| -1a | -0.999a | -0.998a | 0.592 | 0.997a | -0.995 | -0.999a |
| DDIT3
expression |
|
| 0.999a | 0.999a | -0.588 | -0.997 | 0.995 | 0.999a |
| BAX
expression |
|
|
| 0.994 | -0.631 | -1a | 0.999a | 0.996 |
| ATF4
expression |
|
|
|
| -0.546 | -0.991 | 0.988 | 1a |
| eIF2α
expression |
|
|
|
|
| 0.651 | -0.668 | -0.556 |
| Bcl-2
expression |
|
|
|
|
|
| -1a | -0.993 |
| GADD34
expression |
|
|
|
|
|
|
| 0.99 |
Discussion
The present study evaluated the efficacy of the
investigated small-molecule PERK inhibitor LDN-87357 in an
experimental PD in vitro model using the human neuroblastoma
SH-SY5Y cell line. The SH-SY5Y neuroblastoma cell line was used as
it is one of the most commonly used in vitro models in PD
research. SH-SY5Y is a subclone of the SK-N-SH line and it exhibits
numerous characteristics of dopaminergic neurons, such as tyrosine
hydroxylase and dopamine transporter expression (41,43).
Other types of cells that can be used as in vitro models of
PD include, the human HEK293, H4 and LUHMES cell lines, and rat
(PC12, N27 and CSM14.1) and mouse (MN9D, Cath.a and CAD) derived
cell lines (44–46). Primary dopaminergic neurons
isolated from brain specimens or neurons derived from induced
pluripotent stem cells (iPSCs) may also be utilized to mimic PD
pathology in vitro (44,45).
In contrast to the other aforementioned cellular models, SH-SY5Y is
human-derived, dopaminergic, easily available and relatively easy
to maintain in an in vitro culture system (41,43).
The aforementioned features led to the selection of this cell line
for the experiments performed in the present study.
We have previously demonstrated that treatment of
SH-SY5Y cells with 500 nM Th for 2 h evoked ER stress conditions,
using western blotting, which demonstrated that the protein
expression level of p-eIF2α was significantly elevated compared
with SH-SY5Y control cells not treated with any of the compounds
(47). In the present study, the
response of SH-SY5Y cells ER stress conditions induced by Th, a
specific ER stress activator, was demonstrated. Th is a
non-competitive inhibitor of sarco/ER Ca2+-ATPase, that
induces transient increases of intracellular free calcium levels;
such alterations in Ca2+ levels serve an important role
in PD pathogenesis (48,49). Treatment of SH-SY5Y cells with Th
results in the induction of ER stress conditions (with elevated
levels of ER stress markers, such as BiP, p-PERK/p-eIF2α, ATF4,
CHOP, IRE1α/XBP1s and ATF6) and mitochondrial dysfunction, both of
which have been implicated in PD pathology (50–54).
It has been reported that continued exposure of dopaminergic cells
to Th ultimately leads to apoptotic cell death and
neurodegeneration (55).
Interestingly, Th was previously reported to increase the number of
αS oligomers and induce αS aggregation, which is a characteristic
feature of PD (56,57).
In the present study, LDN-87357 was demonstrated to
be effective in the PD model used, at the cellular level: treatment
with LDN-87357 in Th-induced ER stress conditions resulted in a
significant decrease in the mRNA expression levels of pro-apoptotic
ER stress marker genes, including DDIT3, Bax, ATF4 and
GADD34, and a significant increase in the mRNA expression
level of the anti-apoptotic Bcl-2 gene. Furthermore, the XTT
assay did not demonstrate any cytotoxic effect in SH-SY5Y cells at
any concentration of LDN-87357, at any incubation period. Moreover,
the colorimetric caspase-3 assay in LDN-87357 treated cells did not
demonstrate any significant increase in apoptotic cell death, and
treatment with Th and LDN-87357 resulted in a significant decline
in the apoptotic rate, compared with treatment with Th alone.
PI staining demonstrated that LDN-87357 treatment
had no significant effect on the cell cycle distribution of SH-SY5Y
cells. Under ER stress conditions induced by Th, LDN-87357
treatment resulted in a significant decrease in the proportion of
cells at G2/M phase, with a simultaneous increase in the G0/1
phase, compared with cells treated with Th only. Furthermore, cell
viability was negatively correlated with DDIT3, BAX, ATF4
gene expression and positively correlated with Bcl-2 gene
expression. Moreover, there was also a positive correlation between
the arrest of the cell cycle in the G2/M phase and the expression
of DDIT3 and BAX genes.
Therefore, it appears that LDN-87357 may
significantly contribute to the limitation of the negative
consequences of ER stress conditions, particularly those associated
with cell apoptosis, which is a major factor in PD pathogenesis and
progression (22).
Previous studies have also reported the properties
of another, related small-molecule PERK inhibitor LDN-0060609 in
numerous cellular models of neurodegenerative diseases. In one
study, LDN-0060609 caused significant inhibition of eIF2α
phosphorylation in an Alzheimer's disease (AD) model based on
phenotype 1 rat normal astrocytes from diencephalon (DI TNC1) under
Th-induced ER stress. Moreover, LDN-0060609 did not induce any
significant increase in the apoptotic rate and had no remarkable
effect on cell cycle distribution in the DI TNC1 cell line, nor did
it demonstrate any toxic effect on cell viability (58). Another study examined the activity
of LDN-0060609 in a mouse neuron CATH.a cell line used as an in
vitro AD model, under ER stress conditions. The compound
significantly reduced apoptosis by decreasing the protein
expression level of CHOP without any cytotoxic effect (47).
LDN-0060609 was also reported to trigger marked
inhibition of p-eIF2α expression in human trabecular meshwork (HTM)
cells, an in vitro model for primary open angle glaucoma,
treated with Th. Furthermore, no cytotoxic or genotoxic outcomes
were reported in HTM cells at any concentration or incubation
period. Moreover, the pharmacological effectiveness was confirmed
by significant reversal of the negative effects of ER stress
conditions induced by Th, demonstrated by increased HTM cell
viability and reduced DNA damage. LDN-0060609 was also reported to
restore normal cell morphology and increase proliferation in a
Th-treated HTM cell line (59).
Furthermore, LDN-0060609 was also reported to have caused a
remarkable inhibition of p-eIF2α expression in SH-SY5Y cells,
without triggering any toxic effect (47). These findings indicated the value
of small-molecule PERK inhibitors as novel treatment options for
neurodegenerative diseases. Extended research at the cellular level
on the properties of new PERK inhibitors, such as LDN-87357,
particularly regarding PD and other globally-important
neurodegenerative diseases, may lead to the development of novel
therapeutic approaches. The results of the present study are
promising and may contribute to the growing body of knowledge
regarding the use of PERK in this field. Novel neuroprotective
compounds such as the small-molecule PERK inhibitor LDN-87357,
which can selectively target pro-apoptotic molecules and pathways
require further study.
Protein misfolding and aggregation is a key
molecular mechanism widely-known to underlie the neurodegeneration
process. As neurons are sensitive to protein misfolding, the
resulting ER dysfunctions, ER stress and UPR activation serve a
crucial role in the molecular pathogenesis of neurodegenerative
diseases (21). An in vivo
study in an A53T (missense SNCA gene mutation) transgenic
mouse model demonstrated co-occurring αS pathology and UPR
induction, supported inter alia by increased accumulation of
polyubiquitin chains associated with ER stress (60). In a previous study which used
differentiated rat sympathetic-like neuron cells (PC12), A53 αS
overexpression was primarily associated with increased ROS
production and impaired proteasome function; consequent ER stress
induction was detected by the up-regulation of the eIF2α and ER
stress-related genes: glycine rich protein 17 and DNA
damage-inducible gene 153 (61).
The PERK-mediated branch of the UPR pathway is known
to be associated with several neurodegenerative entities, such as
PD (24,25,32),
Alzheimer's disease (21,62) and prion disease (21,63).
Numerous post-mortem studies of PD patients have confirmed that the
protein expression levels of ER stress markers, especially of the
PERK-dependent branch of UPR, are elevated in PD brain tissue
samples (64). For instance,
Hoozemans et al (33)
reported greater immunoreactivity to the p-PERK and p-eIF2α in
neuromelanin-containing dopaminergic neurons from the SNpc
region, compared with controls. Moreover, in dopaminergic neurons,
the immunoreactivities of the p-PERK and αS were colocalized.
Baek et al (65) reported a significant increase in
the mRNA expression level of GRP78 in the prefrontal and
parietal cortex, caudate nucleus and cingulate gyrus of PD brains,
which was in contrast with the GRP78 protein expression level,
which was significantly decreased. These findings indicated that
regulation of GRP78, a crucial chaperone for the maintenance of
proteostasis, was impaired in the course of PD (65). Moreover, further post-mortem
research strongly supported the role of PERK-mediated pathway in PD
pathogenesis and reported that CHOP mRNA and protein
expression levels were upregulated in the SNpc region
(66).
However, several studies have reported interactions
between αS and various UPR-related proteins. Direct interactions
between αS aggregates and GRP78, associated with UPR activation,
have been reported in both in vitro and in vivo
models (17,35). Furthermore, downregulation of GRP78
was reported to result in a decline in exogenous αS activity, which
suggested this specific ER chaperone may be a primary target of αS.
Activation of the signaling cascade by GRP78 affected the
morphology and dynamics of the neuronal cytoskeleton, and led to
deficits in synaptic function; events which directly precede the
neurodegeneration process (67).
Credle et al (68) reported that αS overload impaired
the function of the cytoprotective factor, ATF6, which is an ER
transmembrane protein and one of the three components of the UPR.
ATF6 activation requires its transfer to coat protein complex II
(COPII). It has been reported that αS inhibits ER stress-mediated
ATF6 processing via COPII-mediated ER-Golgi transit. As a result,
the pro-apoptotic signaling intensifies, while ER-associated
degradation activity declines. ER-Golgi trafficking could be
damaged by direct interactions between the important vesicular
transit regulator ras associated binding 1 (RAB1) GTPase and αS. As
a result of impaired protein maturation, ER stress conditions are
induced. Furthermore, elevated expression of RAB1 was reported to
result in the loss of dopaminergic neuronal protection in in
vivo models of PD (69,70).
Paiva et al (71) reported that aggregated A30P αS
(with a missense mutation in SNCA) may upregulate the
expression of the collagen type IV alpha 1 chain gene which encodes
collagen IV, which is an important secretory cargo in the Golgi
body. Consequently, altered ER/Golgi morphology and increased
vulnerability to ER stress conditions in dopaminergic neurons have
been reported.
Another well-established ER stress regulator
associated with UPR activation is calcium homeostasis in the ER,
which is also a major cofactor for chaperone function. An in
vivo study on mice with knockout of the CalbindinD9k
gene, which encodes a calcium binding protein, identified elevated
intracellular calcium levels and αS overload, as well as ER-stress
mediated apoptosis, in dopaminergic neurons (72). Furthermore, the calcium level
appears to be disrupted by activation of the SERCA, ER calcium
pump, due to αS aggregates. Subsequently, calcium reuptake by the
mitochondria, which resulted in a significant increase in calcium
level, enhanced ROS production and neural cell sensitization to
apoptosis (36,73).
The chronically-activated PERK-dependent signaling
pathway is regarded as a direct pharmacological target for
neurodegenerative diseases, and it has been previously evaluated in
numerous research models (74),
including in vivo models, such as a mouse neurotoxin-based
PD model (75), and frontotemporal
dementia (76) and prion disease
(63) models. The selective,
first-in-class PERK inhibitor,
7-Methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine
(GSK2606414), is characterized by good BBB penetration and good
bioavailability when administered orally (77). The compound has been tested in
in vivo studies, including neurodegenerative diseases such
as frontotemporal dementia (76)
and prion disease (63) models
with promising results. GSK2606414 has been reported to protect
from further neuronal damage and reduce neurotoxic damage. Mercado
et al (75) reported that
GSK2606414 effectively inhibited the PERK-mediated pathway in a
mouse neurotoxin-based PD model, after experimental induction of ER
stress. The investigated compound protected dopaminergic neurons in
the SNpc region, improved motor performance, and increased
dopamine levels and the expression of synaptic proteins, such as
synaptosomal-associated protein and vesicle-associated membrane
protein 2. However, it should be noted that apart from its
neuroprotective activity, treatment with GSK2606414, was associated
with cytotoxicity and side effects, such as body weight loss,
pancreatic toxicity and hyperglycemia in the tested animals
(63,75).
Another UPR-targeting compound, the eIF2α
phosphatase inhibitor Salubrinal, has been evaluated in
experimental models of certain neurodegenerative processes such as
AD (78) and traumatic brain
injury (79). Salubrinal has also
been evaluated in numerous PD experimental models with favorable
results (60,61,80–82).
Salubrinal use was reported to restore motor function in a mouse PD
model with αS overexpression through increase of the level of
p-eIF2α by growth arrest and GADD34 (60). It also upregulated ATF4 expression
in the SH-SY5Y cell line (80).
Another study reported that the neuroprotective
effect of Salubrinal may be associated with reduced IκB kinase
activation, IκB degradation, and the resulting activation of
nuclear factor-kappa B (NF-κB), rather than with the direct
inhibition of the UPR pathway (83). These findings have been supported
by a recent study, where Salubrinal diminished motor impairments
and dopamine-related behavioral deficits in an intranigral
lipopolysaccharide-induced hemi-PD rat model; however, the findings
indicated that the beneficial effect could be related to a decrease
in the expression of numerous factors, such as inducible nitric
oxide synthase, cyclooxygenase-2 or NF-κB, as well as with the
attenuation of neuroinflammation processes (82).
An alternative neuroprotective compound that has
been evaluated in another neurodegenerative entity, prion disease,
is the small-molecule integrated stress response inhibitor (ISRIB).
ISRIB restored translation downstream of eIF2α in treated animals
(84). Mechanistically, ISRIB
partially restored global protein synthesis under ER stress
conditions compared with GSK2606414, which completely restored the
expression of CHOP and global protein synthesis. However, ISRIB had
no toxic effect on pancreatic cells (85). These findings indicate the
potential of PERK-mediated pathway inhibition as a selective target
for achieving neuroprotection with minimized side effects (86).
A major limitation of the present study was the use
of only one experimental model. Further research on LDN-87357
should include other cell lines, which replicate a PD pathology,
such as genetic models and neurotoxin-based models with rotenone,
6-hydroxydopamine or 1-methyl-4-phenyl-1,2,3,6-tetrahydropirydine
(87). Moreover, the results of
the present study do not fully elucidate the mechanism of action of
LDN-87357 and its exact molecular target in the PERK-dependent
signaling pathway. Further investigation is required to evaluate
inter alia, the expression of specific PERK-mediated pathway
marker proteins. Lastly, in vivo studies involving PD animal
models are needed to determine the pharmacokinetic and
pharmacodynamic properties of LDN-87357.
Presently, the pathogenesis of PD and its involved
molecular pathways are not fully understood, and current treatment
strategies remain insufficient, as they focus only on progression
of the disease. The results of the present study provide further
details of the association between the ER stress-mediated
activation of the PERK-dependent UPR signaling pathway and PD
pathogenesis at the molecular level. Selective, small-molecule
inhibitors of the UPR components, especially those targeting PERK,
constitute an attractive option for the development of novel PD
treatment strategies. Such inhibitors exhibit numerous
neuroprotective effects such as chronic terminal ER stress
prevention, apoptotic cell death reduction, neuroinflammation
decrease, synaptic function restoration and neuronal plasticity
stimulation. Previous studies have also reported that
small-molecule inhibitors are characterized by good BBB penetration
and bioavailability. Therefore, targeting the components of the UPR
signaling pathway using small-molecule inhibitors, like the PERK
inhibitor LDN-87357, may contribute to development of novel
therapeutic strategies against PD.
Acknowledgements
Not applicable.
Funding
This research was funded by The Medical University of Lodz,
Poland (grant no. 564/5-000-00/564-20-057), and The Polish National
Science Centre (NCN): OPUS grant (grant no. 2016/21/B/NZ5/01411)
and the PRELUDIUM BIS 3 grant (grant no. 2021/43/O/NZ5/02068).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
IM and EK conceptualized the present study. IM and
WRK were responsible for the methodology, and IM and EK were
responsible for formal analysis. The investigation was performed by
WL, WRK, GG and NS. IM and WL provided the resources used. WL, WRK,
GG and NS wrote the original draft, and IM and EK reviewed and
edited the manuscript. WRK and GG performed visualization of the
data. IM and EK supervised the project and IM was responsible for
project administration and funding acquisition. All authors have
read and approved the final version of the manuscript. IM, WRK and
GG confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Balestrino R and Schapira AHV: Parkinson
disease. Eur J Neurol. 27:27–42. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aarsland D, Batzu L, Halliday GM, Geurtsen
GJ, Ballard C, Ray Chaudhuri K and Weintraub D: Parkinson
disease-associated cognitive impairment. Nat Rev Dis Primers.
7:472021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Twelves D, Perkins KS and Counsell C:
Systematic review of incidence studies of Parkinson's disease. Mov
Disord. 18:19–31. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Simon DK, Tanner CM and Brundin P:
Parkinson disease epidemiology, pathology, genetics, and
pathophysiology. Clin Geriatr Med. 36:1–12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dorsey ER, Sherer T, Okun MS and Bloem BR:
The emerging evidence of the parkinson pandemic. J Parkinsons Dis.
8 (Suppl 1):S3–S8. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prakash KG, Bannur BM, Chavan MD, Saniya
K, Sailesh KS and Rajagopalan A: Neuroanatomical changes in
Parkinson's disease in relation to cognition: An update. J Adv
Pharm Technol Res. 7:123–126. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Comi C, Magistrelli L, Oggioni GD,
Carecchio M, Fleetwood T, Cantello R, Mancini F and Antonini A:
Peripheral nervous system involvement in Parkinson's disease:
Evidence and controversies. Parkinsonism Relat Disord.
20:1329–1334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cacabelos R: Parkinson's disease: From
pathogenesis to pharmacogenomics. Int J Mol Sci. 18:5512017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jankovic J: Parkinson's disease: Clinical
features and diagnosis. J Neurol Neurosurg Psychiatry. 79:368–376.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tolosa E, Garrido A, Scholz SW and Poewe
W: Challenges in the diagnosis of Parkinson's disease. Lancet
Neurol. 20:385–397. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aarsland D, Creese B, Politis M, Chaudhuri
KR, Ffytche DH, Weintraub D and Ballard C: Cognitive decline in
Parkinson disease. Nat Rev Neurol. 13:217–231. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bloem BR, Okun MS and Klein C: Parkinson's
disease. Lancet. 397:2284–2303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Day JO and Mullin S: The genetics of
Parkinson's disease and implications for clinical practice. Genes
(Basel). 12:10062021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Antony PM, Diederich NJ, Kruger R and
Balling R: The hallmarks of Parkinson's disease. FEBS J.
280:5981–5993. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Belvisi D, Pellicciari R, Fabbrini G,
Tinazzi M, Berardelli A and Defazio G: Modifiable risk and
protective factors in disease development, progression and clinical
subtypes of Parkinson's disease: What do prospective studies
suggest? Neurobiol Dis. 134:1046712020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Noyce AJ, Bestwick JP, Silveira-Moriyama
L, Hawkes CH, Giovannoni G, Lees AJ and Schrag A: Meta-analysis of
early nonmotor features and risk factors for Parkinson disease. Ann
Neurol. 72:893–901. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Colla E: Linking the endoplasmic reticulum
to Parkinson's disease and Alpha-Synucleinopathy. Front Neurosci.
13:5602019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Malpartida AB, Williamson M, Narendra DP,
Wade-Martins R and Ryan BJ: Mitochondrial dysfunction and mitophagy
in Parkinson's disease: From mechanism to therapy. Trends Biochem
Sci. 46:329–343. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trist BG, Hare DJ and Double KL: Oxidative
stress in the aging substantia nigra and the etiology of
Parkinson's disease. Aging Cell. 18:e130312019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hou X, Watzlawik JO, Fiesel FC and
Springer W: Autophagy in Parkinson's disease. J Mol Biol.
432:2651–2672. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ghemrawi R and Khair M: Endoplasmic
reticulum stress and unfolded protein response in neurodegenerative
diseases. Int J Mol Sci. 21:61272020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsujii S, Ishisaka M and Hara H:
Modulation of endoplasmic reticulum stress in Parkinson's disease.
Eur J Pharmacol. 765:154–156. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ni M and Lee AS: ER chaperones in
mammalian development and human diseases. FEBS Lett. 581:3641–3651.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mercado G, Castillo V, Soto P and Sidhu A:
ER stress and Parkinson's disease: Pathological inputs that
converge into the secretory pathway. Brain Res. 1648:626–632. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martinez A, Lopez N, Gonzalez C and Hetz
C: Targeting of the unfolded protein response (UPR) as therapy for
Parkinson's disease. Biol Cell. 111:161–168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Teske BF, Wek SA, Bunpo P, Cundiff JK,
McClintick JN, Anthony TG and Wek RC: The eIF2 kinase PERK and the
integrated stress response facilitate activation of ATF6 during
endoplasmic reticulum stress. Mol Biol Cell. 22:4390–4405. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jaud M, Philippe C, Van Den Berghe L,
Ségura C, Mazzolini L, Pyronnet S, Laurell H and Touriol C: The
PERK branch of the unfolded protein response promotes DLL4
expression by activating an alternative translation mechanism.
Cancers (Basel). 11:1422019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saito A and Imaizumi K: The broad spectrum
of signaling pathways regulated by unfolded protein response in
neuronal homeostasis. Neurochem Int. 119:26–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gorbatyuk MS, Shabashvili A, Chen W,
Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N and
Gorbatyuk OS: Glucose regulated protein 78 diminishes
alpha-synuclein neurotoxicity in a rat model of Parkinson disease.
Mol Ther. 20:1327–1337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rozpedek W, Pytel D, Mucha B, Leszczynska
H, Diehl JA and Majsterek I: The role of the
PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression
during endoplasmic reticulum stress. Curr Mol Med. 16:533–544.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rozpedek-Kaminska W, Siwecka N,
Wawrzynkiewicz A, Wojtczak R, Pytel D, Diehl JA and Majsterek I:
The PERK-dependent molecular mechanisms as a novel therapeutic
target for neurodegenerative diseases. Int J Mol Sci. 21:21082020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hoozemans JJ, van Haastert ES, Eikelenboom
P, de Vos RA, Rozemuller JM and Scheper W: Activation of the
unfolded protein response in Parkinson's disease. Biochem Biophys
Res Commun. 354:707–711. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gully JC, Sergeyev VG, Bhootada Y,
Mendez-Gomez H, Meyers CA, Zolotukhin S, Gorbatyuk MS and Gorbatyuk
OS: Up-regulation of activating transcription factor 4 induces
severe loss of dopamine nigral neurons in a rat model of
Parkinson's disease. Neurosci Lett. 627:36–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bellucci A, Navarria L, Zaltieri M,
Falarti E, Bodei S, Sigala S, Battistin L, Spillantini M, Missale C
and Spano P: Induction of the unfolded protein response by
α-synuclein in experimental models of Parkinson's disease. J
Neurochem. 116:588–605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Betzer C, Lassen LB, Olsen A, Kofoed RH,
Reimer L, Gregersen E, Zheng J, Calì T, Gai WP, Chen T, et al:
Alpha-synuclein aggregates activate calcium pump SERCA leading to
calcium dysregulation. EMBO Rep. 19:e446172018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jankovic J and Tan EK: Parkinson's
disease: Etiopathogenesis and treatment. J Neurol Neurosurg
Psychiatry. 91:795–808. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oertel W and Schulz JB: Current and
experimental treatments of Parkinson disease: A guide for
neuroscientists. J Neurochem. 139 (Suppl 1):S325–S337. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pytel D, Seyb K, Liu M, Ray SS, Concannon
J, Huang M, Cuny GD, Diehl JA and Glicksman MA: Enzymatic
characterization of ER Stress-dependent kinase, PERK, and
development of a high-throughput assay for identification of PERK
inhibitors. J Biomol Screen. 19:1024–1034. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bandyopadhyay S, Ni J, Ruggiero A, Walshe
K, Rogers MS, Chattopadhyay N, Glicksman MA and Rogers JT: A
high-throughput drug screen targeted to the 5′untranslated region
of Alzheimer amyloid precursor protein mRNA. J Biomol Screen.
11:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xicoy H, Wieringa B and Martens GJ: The
SH-SY5Y cell line in Parkinson's disease research: A systematic
review. Mol Neurodegener. 12:102017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xie B, Lin F, Peng L, Ullah K, Wu H, Qing
H and Deng Y: Methylglyoxal increases dopamine level and leads to
oxidative stress in SH-SY5Y cells. Acta Biochim Biophys Sin
(Shanghai). 46:950–956. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Slanzi A, Iannoto G, Rossi B, Zenaro E and
Constantin G: In vitro models of neurodegenerative diseases. Front
Cell Dev Biol. 8:3282020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Falkenburger BH, Saridaki T and Dinter E:
Cellular models for Parkinson's disease. J Neurochemistry. 139
(Suppl 1):S121–S130. 2016. View Article : Google Scholar
|
|
46
|
Bai X and Strong R: Expression of
synaptophysin protein in different dopaminergic cell lines. J
Biochem Pharmacol Res. 2:185–190. 2014.PubMed/NCBI
|
|
47
|
Rozpedek W, Pytel D, Diehl JA and
Majsterek I: Niskoczasteczkowe inhibitory szlaku adaptacyjnej
odpowiedzi na stres zaleznego od kinazy PERK jako nowatorska
strategia terapeutyczna w leczeniu choroby Alzheimera. Pol Merkur
Lekarski. 46:9–15. 2019.(In Polish). PubMed/NCBI
|
|
48
|
Rivero-Rios P, Gomez-Suaga P, Fdez E and
Hilfiker S: Upstream deregulation of calcium signaling in
Parkinson's disease. Front Mol Neuroscience. 7:532014.PubMed/NCBI
|
|
49
|
Sun Y, Selvaraj S, Pandey S, Humphrey KM,
Foster JD, Wu M, Watt JA, Singh BB and Ohm JE: MPP+
decreases store-operated calcium entry and TRPC1 expression in
Mesenchymal Stem Cell derived dopaminergic neurons. Sci Rep.
8:117152018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Brodnanova M, Hatokova Z, Evinova A,
Cibulka M and Racay P: Differential impact of imipramine on
thapsigargin- and tunicamycin-induced endoplasmic reticulum stress
and mitochondrial dysfunction in neuroblastoma SH-SY5Y cells. Eur J
Pharmacol. 902:1740732021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Panagaki T, Michael M and Holscher C:
Liraglutide restores chronic ER stress, autophagy impairments and
apoptotic signalling in SH-SY5Y cells. Sci Rep. 7:161582017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Koo HJ, Piao Y and Pak YK: Endoplasmic
reticulum stress impairs insulin signaling through mitochondrial
damage in SH-SY5Y cells. Neurosignals. 20:265–280. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chung H, Chung HY, Bae CW, Kim CJ and Park
S: Ghrelin suppresses tunicamycin- or thapsigargin-triggered
endoplasmic reticulum stress-mediated apoptosis in primary cultured
rat cortical neuronal cells. Endocr J. 58:409–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Dibdiakova K, Saksonova S, Pilchova I,
Klacanova K, Tatarkova Z and Racay P: Both thapsigargin- and
tunicamycin-induced endoplasmic reticulum stress increases
expression of Hrd1 in IRE1-dependent fashion. Neurol Res.
41:177–188. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ullrich C and Humpel C: The pro-apoptotic
substance thapsigargin selectively stimulates re-growth of brain
capillaries. Curr Neurovasc Res. 6:171–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Goodwin J, Nath S, Engelborghs Y and
Pountney DL: Raised calcium and oxidative stress cooperatively
promote alpha-synuclein aggregate formation. Neurochem Int.
62:703–711. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ito S, Nakaso K, Imamura K, Takeshima T
and Nakashima K: Endogenous catecholamine enhances the dysfunction
of unfolded protein response and alpha-synuclein oligomerization in
PC12 cells overexpressing human alpha-synuclein. Neurosci Rese.
66:124–130. 2010. View Article : Google Scholar
|
|
58
|
Rozpedek W, Pytel D, Poplawski T, Walczak
A, Gradzik K, Wawrzynkiewicz A, Wojtczak R, Mucha B, Diehl JA and
Majsterek I: Inhibition of the PERK-dependent unfolded protein
response signaling pathway involved in the pathogenesis of
Alzheimer's disease. Curr Alzheimer Res. 16:209–218. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rozpedek-Kaminska W, Galita G, Siwecka N,
Carroll SL, Diehl JA, Kucharska E, Pytel D and Majsterek I: The
potential role of small-molecule PERK inhibitor LDN-0060609 in
primary open-angle glaucoma treatment. Int J Mol Sci. 22:44942021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Colla E, Coune P, Liu Y, Pletnikova O,
Troncoso JC, Iwatsubo T, Schneider BL and Lee MK: Endoplasmic
reticulum stress is important for the manifestations of
alpha-synucleinopathy in vivo. J Neurosci. 32:3306–3320. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Smith WW, Jiang H, Pei Z, Tanaka Y, Morita
H, Sawa A, Dawson VL, Dawson TM and Ross CA: Endoplasmic reticulum
stress and mitochondrial cell death pathways mediate A53T mutant
alpha-synuclein-induced toxicity. Hum Mol Genet. 14:3801–3811.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Smedley GD, Walker KE and Yuan SH: The
role of PERK in understanding development of neurodegenerative
diseases. Int J Mol Sci. 22:81462021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Moreno JA, Halliday M, Molloy C, Radford
H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA
and Mallucci GR: Oral treatment targeting the unfolded protein
response prevents neurodegeneration and clinical disease in
prion-infected mice. Sci Transl Med. 5:206ra1382013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Costa CAD, Manaa WE, Duplan E and Checler
F: The endoplasmic reticulum stress/unfolded protein response and
their contributions to Parkinson's disease physiopathology. Cells.
9:24952020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baek JH, Mamula D, Tingstam B, Pereira M,
He Y and Svenningsson P: GRP78 level is altered in the brain, but
not in plasma or cerebrospinal fluid in Parkinson's disease
patients. Front Neurosci. 13:6972019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Selvaraj S, Sun Y, Watt JA, Wang S, Lei S,
Birnbaumer L and Singh BB: Neurotoxin-induced ER stress in mouse
dopaminergic neurons involves downregulation of TRPC1 and
inhibition of AKT/mTOR signaling. J Clin Invest. 122:1354–1367.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bellani S, Mescola A, Ronzitti G, Tsushima
H, Tilve S, Canale C, Valtorta F and Chieregatti E: GRP78
clustering at the cell surface of neurons transduces the action of
exogenous alpha-synuclein. Cell Death Differ. 21:1971–1983. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Credle JJ, Forcelli PA, Delannoy M, Oaks
AW, Permaul E, Berry DL, Duka V, Wills J and Sidhu A:
α-Synuclein-mediated inhibition of ATF6 processing into COPII
vesicles disrupts UPR signaling in Parkinson's disease. Neurobiol
Dis. 76:112–125. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cooper AA, Gitler AD, Cashikar A, Haynes
CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al:
Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron
loss in Parkinson's models. Science. 313:324–328. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gitler AD, Bevis BJ, Shorter J, Strathearn
KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC,
McCaffery JM, et al: The Parkinson's disease protein
alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad
Sci USA. 105:145–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Paiva I, Jain G, Lázaro DF, Jerčić KG,
Hentrich T, Kerimoglu C, Pinho R, Szegő ÈM, Burkhardt S, Capece V,
et al: Alpha-synuclein deregulates the expression of COL4A2 and
impairs ER-Golgi function. Neurobiol Dis. 119:121–135. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Jung EM, Yoo YM, Park SY, Ahn C, Jeon BH,
Hong EJ, Kim WY and Jeung EB: Calbindin-D9k is a novel risk gene
for neurodegenerative disease. Cell Physiol Biochem. 54:438–456.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Giorgi C, Bonora M, Sorrentino G,
Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di
Virgilio F, Zito E, et al: p53 at the endoplasmic reticulum
regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci
USA. 112:1779–1784. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kovaleva V and Saarma M: Endoplasmic
reticulum stress regulators: New drug targets for Parkinson's
disease. J Parkinsons Dis. 11 (Suppl 2):S219–S228. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mercado G, Castillo V, Soto P, López N,
Axten JM, Sardi SP, Hoozemans JJM and Hetz C: Targeting PERK
signaling with the small molecule GSK2606414 prevents
neurodegeneration in a model of Parkinson's disease. Neurobiol Dis.
112:136–148. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Radford H, Moreno JA, Verity N, Halliday M
and Mallucci GR: PERK inhibition prevents tau-mediated
neurodegeneration in a mouse model of frontotemporal dementia. Acta
Neuropathol. 130:633–642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Axten JM, Medina JR, Feng Y, Shu A,
Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et
al: Discovery of
7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p
yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective
first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic
reticulum kinase (PERK). J Med Chem. 55:7193–7207. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
O'Connor T, Sadleir KR, Maus E,
Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B, Bembinster LA,
Lammich S, et al: Phosphorylation of the translation initiation
factor eIF2alpha increases BACE1 levels and promotes
amyloidogenesis. Neuron. 60:988–1009. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang ZF, Gao C, Chen W, Gao Y, Wang HC,
Meng Y, Luo CL, Zhang MY, Chen G, Chen XP, et al: Salubrinal offers
neuroprotection through suppressing endoplasmic reticulum stress,
autophagy and apoptosis in a mouse traumatic brain injury model.
Neurobiol Learn Mem. 161:12–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Wu L, Luo N, Zhao HR, Gao Q, Lu J, Pan Y,
Shi JP, Tian YY and Zhang YD: Salubrinal protects against
rotenone-induced SH-SY5Y cell death via ATF4-parkin pathway. Brain
Res. 1549:52–62. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Gupta S, Mishra A and Singh S: Cardinal
role of eukaryotic initiation factor 2 (eIF2α) in progressive
dopaminergic neuronal death & DNA fragmentation: Implication of
PERK:IRE1α:ATF6 axis in Parkinson's pathology. Cell Signal.
81:1099222021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Cankara FN, Kuş MS, Günaydın C, Şafak S,
Bilge SS, Ozmen O, Tural E and Kortholt A: The beneficial effect of
salubrinal on neuroinflammation and neuronal loss in intranigral
LPS-induced hemi-Parkinson disease model in rats. Immunopharmacol
Immunotoxicol. 44:168–177. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Huang X, Chen Y, Zhang H, Ma Q, Zhang YW
and Xu H: Salubrinal attenuates β-amyloid-induced neuronal death
and microglial activation by inhibition of the NF-κB pathway.
Neurobiol Aging. 33:1007.e9–e17. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sidrauski C, Tsai JC, Kampmann M, Hearn
BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang
EL, et al: Pharmacological dimerization and activation of the
exchange factor eIF2B antagonizes the integrated stress response.
Elife. 4:e073142015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Halliday M, Radford H, Sekine Y, Moreno J,
Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer
PM, et al: Partial restoration of protein synthesis rates by the
small molecule ISRIB prevents neurodegeneration without pancreatic
toxicity. Cell Death Dis. 6:e16722015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hughes D and Mallucci GR: The unfolded
protein response in neurodegenerative disorders-therapeutic
modulation of the PERK pathway. FEBS J. 286:342–355. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Chia SJ, Tan EK and Chao YX: Historical
perspective: Models of Parkinson's Disease. Int J Mol Sci.
21:24642020. View Article : Google Scholar : PubMed/NCBI
|