Introduction
The intestinal epithelial barrier is one of the most
important immune barriers of defense against invasive symbiotic
bacteria and intestinal pathogens (1). Damage to the intestinal epithelial
barrier may lead to the translocation of bacteria and metabolites
into the bloodstream and tissues (2,3). In
severe cases, this translocation may trigger systemic inflammation
and multiple organ dysfunction syndrome (4). Numerous pathological states, such as
ischemia reperfusion injury, inflammation and cancers, such as
liver or pancreatic cancer (5),
cause damage to the intestinal mucosal barrier (6), leading to complexities in clinical
practice and poor patient prognosis (7). Therefore, current research is focused
on exploring novel methods for protecting and repairing intestinal
barrier function.
Octreotide (OCT) is a synthetic octapeptide
derivative of natural somatostatin (8). Results of previous studies
demonstrated that OCT may prevent diarrhea caused by chemotherapy,
intestinal injury caused by severe pancreatitis and inflammatory
bowel disease (IBD) (9–11). Thus, OCT may exhibit potential in
the treatment of intestinal injury.
Somatostatin receptors (SSTRs) are G-protein-coupled
receptors that are widely expressed on cell membranes in the human
brain and kidney (12,13), and in colon tissue (14). Numerous previous studies on SSTRs
have focused on their use in preventing tumor cell proliferation
(15–17). Somatostatin binds to SSTRs to exert
effects on cells (12,18). Results of previous studies revealed
that out of the five subtypes of SSTRs, subtypes 2, 3 and 5 exhibit
a high affinity for OCT (8,19).
Although somatostatin protects the intestinal mucosal barrier by
regulating the expression of tight junction (TJ) proteins (8,20–23),
the specific molecular mechanisms remain to be fully
elucidated.
Autophagy is a process in which cytoplasmic proteins
or organelles are phagocytosed into vesicles that fuse with
lysosomes. Following the formation of autophagy lysosomes, all
contents of the lysosome are degraded (24–26).
Physiologically, autophagy is required for cell metabolism and the
renewal of intracellular organelles. Results of previous studies
demonstrated that autophagy in intestinal epithelial cells is
directly associated with TJ and intestinal epithelial barrier
function (27,28).
Lipopolysaccharide (LPS) is unique to the outer
membrane of Gram-negative bacteria and is involved in intestinal
epithelial innate immunity (29).
LPS triggers an inflammatory signaling cascade to induce TJ
dysfunction, resulting in intestinal epithelial barrier dysfunction
(30,31). Therefore, the present study aimed
to explore the effects of OCT on autophagy and SSTR function using
LPS-induced Caco2 cells. In addition, the effects of OCT on TJ and
intestinal mucosal barrier function were investigated by western
blot and reverse transcription-quantitative (RT-q)PCR. The present
study provides novel insight into potential methods for the
protection and repair of the intestinal epithelial barrier.
Furthermore, results of the present study may provide a novel
theoretical basis for the treatment of intestinal mucositis in
clinical practice.
Materials and methods
Cell culture and treatment
The human colon adenocarcinoma cell lines Caco2 and
Sw480 were obtained from Professor Zunling Li and the identity of
the cell lines was confirmed by short tandem repeat sequencing.
Cells were initially purchased from the American Type Culture
Collection. Caco2 cells were cultured in minimum essential medium
(including non-essential amino acid; cat. no. PM150410;
Sigma-Aldrich; Merck KGaA) containing 20% fetal bovine serum
(Thermo Fisher Scientific, Inc.) and stored at 37°C under 5%
CO2 with saturated humidity. Sw480 cells were cultured
in DMEM (cat. no. RNBL7920; Sigma-Aldrich; Merck KGaA) containing
10% fetal bovine serum and stored under the same conditions. They
were passaged once every other day and cells in the logarithmic
growth phase were used for experimental research.
OCT and LPS treatment in vitro
The treatment method for LPS (cat. no. L4391;
Sigma-Aldrich; Merck KGaA) for the two cell lines was performed by
adding it to the corresponding culture medium with a final
concentration of 100, 50, 10, 1 and 0.1 µg/ml for different
durations. OCT (cat. no. HY-17365; MedChemExpress) with a final
concentration of 10, 20 or 50 µM was added to the corresponding
culture medium 2 h before the addition of LPS. Caco2 cells were
inoculated into a 96-well plate with an initial density of
5×104 cells/well and Sw480 were inoculated with an
initial density of 6×104 cells/well. In subsequent
experiments, the cells were divided into three groups: Control, 50
µg/ml LPS and 10 µM OCT. The control group was left untreated. For
the LPS + OCT group, cells were first pretreated with OCT for 2 h
and then coincubated with LPS for 24 h, and then various indicators
were detected.
Interference with SSTR2 using small
interfering (si)RNA
Cells were inoculated into a six-well plate at a
density of 5×105 cells/per well and they were allowed to
adhere to the bottom of the wells and grow to 50–70% confluence.
The instructions provided by the manufacturer of GP-transfect mate
(Suzhou Jima Gene, Co., Ltd.) were followed to transfect the cells
and the medium was changed 4–6 h after transfection. The total RNA
or protein were extracted for detection at 48–72 h after
transfection. The siRNA sequences were synthesized by Jima Gene
Co., Ltd. The siRNA sets were as follows: Negative Control,
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(anti-sense); SSTR2-Homo-1097 5′-GCUCCUCUAAGAGGAAGAATT-3′ (sense)
and 5′-UUCUUCCUCUUAGAGGAGCTT-3′ (anti-sense); SSTR2-Homo-1264
5′-GUCCUCACCUAUGCUAACAAT-3′ (sense) and 5′-UGUUAGCAUAGGUGAGGACTT-3′
(anti-sense); SSTR2-Homo-1049 5′-GCUACCUGUUCAUUAUCAUTT-3′ (sense)
and 5′-AUGAUAAUGAACAGGUAGCTT-3′ (anti-sense).
Cell viability assay
A CCK-8 kit (cat. no. C0038; Beyotime Institute of
Biotechnology) was used to detect the viability of the cells
treated with the drug. Cells with a density of 5×104
cells/well were seeded into a 96-well plate and cultured for 24 h
until they exhibited adherent growth according to the instructions
in the manual, and the cells were then stimulated with drugs for 24
h at 37°C. Subsequently, the cells were incubated with CCK-8
solution for 1 h at 37°C and the absorbance at 450 nm was detected
with a microplate reader. Each experimental condition in each group
was set up in three wells. Although the control group had three
repeated experiments, it has been normalized as a reference
standard for activity calculation.
Western blot analysis
First, cells were cultured in six-well plates at a
density of 1×106 cells/well. After 24 h of cell adhesion
and growth, LPS and OCT were added to pretreat the cells. The
whole-cell protein was extracted according to the instructions
provided by the RIPA manufacturer, and the cell pellet was
resuspended using a RIPA mixture (cat. no. R0020; Beyotime
Institute of Biotechnology) containing PMSF (cat. no. P0100) and
phosphatase inhibitor (cat. no. P1082; Beyotime Institute of
Biotechnology), followed by 30 min of incubation in an ice bath,
during which repeated pipetting was performed every 5 min to ensure
full cells lysis. Following centrifugation at 12,000 × g for 20
min, the extracted protein was obtained as the supernatant. The
protein concentration was then measured by using a Nanodrop 2000c
(Thermo Fisher Scientific, Inc.). Protein samples (10–20 µg) were
then separated on 10 or 12% gels using SDS-PAGE. The protein in the
gel was then transferred to a nitrocellulose membrane and the
nonspecific binding sites on the membrane were blocked for 2 h at
room temperature with 5% non-fat milk. The membranes were incubated
with primary antibody overnight at 4°C, and subsequently, they were
incubated with horseradish peroxidase-labeled secondary antibody
for 1 h at room temperature. Finally, the enhanced luminescent
agent A solution and stabilizer B solution (cat. no.
BL520B1/BL520B2; Biosharp) were mixed in a 1:1 ratio to visualize
the protein bands. The primary antibodies included the following:
Antibodies against microtubule-associated protein 1 light chain 3B
(LC3; cat. no. ab192890; 1:1,500 dilution; Abcam), zona occludens 1
(zo-1; cat. no. 10019107; 1:1,500 dilution; Proteintech Group,
Inc.), GAPDH (cat. no. AF7021; 1:8,000 dilution; Affinity
Biosciences), SSTR2 (cat. no. YT-5740), SSTR3 (cat. no. YN-2540),
SSTR5 (cat. no. YN-2541; all 1:1,000 dilution; ImmunoWay
Biotechnology), goat-anti mouse (cat. no. orb229658; 1:8,000
dilution; Biorbyt) and goat anti-rabbit (cat. no. ZB-2301; 1:8,000
dilution; Zhongshan Jinqiao Biotechnology Co., Ltd.). The intensity
of the bands was quantified using ImageJ software version 1.49
(National Institutes of Health).
LC3 double label adenovirus
transfection
The target cells were inoculated onto a 24-well
plate at a concentration of 1×105 cells/well, and it was
ensured that the cell convergence rate is between 50 and 70% when
cells were transfected with the Ad-monomeric red fluorescence
protein (mRFP)-green fluorescence protein (GFP)-LC3 adenovirus
(Hanheng Biotechnology Co., Ltd.) the next day. According to the
instructions and technical guidance, the virus was added to the
culture medium, left to incubate for 3 h at 37°C, and the medium
was then replaced with fresh culture medium. After 24 h, GFP and
RFP expression may be observed, and cell fixation, sealing
(mounting medium antifading; Beijing Solarbio Technology Co., Ltd.)
and imaging analysis may be performed from 36 to 48 h. Laser
confocal microscopy imaging (Leica Microsystems GmbH) was used to
capture and manually count autophagic dots. Images were captured
using a ×40 objective and the experiment was repeated three times;
during each repetition, three views were selected to count the
fluorescent dots.
Immunofluorescence
Cells were diluted to a density of 5×104
after cell counting and seeded on cover glasses in a 24-well plate
in advance. After transfecting cells in a 24-well plate with
Ad-mRFP-GFP-LC3 adenovirus for 48 h, they were fixed with 4%
paraformaldehyde for 15 min at room temperature and then washed
three times with PBS. The supernatant was then discarded and
sterilized forceps were used to remove the cover glasses. Using a
drop of mounting medium with antifading (cat. no. 20210427; Beijing
Solarbio Technology Co., Ltd.) the cover glasses were mounted on
slides. Digital image acquisition was performed using laser
confocal microscopy (Leica Microsystems GmbH).
RT-qPCR
Cells were seeded into six-well plates at a density
of 1×106 cells/well, allowed to attach to the bottom of
the wells and then incubated with the indicated concentrations of
OCT and/or LPS. Total RNA was extracted using RNAiso plus (cat. no.
AM33539A; Takara Biotechnology Co., Ltd.) according to the
instructions in the manual. RT was then performed using the Evo
M-MLV RT Mix kit (cat. no. AG11728) and 500 ng of RNA quantified by
the Nanodrop system 2000c (Thermo Fisher Scientific, Inc.) was
reverse-transcribed into cDNA. The next step was real-time PCR
quantification of tight ligation gene mRNA using SYBR Advantage
qPCR Premix (cat. no. AG11701; Takara Bio, Inc.). A two-step
program was chosen for qPCR. In step 1, the temperature was set to
95°C for 30 sec for 1 cycle. In step 2, the temperature was set to
95°C for 5 sec and 60°C for 30 sec for 40 cycles. The relative mRNA
expression was determined by the 2−∆∆Cq calculation
method (32). GAPDH was used as a
housekeeping gene for mRNA. The target primer sequences were
synthesized by Sangon Biotech. The primer sets were as follows:
β-actin forward, 5′-CCTGGACTTCGAGCAAGAGATGG-3′ and reverse,
5′-CAGGAAGGAAGGCTGGAAGAGTG-3′; GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′;
TNF-α forward, 5′-CCTCTCTCTAATCAGCCCTCTG-3′ and reverse,
5′-GAGGACCTGGGAGTAGATGAG-3′; IL-6 forward,
5′-ACTCACCTCTTCAGAACGAATTG-3′ and reverse,
5′-CCATCTTTGGAAGGTTCAGGTTG-3′; ZO-1 forward,
5′-GCGGATGGTGCTACAAGTGATG-3′ and reverse,
5′-GCCTTCTGTGTCTGTGTCTTCATAG-3′; occludin (OCLN) forward,
5′-TACGGAAGTGGCTATGGCTATGG-3′ and reverse,
5′-CTTTGCTGCTCTTGGGTCTGTATAG-3′; and claudin (CLDN)1 forward,
5′-TGGTGGTTGGCATCCTCCTG-3′ and reverse,
5′-TCATCGTCTTCCAAGCACTTCATAC-3′.
Trans-epithelial electrical resistance
(TEER)
The TEER measurements across Caco-2 cell monolayers
were performed using a Millicell ERS instrument (EMD Millipore).
Cells were seeded with a density of 1×104 per well in a
24-well Transwell plate (cat. no. 02822019; Corning, Inc.),
ensuring that the liquid levels on the apicl (AP) sides and
basolateral (BL) sides were at level. The fluid was changed every
other day until the cells formed a tight junction on the 21st day.
Before using the resistance meter, it was cleaned and set to zero
with alcohol and PBS. The positive and negative electrodes were
inserted into the orifice plate according to the manufacturer's
instructions until the resistance meter was able to read smoothly
and count. The resistance value Ω and percentage of each well were
calculated according to a formula, with 3 composite wells in each
group to reduce experimental errors. The TEER values of these cells
after treatment were recorded. Resistance due to the cell
monolayers was determined in the presence and the absence of OCT
after subtracting the contribution of the blank filter. TEER was
calculated as follows: TEER=(R1-R0) × A (Ω), where R1 and R0
represent the TEER readings from the wells with cells and the
no-cell background wells, respectively. A (cm2)
represents the surface area of the cell monolayer on the
insert.
The percentage change in TEER was calculated as
follows: TEER%=TEERtest/TEERinitial ×100.
Cell permeability
The cells were seeded into 24-well Transwell plates
at a density of 1×105 cells/well. Cells were cultured to
simulate the tight junction structure of small intestinal
epithelial cells. Prior to detection, Hank's balanced salt solution
containing 1 mg/ml FITC-Dextran4000 (FD4; cat. no. HY-128868A;
MedChemExpress) was added to the AP side of the cells and PBS was
added to the BL side. FD4 (0.1 mg/ml) was added to the basal media
in the Transwell chamber. Media were collected from the Transwell
insert after 3 h. The fluorescence signal (excitation at 485 nm and
emission at 538 nm) was measured and the FD4 concentration was
calculated based on fluorescence intensity.
Transmission electron microscopy
(TEM)
Observation of Caco-2 cell autophagosomes was
performed using a Leica TEM (Leica Microsystems GmbH). Cells were
collected in 1.5-ml centrifuge tubes, fixed with glutaraldehyde
(cat. no. G6257; Merck & Co., Inc.) solution overnight at 4°C
after two washes of PBS, followed by fixation with 1% osmic acid
for 1–2 h at 4°C, three washes with PBS and gradient dehydration
for 15 min per gradient. The next step was to use a gradient
permeation of the embedding agent, followed by a 37°C permeation of
the pure embedding agent overnight. After heating and
polymerization at 70°C for at least 24 h, the sections were double
stained with lead citrate and acetic acid peroxide oil, and then
observed using a Leica TEM (Leica Microsystems GmbH).
Statistical analysis
All experiments were conducted three parallel
experiments. The results in each figure are expressed as the mean ±
standard error of the mean. GraphPad Prism 8 software (GraphPad;
Dotmatics) was used for statistical analysis. P-values were
calculated using one-way analysis of variance with Tukey's post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
OCT prevents LPS-induced intestinal
epithelial cell injury in Caco2 and Sw480 cells
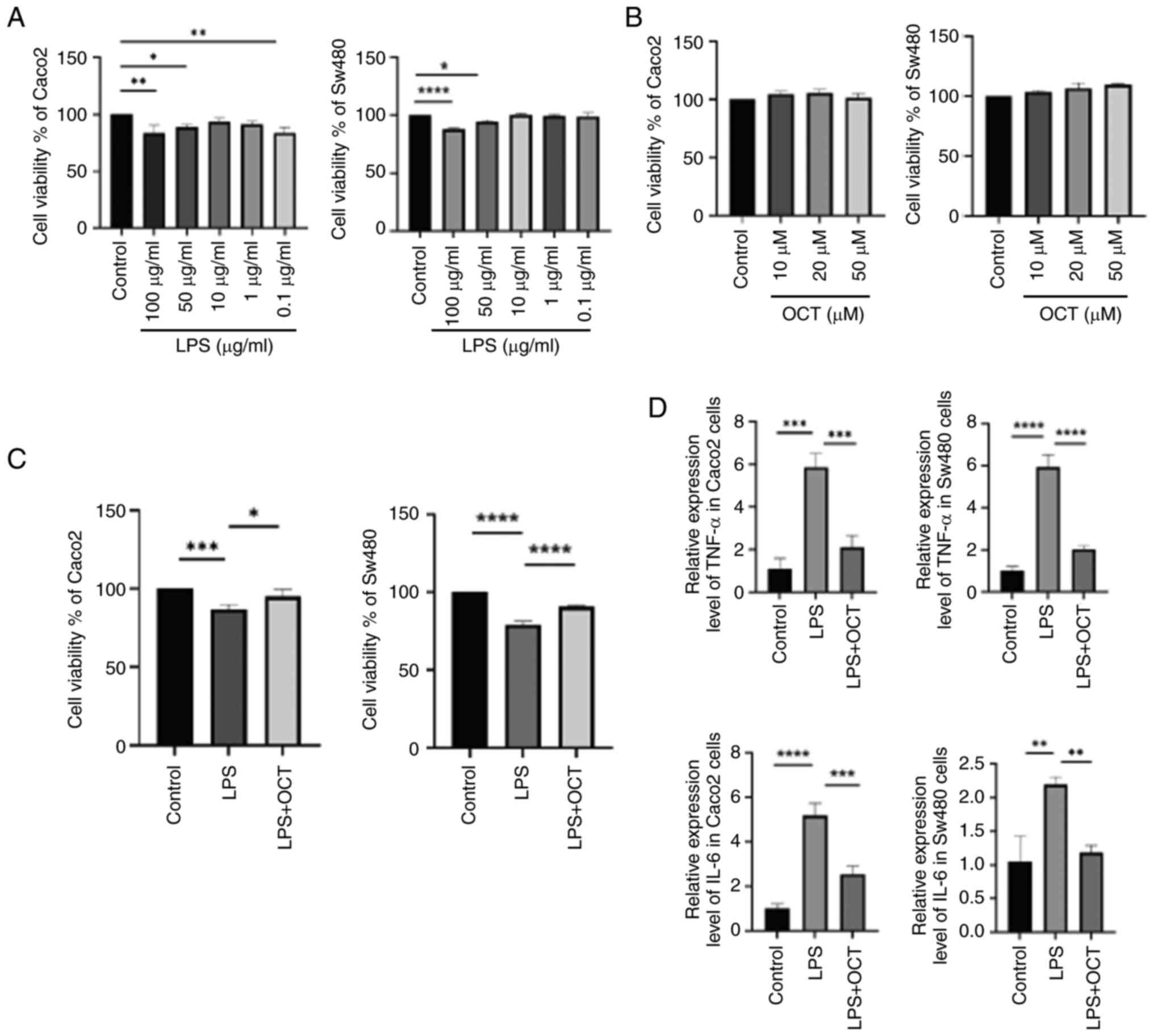
The effects of different concentrations of LPS on
cell viability were measured in both Caco2 and Sw480 cells. The
results indicated that after 24 h of incubation, 100 and 50 µg/ml
LPS markedly decreased the cell viability in these two cell lines
(Fig. 1A). A lower concentration
of 50 µg/ml LPS was chosen for subsequent experiments. To determine
whether OCT exerted any effects on the cells, they were incubated
with 10, 20 and 50 µM OCT for 24 h, and cell viability was
evaluated. The results suggested that OCT had no significant effect
on the viability of these two cell lines (Fig. 1B). According to previous referenced
results, 10 µM OCT is often used as the optimal concentration for
processing cells (33). Thus, 50
µg/ml LPS and 10 µM OCT were selected for use in subsequent
experiments, and Caco2 and Sw480 cells were both pre-treated with
10 µM OCT 2 h prior to treatment with 50 µg/ml LPS for 24 h. Of
note, pre-treatment with 10 µM OCT significantly improved the cell
viability compared with that of cells treated with LPS alone
(Fig. 1C). TNF-α and IL-6 are two
important pro-inflammatory cytokines that are significantly
increased in IBD and the expression of these gene was measured
using RT-qPCR. As indicated in Fig.
1D, LPS significantly induced the secretion of TNF-α and IL-6
in Caco2 and Sw480 cells. In addition, OCT inhibited the effects of
LPS on TNF-α and IL-6 expression. Collectively, these results
suggested that OCT may attenuate LPS-induced intestinal epithelial
cell injury and inflammation in vitro.
OCT inhibits intestinal epithelial
cell Caco2 damage by regulating SSTR2
To investigate which SSTR subtype has a role in the
OCT-mediated protection of intestinal epithelial cells, the protein
expression levels of SSTR2, −3 and −5 were examined. The results
demonstrated that the expression levels of SSTR2 were significantly
increased following OCT treatment, while no significant differences
in SSTR3 and SSTR5 expression were observed (Fig. 2A and B). Thus, it was hypothesized
that OCT may protect intestinal epithelial cells through binding to
SSTR2. SSTR2 knockdown was subsequently performed using siRNA
transfection (Fig. 2C). The
highest level of transfection efficiency was observed following
transfection with siRNA-1264 thus, siRNA-1264 was selected for use
in subsequent experiments. The results demonstrated that OCT
treatment did not reverse the LPS-induced reduction in Caco2 and
Sw480 cell viability following SSTR2 knockdown (Fig. 2D). Furthermore, in SSTR2 knockdown
cells, OCT treatment did not reverse the LPS-induced increase in
pro-inflammatory cytokine expression levels (Fig. 2E).
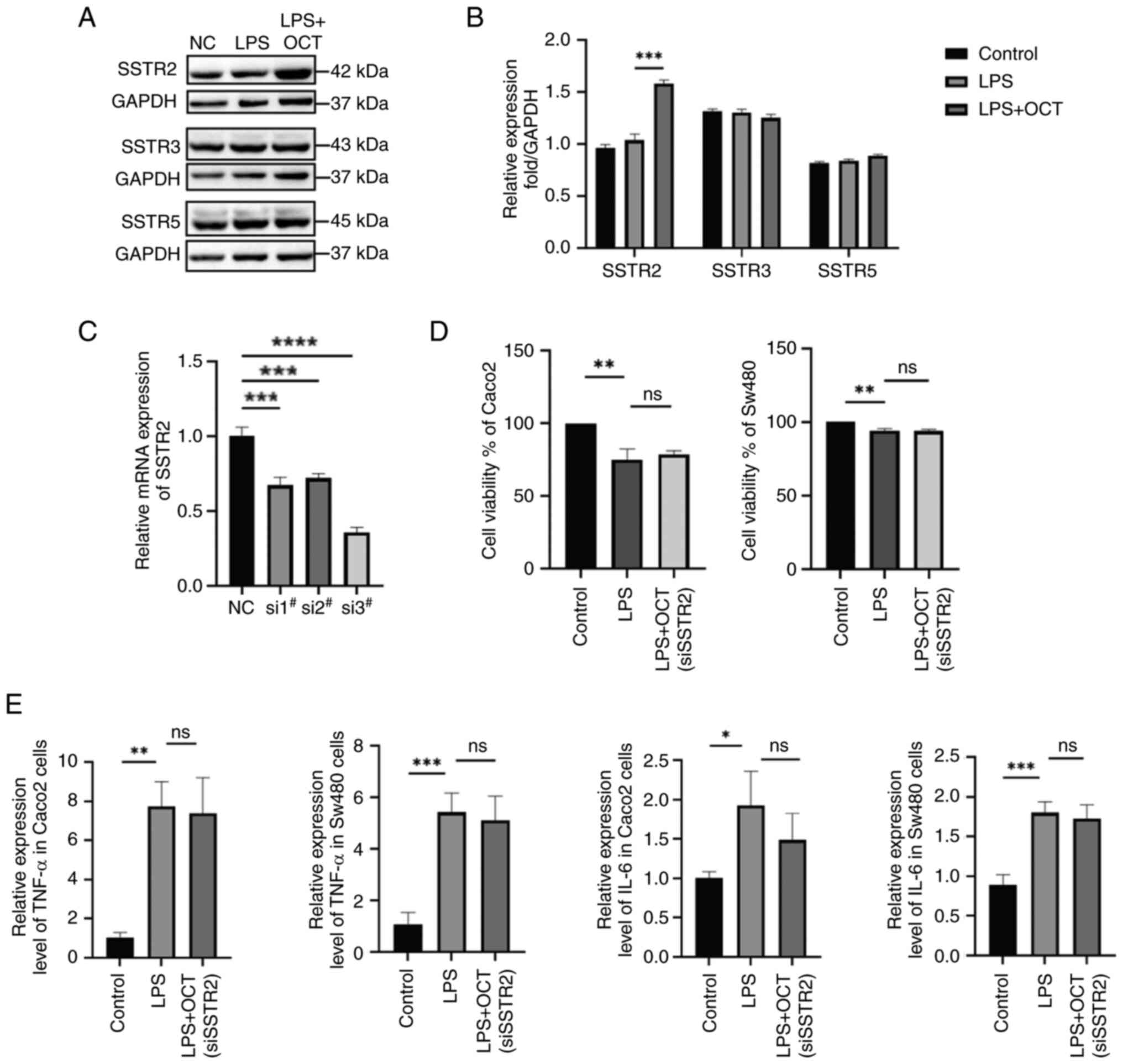 | Figure 2.OCT alleviates intestinal epithelial
cell damage by regulating SSTR2. (A and B) Caco2 cells were
pretreated with 10 µM OCT for 2 h and then stimulated with 50 µg/ml
LPS for 24 h. The protein expression levels of SSTR2, SSTR3 and
SSTR5 were determined by western blot analysis. (A) Representative
western blots and (B) quantified expression levels. (C) Detection
of SSTR2 gene interference efficiency by RT-qPCR. (D) The Cell
Counting Kit-8 assay was used to detect the viability of Caco2
cells (left) and Sw480 (right) treated with LPS (50 µg/ml) and OCT
(10 µM) after interfering with SSTR2 expression. (E) TGF-α and IL-6
in Caco2 and Sw480 cells were determined by RT-qPCR. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001 as indicated; ns, no
significance. RT-qPCR, reverse transcription-quantitative PCR; LPS,
lipopolysaccharide; OCT, Octreotide; NC, negative control; si,
small interfering RNA; SSTR, somatostatin receptor. |
OCT protects against LPS-induced
intestinal epithelial barrier dysfunction in Caco2 cells
To further explore the effects of OCT on LPS-induced
intestinal epithelial barrier dysfunction, the integrity and
permeability of the intestinal epithelium were evaluated using TEER
and FD4 assays in Caco2 cells. After 24 h of incubation, the
resistance and permeability of cells in the control group remained
at a stable level. In addition, the results revealed a significant
decrease in resistance and a significant increase in FD4
permeability following LPS treatment. It was also demonstrated that
cells pre-treated with OCT exhibited higher TEER values (Fig. 3A) and lower levels of FD4
permeability (Fig. 3B) as compared
with cells treated with LPS alone. Following SSTR2 knockdown, OCT
pre-treatment did not rescue TEER values or FD4 permeability in
Caco2 cells, suggesting that OCT may preserve monolayer integrity
in Caco2 cells via SSTR2.
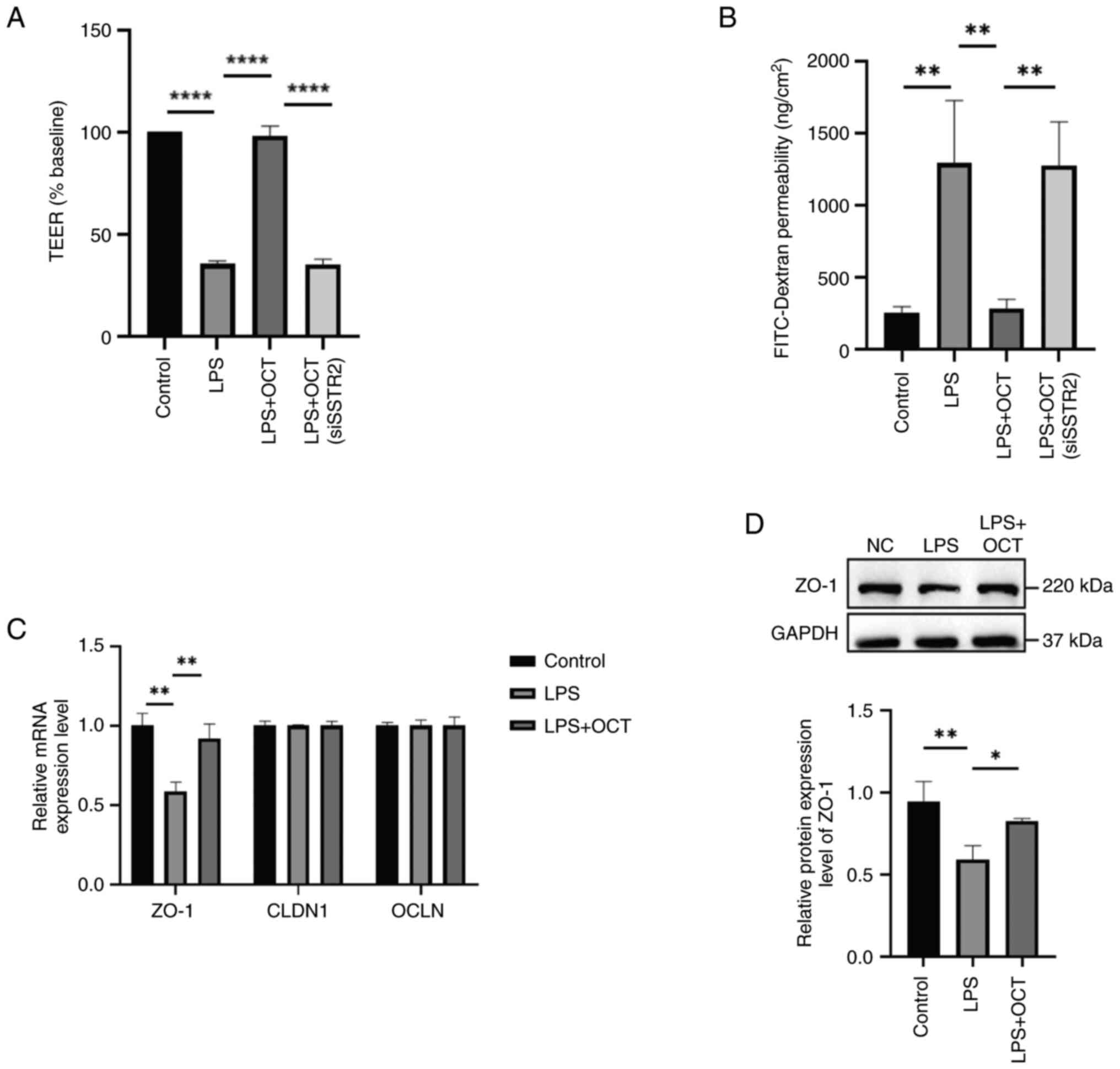 | Figure 3.OCT protects against intestinal
epithelial barrier dysfunction induced by LPS in Caco2 cells. (A
and B) Caco2 cells were pretreated with 10 µM OCT for 2 h and then
stimulated with 50 µg/ml LPS for 24 h. (A) TEER and (B)
FITC-Dextran-4 flux were measured to evaluate the paracellular
permeability. (C) Caco2 cells were pretreated with 10 µM OCT for 2
h and then stimulated with 50 µg/ml LPS for 24 h. The mRNA and
protein expression levels of OCLN, CLDN1 and zo-1 were determined
by reverse transcription-quantitative PCR and western blot
analysis. (D) Caco2 cells were pretreated with 10 µM OCT for 2 h
and then stimulated with 50 µg/ml LPS for 24 h. The protein
expression levels of zo-1 were determined by western blot analysis.
*P<0.05, **P<0.01, ****P<0.0001 as indicated. LPS,
lipopolysaccharide; OCT, Octreotide; NC, negative control; si,
small interfering RNA; SSTR, somatostatin receptor; CLDN, claudin;
zo-1, zona occludens 1; OCLN, occludin; TEER, trans-epithelial
electrical resistance. |
RT-qPCR was used to investigate the expression
levels of TJ proteins (Fig. 3C).
The results suggested that the expression level of zo-1 was
decreased after treatment with LPS; however, it was rescued after
the combined treatment with LPS and OCT. No similar changes were
observed in the expression of the other two tight junction
molecules. Subsequently, zo-1 protein expression levels were
evaluated using western blotting (Fig.
3D), and the results obtained were comparable with those
observed using RT-qPCR.
OCT maintains basal levels of
autophagy in Caco2 cells through SSTR2
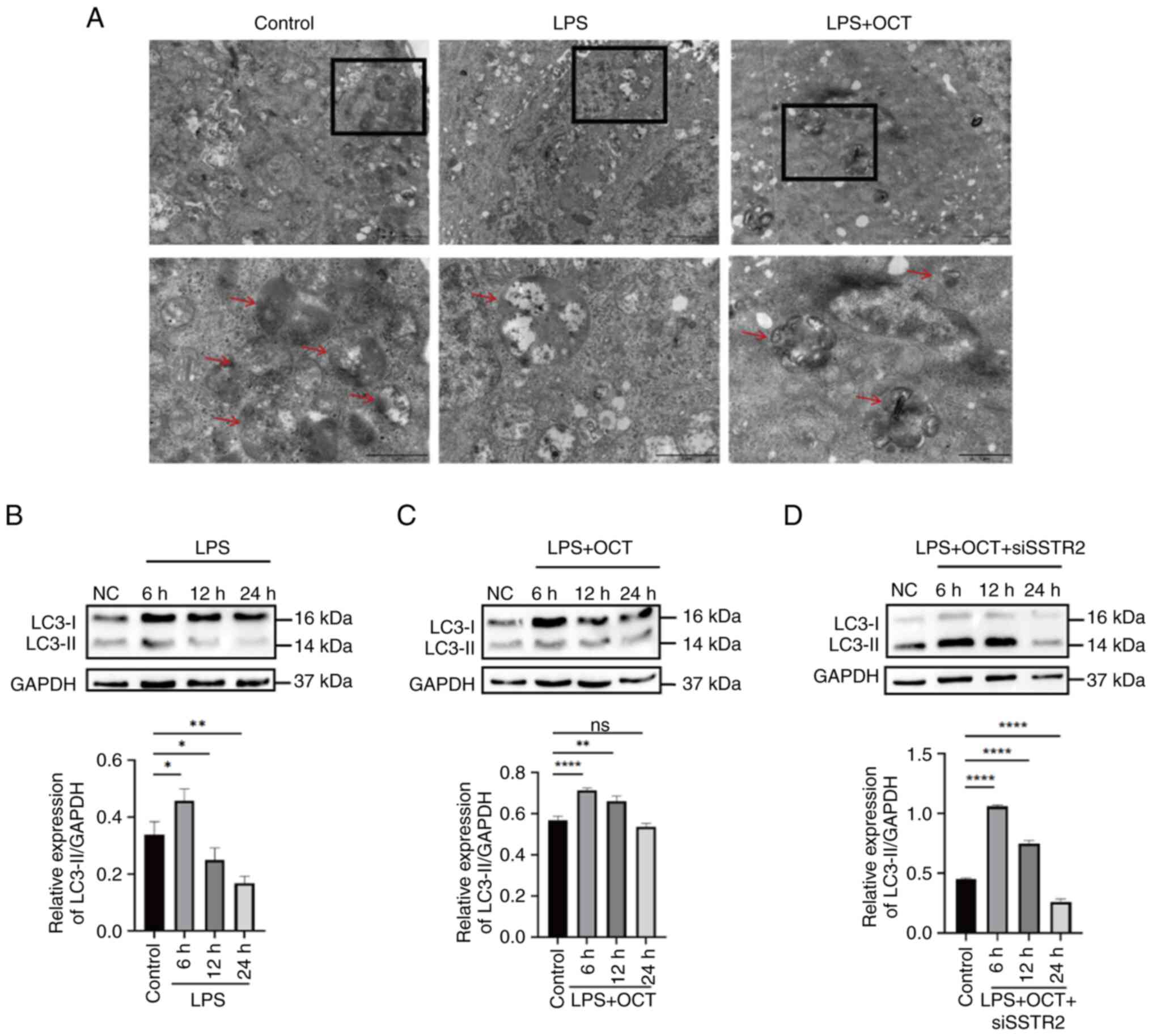
Previous studies revealed that autophagy has an
important role in maintaining the intestinal epithelial barrier by
regulating TJ proteins (34,35).
To determine the protective mechanisms of OCT in intestinal TJ
barrier function, the formation of autophagosomes and
autophagolysosomes in Caco2 cells was observed using TEM. The
results indicated that intestinal epithelial cells exhibited a
basal level of autophagy under normal physiological conditions;
however, autophagy levels were significantly decreased following
LPS treatment for 24 h. By contrast, levels of autophagy returned
to baseline following treatment with OCT (Fig. 4A). Collectively, these results
suggested that autophagy in Caco2 cells may have a role in the
OCT-induced protection of the intestinal epithelial barrier.
As autophagy is a dynamic process, changes in the
levels of autophagy were observed at different time-points. Results
of the western blot analysis revealed that the ratio of
LC3-II/GAPDH expression reached a maximal level after treatment for
6 h, and thereafter, the expression levels were reduced following
prolonged LPS treatment. Low levels of LC3-II expression were
observed in Caco2 cells following incubation for 24 h (Fig. 4B). Of note, the ratio of
LC3-II/GAPDH expression also declined following OCT treatment and
returned to baseline following incubation for 24 h (Fig. 4C). However, following SSTR2
knockdown, OCT treatment did not restore the levels of autophagy in
Caco2 cells (Fig. 4D).
OCT regulates changes in autophagy in
Caco2 cells
Caco2 cells were transfected with the
Ad-mRFP-GFP-LC3 adenovirus to assess potential changes in
autophagy. In this system, the GFP signal is quenched in the acidic
environment of lysosomes, whilst the mRFP signal remains stable.
Therefore, autolysosomes and autophagosomes are labelled red or
yellow, respectively (36).
Utilizing this fluorescence peculiarity, the autophagy flux in each
drug treatment group was monitored (Fig. 5A). The numbers of yellow and red
dots were significantly increased following LPS treatment alone,
reaching a maximal value after 6 h, before declining after 24 h
(Fig. 5B). Pre-treatment with OCT
also increased the numbers of yellow and red dots after 6 h, and
these levels returned to baseline after 24 h (Fig. 5C). Of note, these results were
comparable with those obtained in the western blot analysis of
LC3-II protein expression (Fig. 4B and
C).
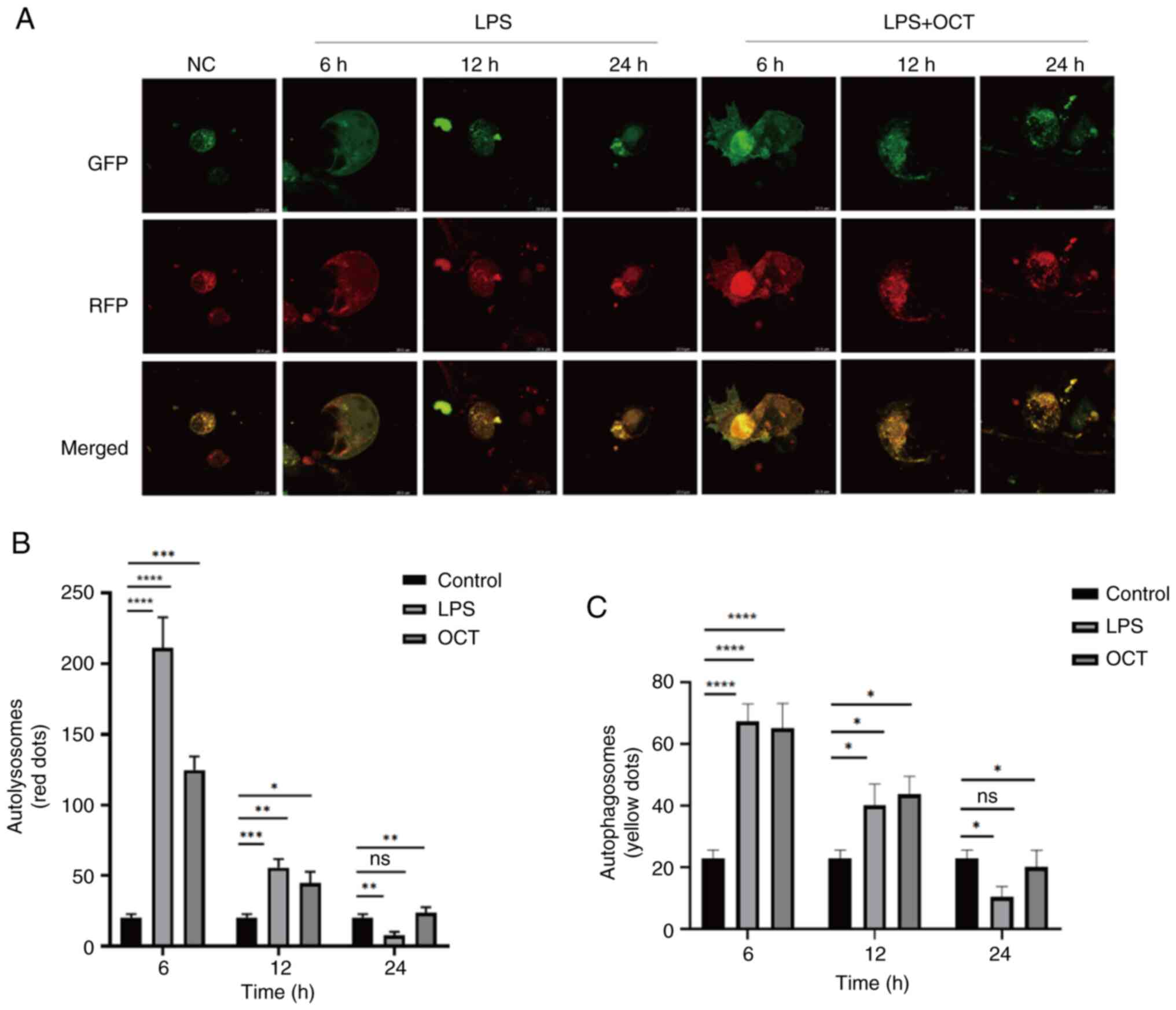 | Figure 5.OCT regulates autophagy flow
alteration in Caco2 cells. (A) Cells infected with autophagy
double-labeled adenovirus were treated for different durations,
followed by only 50 µg/ml LPS or incubation with 50 µg/ml LPS and
10 µM OCT stimulation for 24 h. Leica laser confocal microscopy was
performed to detect changes in autophagy flow. The magnification of
the microscope is ×40. (B) The number of yellow fluorescence spots
after merging of GFP and RFP (autophagosomes) in autophagic flow
was counted. (C) The number of red fluorescence spots after merging
of GFP and RFP (autophagolysosomes) in autophagic flow was counted.
*P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 as
indicated; ns, no significance. LPS, lipopolysaccharide; OCT,
Octreotide; NC, negative control; si, small interfering RNA; SSTR,
somatostatin receptor; GFP, green fluorescence protein; RFP, red
fluorescence protein. |
Collectively, these results suggested that OCT may
protect against LPS-induced intestinal epithelial barrier
dysfunction via SSTR2. These changes may be closely associated with
autophagy in intestinal epithelial cells.
Discussion
Somatostatin analogues are widely used in clinical
practice (37) and are considered
a safe and effective treatment option for acromegaly and acute
pancreatitis (38). OCT, a
somatostatin analogue, reduces damage to the intestinal mucosal
barrier (39), prevents
chemotherapy-induced diarrhea and other refractory diarrhea
(40), and alleviates IBD and
intestinal mucosal barrier injury (41). However, the mechanisms underlying
the protective effects of OCT in intestinal epithelial cells have
remained elusive. Therefore, further investigations are required to
determine the regulatory mechanism of OCT in intestinal epithelial
cells and to explore novel targets for the treatment of diseases
that cause intestinal mucosal damage.
LPS treatment of Caco2 cells is widely used to
simulate intestinal mucositis in vitro (31,32,42).
The present study aimed to determine the effects of OCT in
LPS-treated Caco2 cells, using Sw480 cells for confirmation Results
of the present study revealed that the LPS-induced reduction in
cell viability was inhibited following OCT pre-treatment in Caco2
and Sw480 cells, suggesting that OCT may exert a protective role on
the intestinal inflammatory environment. However, the role of OCT
in the protection of intestinal epithelial cell injury remains
unclear.
Somatostatin exerts its biological effects through
interacting with SSTRs, which belong to the G-protein-coupled
receptor superfamily of receptors (43,44).
To date, five SSTR subtypes have been identified, namely SSTR1-5,
with all five subtypes widely expressed in human tissues (14,43).
Results of a previous study revealed that SSTR2 methylation may act
as a prognostic indicator in colon cancer (45). SSTR1 and SSTR2 are expressed at
high levels in the colon of patients with IBD and ulcerative
colitis (46,47). Of note, OCT binds to SSTR2 and
SSTR5 with high affinity, and to SSTR3 with a low affinity
(48). OCT does not bind to SSTR1
or SSTR4 (8). Giuliani (48) previously reported that long-acting
injectable SSTR ligands, OCT and Lantenside, are suitable as
first-line treatment options for patients with acromegaly (49). In addition, Valencak et al
(23) reported that DOTA-Tyr OCT
is commonly used in clinical practice for the treatment of
neuroendocrine tumors, and exerts effects via binding with SSTR2
(23). In addition, results of a
previous study demonstrated that OCT may be used to effectively
treat patients with thymic epithelial tumors expressing SSTR2
(50). Somatostatins are used for
the inhibition of inflammatory responses in clinical practice
(51). Of note, the 2A subtype of
SSTRs is used in the treatment of IBD, and the regulation of nerve
transmission, proliferation and apoptosis (46). According to previous literature
research, knockdown of SSTR is mostly concentrated in tumor model
studies (52,53). For example, knockout of SSTR
subtypes can improve the sensitivity of the body to induce
neurological diseases (54).
Downregulation of SSTR expression can promote the migration and
invasion of cancer cells (55);
however, knockdown models of SSTR are rare in inflammatory
diseases. In the present cell experiments, the expression of SSTR2
was interfered with and there was no impact on other cellular
functions. Thus, the potential protective effects of OCT in
intestinal epithelial cells were explored in the present study.
Results of the present study revealed that SSTR2 expression was
significantly increased following OCT treatment in Caco2 cells. In
addition, the LPS-induced reduction in cell viability was not
reversed following OCT pre-treatment and SSTR2 knockdown in Caco2
and Sw480 cells, suggesting that the protective effects of OCT were
inhibited following SSTR2 knockdown. Collectively, these results
suggested that OCT may attenuate LPS-induced intestinal epithelial
injury through regulation of SSTR2.
Damage to intestinal epithelial cells is directly
associated with the function of the intestinal mucosal barrier
(56). The defensive role of the
intestinal epithelial barrier is dependent on intercellular TJs
(57). Of note, formation of TJ
protein complexes, including OCLN and zo-1, is crucial for
maintaining the intestinal mucosal barrier (1). In addition, results of a previous
study revealed that TJ destruction and high levels of mucosal
permeability are induced by LPS (58). Results of a previous study revealed
that somatostatins may mediate recovery from LPS-induced intestinal
epithelial barrier dysfunction through regulation of CLDN4
(59). However, the specific
molecular mechanisms or signaling pathways involved were not
revealed. Results of the present study demonstrated that OCT
treatment reversed intestinal barrier dysfunction in LPS-treated
Caco2 cells, as evidenced by elevated TEER values, decreased FD4
flux and increased zo-1 protein expression. However, the effects of
OCT were reversed following SSTR2 knockdown. These results
suggested that OCT may preserve intestinal mucosal barrier function
and protect intestinal epithelial cells via SSTR2.
Intestinal mucosal barrier function, particularly TJ
function, is closely associated with autophagy. Results of a
previous study revealed that autophagy plays an important role in
maintaining intestinal epithelial barrier function through
alteration of TJ protein dynamics (27). TJ dysfunction in the intestinal
epithelium and defective autophagy are factors that potentiate IBD
(60). Thus, research is focused
on the potential interaction between autophagy and TJ proteins.
Results of a previous study revealed that autophagy induced the
degradation of CLDN1, which reduced the permeability of the
intestinal epithelial TJ (61).
Furthermore, autophagy is closely associated with CLDN proteins
(62). Results of a previous study
revealed that CLDN proteins are associated with increases in
intestinal TJ permeability, which has a role in the intestinal
pathological process (63).
Autophagy is an evolutionary mechanism that degrades
cytoplasmic components, and is essential for a variety of
physiological and pathological processes. Autophagy is associated
with the occurrence and development of IBD (60), although the underlying mechanisms
remain elusive. Results of a previous study demonstrated that
defects in autophagy may exacerbate intestinal inflammation in
genetically-modified mouse models (64). The results indicated the protective
effects of autophagy in intestinal epithelial cells. Intestinal
epithelial cells lacking autophagy may cause epithelial barrier
dysfunction. In addition, restoration of the autophagic process may
ameliorate intestinal inflammation both in humans and mouse models
(65). Therefore, autophagy
exhibits potential as a target for regulating inflammatory response
in intestinal epithelial cells. To further investigate the
protective mechanisms of OCT mediated by SSTR2 in intestinal
epithelial cells, autophagy was observed in the present study.
A basal level of autophagy is typically required for
the maintenance of cell physiology. However, levels of autophagy
are increased in response to numerous pathological processes, such
as hypoxia, inflammation, infection and tumor formation, where
cells attempt to survive against stress responses (66). Foerster et al (67) previously proposed that the
crosstalk between cell stress pathways and autophagy may restore
intestinal homeostasis (67).
Thus, basal levels of autophagy are required for cell
homeostasis.
Results of the present study revealed that the
number of autophagolysosomes and autophagosomes was decreased
following LPS treatment in Caco2 cells, whereas the pretreatment of
OCT blunted the effects induced by LPS. Thus, autophagy may have a
key role in the OCT-mediated protection of intestinal epithelial
cells.
Of note, >20 autophagy-related genes (ATGs) have
a role in autophagy. LC3 belongs to the ATG8 family and is a
labeled protein molecule on the membrane of autophagosomes in
higher eukaryotes. Following the formation of autophagosomes, LC3-I
is coupled with phosphatidylethanolamine to form LC3-II, and
subsequently localized in the inner and outer membranes of
autophagosomes (68). LC3-II
remains stable on the autophagosome membrane until it fuses with
the lysosome, meaning that the level of LC3-II expression is
indicative of the number of autophagosomes. Wang et al
(69) previously reported that the
extent of transformation from LC3-I to LC3-II was increased as IBD
severity increased in patients, suggesting that the severity of
intestinal inflammation is associated with autophagy levels
(69). The most common methods
used for autophagy detection are western blot analysis of LC3
conversion (LC3-II/LC3-I) and the observation of LC3 point-like
aggregates using fluorescence microscopy (70). In the present study, to determine
the effects of OCT on autophagy, changes in LC3 dynamics in Caco2
cells were monitored from 0 to 24 h. The results demonstrated that
LC3-II protein expression levels were increased to the maximal
level after treatment with LPS for 6 h, which may be indicative of
a self-protective mechanism in intestinal epithelial cells when
responding to pathological stress. After 12 h of LPS treatment,
LC3-II protein expression levels were reduced and levels of
autophagy reached the lowest point following LPS treatment for 24
h. However, levels of autophagy returned to basal levels following
pre-treatment with OCT, highlighting that physiological autophagy
was maintained following OCT treatment. In addition, results of the
present study revealed that the regulatory effects of OCT on
autophagy were inhibited following SSTR2 knockdown.
OCT is a synthetic analogue of somatostatin and
exerts effects in cells by binding to SSTRs. Results of the present
study revealed that basal autophagy levels in Caco2 cells are
associated with OCT-mediated SSTR2 activation, leading to increased
cell viability and improvements in barrier function.
In conclusion, the present study suggested that
OCT-mediated SSTR2 activation may preserve intestinal mucosal
barrier function by regulating autophagy in intestinal epithelial
cells. Thus, OCT exhibits potential in the treatment of intestinal
inflammation. In addition, the present study may provide a novel
theoretical basis for the treatment of intestinal mucosal injury
caused by various pathological states. However, the present study
has limitations. For instance, further investigations into the
molecular mechanisms underlying SSTR activation are required to
identify the upstream components of the SSTR signaling pathway. In
addition, the regulatory effects of OCT on the SSTR signaling
pathway require further verification in vivo.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 82000501), the Science and
Technology Innovation Development Plan Project of Yantai (grant no.
2022YD068) and Xu Rongxiang Regenerative Medicine Research Program
of Binzhou Medical University (grant no. BY2022XRX05).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XL was responsible for the implementation of in
vitro experiments and manuscript writing. YaZ was involved in
experimental data and image processing and draft revision. YuZ
performed the statistical analysis and literature review. XC
assisted with the study design and data analysis. DY helped with
the design of experimental methods, and the literature search and
collation. YL was responsible for the whole idea, project funding
acquisition, experimental and manuscript framework design and
manuscript revision. XL and YL confirm the authenticity of all the
raw data. All authors have read and agreed to the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chelakkot C, Ghim J and Ryu SH: Mechanisms
regulating intestinal barrier integrity and its pathological
implications. Exp Mol Med. 50:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rohr MW, Narasimhulu CA, Rudeski-Rohr TA
and Parthasarathy S: Negative effects of a high-fat diet on
intestinal permeability: A review. Adv Nutr. 11:77–91. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
An J, Liu Y, Wang Y, Fan R, Hu X, Zhang F,
Yang J and Chen J: The role of intestinal mucosal barrier in
autoimmune disease: A potential target. Front Immunol.
13:8717132022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shil A, Olusanya O, Ghufoor Z, Forson B,
Marks J and Chichger H: Artificial sweeteners disrupt tight
junctions and barrier function in the intestinal epithelium through
activation of the sweet taste receptor, T1R3. Nutrients.
12:18622020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Y, Cui W, Li X and Yang H:
Interaction between commensal bacteria, immune response and the
intestinal barrier in inflammatory bowel disease. Front Immunol.
12:7619812021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaminsky LW, Al-Sadi R and Ma TY: IL-1β
and the intestinal epithelial tight junction barrier. Front
Immunol. 12:7674562021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zheng J, Sun Q, Zhang J and Ng SC: The
role of gut microbiome in inflammatory bowel disease diagnosis and
prognosis. United European Gastroenterol J. 10:1091–1102. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burroughs AK and McCormick PA:
Somatostatin and octreotide in gastroenterology. Aliment Pharmacol
Ther. 5:331–341. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
McKay CJ, Imrie CW and Baxter JN:
Somatostatin and somatostatin analogues-are they indicated in the
management of acute pancreatitis? Gut. 34:1622–1626. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li X, Wang Q, Xu H, Tao L, Lu J, Cai L and
Wang C: Somatostatin regulates tight junction proteins expression
in colitis mice. Int J Clin Exp Pathol. 7:2153–2162.
2014.PubMed/NCBI
|
|
11
|
Takano T, Yonemitsu Y, Saito S, Itoh H,
Onohara T, Fukuda A, Takai M and Maehara Y: A somatostatin
analogue, octreotide, ameliorates intestinal ischemia-reperfusion
injury through the early induction of heme oxygenase-1. J Surg Res.
175:350–358. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Klomp MJ, Dalm SU, de Jong M, Feelders RA,
Hofland J and Hofland LJ: Epigenetic regulation of somatostatin and
somatostatin receptors in neuroendocrine tumors and other types of
cancer. Rev Endocr Metab Disord. 22:495–510. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
No authors listed. Somatostatin receptors:
An alternative treatment target for advanced Merkel cell carcinoma.
Br J Dermatol. 184:e32–e52. 2021.PubMed/NCBI
|
|
14
|
Harda K, Szabo Z, Juhasz E, Dezso B, Kiss
C, Schally AV and Halmos G: Expression of somatostatin receptor
subtypes (SSTR-1-SSTR-5) in pediatric hematological and oncological
disorders. Molecules. 25:57752020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lahlou H, Saint-Laurent N, Estève JP,
Eychène A, Pradayrol L, Pyronnet S and Susini C: sst2 Somatostatin
receptor inhibits cell proliferation through Ras-, Rap1-, and
B-Raf-dependent ERK2 activation. J Biol Chem. 278:39356–39371.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zatelli MC, Tagliati F, Taylor JE, Rossi
R, Culler MD and degli Uberti EC: Somatostatin receptor subtypes 2
and 5 differentially affect proliferation in vitro of the human
medullary thyroid carcinoma cell line tt. J Clin Endocrinol Metab.
86:2161–2169. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Colucci R, Blandizzi C, Ghisu N, Florio T
and Del Tacca M: Somatostatin inhibits colon cancer cell growth
through cyclooxygenase-2 downregulation. Br J Pharmacol.
155:198–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shamsi BH, Chatoo M, Xu XK, Xu X and Chen
XQ: Versatile functions of somatostatin and somatostatin receptors
in the gastrointestinal system. Front Endocrinol (Lausanne).
12:6523632021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fleseriu M, Dreval A, Bondar I, Vagapova
G, Macut D, Pokramovich YG, Molitch ME, Leonova N, Raverot G,
Grineva E, et al: Maintenance of response to oral octreotide
compared with injectable somatostatin receptor ligands in patients
with acromegaly: A phase 3, multicentre, randomised controlled
trial. Lancet Diabetes Endocrinol. 10:102–111. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lei S, Cheng T, Guo Y, Li C, Zhang W and
Zhi F: Somatostatin ameliorates lipopolysaccharide-induced tight
junction damage via the ERK-MAPK pathway in Caco2 cells. Eur J Cell
Biol. 93:299–307. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Li X, Geng C, Guo Y and Wang C:
Somatostatin receptor 5 is critical for protecting intestinal
barrier function in vivo and in vitro. Mol Cell Endocrinol.
535:1113902021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liew CW, Vockel M, Glassmeier G, Brandner
JM, Fernandez-Ballester GJ, Schwarz JR, Schulz S, Buck F, Serrano
L, Richter D and Kreienkamp HJ: Interaction of the human
somatostatin receptor 3 with the multiple PDZ domain protein MUPP1
enables somatostatin to control permeability of epithelial tight
junctions. FEBS Lett. 583:49–54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Valencak J, Heere-Ress E, Traub-Weidinger
T, Raderer M, Schneeberger A, Thalhammer T, Aust S, Hamilton G,
Virgolini I and Pehamberger H: Somatostatin receptor scintigraphy
with 111In-DOTA-lanreotide and 111In-DOTA-Tyr3-octreotide in
patients with stage IV melanoma: In-vitro and in-vivo results.
Melanoma Res. 15:523–529. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim KH and Lee MS: Autophagy-a key player
in cellular and body metabolism. Nat Rev Endocrinol. 10:322–337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ganapathy AS, Saha K, Suchanec E, Singh V,
Verma A, Yochum G, Koltun W, Nighot M, Ma T and Nighot P: AP2M1
mediates autophagy-induced CLDN2 (claudin 2) degradation through
endocytosis and interaction with LC3 and reduces intestinal
epithelial tight junction permeability. Autophagy. 18:2086–2103.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu CA, Hou Y, Yi D, Qiu Y, Wu G, Kong X
and Yin Y: Autophagy and tight junction proteins in the intestine
and intestinal diseases. Anim Nutr. 1:123–127. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rathinam VAK, Zhao Y and Shao F: Innate
immunity to intracellular LPS. Nat Immunol. 20:527–533. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang YJ and Wu Q: Sulforaphane protects
intestinal epithelial cells against lipopolysaccharide-induced
injury by activating the AMPK/SIRT1/PGC-1a pathway. Bioengineered.
12:4349–4360. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu XX, Huang XL, Chen RR, Li T, Ye HJ, Xie
W, Huang ZM and Cao GZ: Paeoniflorin prevents intestinal barrier
disruption and inhibits lipopolysaccharide (LPS)-induced
inflammation in Caco-2 cell monolayers. Inflammation. 42:2215–2225.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen G, Ran X, Li B, Li Y, He D, Huang B,
Fu S, Liu J and Wang W: Sodium butyrate inhibits inflammation and
maintains epithelium barrier integrity in a TNBS-induced
inflammatory bowel disease mice model. EBioMedicine. 30:317–325.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Wang S, Gao X, Zhao Y, Li Y, Yang B,
Zhang N and Ma L: Octreotide alleviates autophagy by up-regulation
of MicroRNA-101 in intestinal epithelial cell line Caco-2. Cell
Physiol Biochem. 49:1352–1363. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saha K, Subramenium Ganapathy A, Wang A,
Michael Morris N, Suchanec E, Ding W, Yochum G, Koltun W, Nighot M,
Ma T and Nighot P: Autophagy reduces the degradation and promotes
membrane localization of occludin to enhance the intestinal
epithelial tight junction barrier against paracellular
macromolecule flux. J Crohns Colitis. 17:433–449. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim Y, Lee Y, Heo G, Jeong S, Park S, Yoo
JW, Jung Y and Im E: Modulation of intestinal epithelial
permeability via protease-activated receptor-2-induced autophagy.
Cells. 11:8782022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J,
Qian Y, Kryczek I, Sun D, Nagarsheth N, et al: Fusobacterium
nucleatum promotes chemoresistance to colorectal cancer by
modulating autophagy. Cell. 170:548–563.e16. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hejna M, Schmidinger M and Raderer M: The
clinical role of somatostatin analogues as antineoplastic agents:
Much ado about nothing? Ann Oncol. 13:653–668. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stueven AK, Kayser A, Wetz C, Amthauer H,
Wree A, Tacke F, Wiedenmann B, Roderburg C and Jann H: Somatostatin
analogues in the treatment of neuroendocrine tumors: Past, present
and future. Int J Mol Sci. 20:30492019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vockel M, Breitenbach U, Kreienkamp HJ and
Brandner JM: Somatostatin regulates tight junction function and
composition in human keratinocytes. Exp Dermatol. 19:888–894. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun JX and Yang N: Role of octreotide in
post chemotherapy and/or radiotherapy diarrhea: Prophylaxis or
therapy? Asia Pac J Clin Oncol. 10:e108–e113. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rao S, Viola A, Ksissa O and Fries W:
Ménétrier's disease in a patient with refractory ulcerative
colitis: A clinical challenge and review of the literature. BMJ
Case Rep. 14:e2461372021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang JW, Pan YB, Cao YQ, Wang C, Jiang WD,
Zhai WF and Lu JG: Loganin alleviates LPS-activated intestinal
epithelial inflammation by regulating TLR4/NF-κB and JAK/STAT3
signaling pathways. Kaohsiung J Med Sci. 36:257–264. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Møller LN, Stidsen CE, Hartmann B and
Holst JJ: Somatostatin receptors. Biochim Biophys Acta. 1616:1–84.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Grant M and Kumar U: The role of
G-proteins in the dimerisation of human somatostatin receptor types
2 and 5. Regul Pept. 159:3–8. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li J, Chen C, Bi X, Zhou C, Huang T, Ni C,
Yang P, Chen S, Ye M and Duan S: DNA methylation of CMTM3, SSTR2,
and MDFI genes in colorectal cancer. Gene. 630:1–7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Caruso ML, Di Pinto F, Ignazzi A, Coletta
S, Valentini AM, Cavalcanti E and De Michele F: Increased nerve
twigs in small intestinal mucosa with programmed cell death-ligand
1 and somatostatin receptor type 2A expression in recurrent Crohn
disease: A case report. Medicine (Baltimore). 97:e134922018.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gomes-Porras M, Cárdenas-Salas J and
Álvarez-Escolá C: Somatostatin analogs in clinical practice: A
review. Int J Mol Sci. 21:16822020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Giuliani C: The flavonoid quercetin
induces AP-1 activation in FRTL-5 thyroid cells. Antioxidants
(Basel). 8:1122019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Silverstein JM: Hyperglycemia induced by
pasireotide in patients with Cushing's disease or acromegaly.
Pituitary. 19:536–543. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Roden AC, Rakshit S, Johnson GB, Jenkins
SM and Mansfield AS: Correlation of somatostatin receptor 2
expression, 68Ga-DOTATATE PET scan and octreotide treatment in
thymic epithelial tumors. Front Oncol. 12:8236672022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Periferakis A, Tsigas G, Periferakis AT,
Badarau IA, Scheau AE, Tampa M, Georgescu SR, Didilescu AC, Scheau
C and Caruntu C: Antitumoral and anti-inflammatory roles of
somatostatin and its analogs in hepatocellular carcinoma. Anal Cell
Pathol (Amst). 2021:18400692021.PubMed/NCBI
|
|
52
|
Alexander N, Marrano P, Thorner P, Naranjo
A, Van Ryn C, Martinez D, Batra V, Zhang L, Irwin MS and Baruchel
S: Prevalence and clinical correlations of somatostatin receptor-2
(SSTR2) expression in neuroblastoma. J Pediatr Hematol Oncol.
41:222–227. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lechner M, Schartinger VH, Steele CD, Nei
WL, Ooft ML, Schreiber LM, Pipinikas CP, Chung GT, Chan YY, Wu F,
et al: Somatostatin receptor 2 expression in nasopharyngeal cancer
is induced by Epstein Barr virus infection: Impact on prognosis,
imaging and therapy. Nat Commun. 12:1172021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gonzalez B, Vargas G, Ramirez C, Asa S,
Cheng S, Sandoval C and Mercado M: Cytoplasmic expression of SSTR2
and 5 by immunohistochemistry and by RT/PCR is not associated with
the pharmacological response to octreotide. Endocrinol Nutr.
61:523–530. 2014.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen W, Ding R, Tang J, Li H, Chen C,
Zhang Y, Zhang Q and Zhu X: Knocking Out SST gene of BGC823 gastric
cancer cell by CRISPR/Cas9 enhances migration, invasion and
expression of SEMA5A and KLF2. Cancer Manag Res. 12:1313–1321.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Di Tommaso N, Gasbarrini A and Ponziani
FR: Intestinal barrier in human health and disease. Int J Environ
Res Public Health. 18:128362021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dokladny K, Zuhl MN and Moseley PL:
Intestinal epithelial barrier function and tight junction proteins
with heat and exercise. J Appl Physiol (1985). 120:692–701. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mohammad S and Thiemermann C: Role of
metabolic endotoxemia in systemic inflammation and potential
interventions. Front Immunol. 11:5941502020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Li E, Horn N and Ajuwon KM: EPA and DHA
inhibit endocytosis of claudin-4 and protect against
deoxynivalenol-induced intestinal barrier dysfunction through PPARγ
dependent and independent pathways in jejunal IPEC-J2 cells. Food
Res Int. 157:1114202022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Larabi A, Barnich N and Nguyen HTT: New
insights into the interplay between autophagy, gut microbiota and
inflammatory responses in IBD. Autophagy. 16:38–51. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim J, Choi S, Kim JO and Kim KK:
Autophagy-mediated upregulation of cytoplasmic claudin 1 stimulates
the degradation of SQSTM1/p62 under starvation. Biochem Biophys Res
Commun. 496:159–166. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang Z, Lin P, Chen B, Zhang X, Xiao W, Wu
S, Huang C, Feng D, Zhang W and Zhang J: Autophagy alleviates
hypoxia-induced blood-brain barrier injury via regulation of CLDN5
(claudin 5). Autophagy. 17:3048–3067. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Suzuki T: Regulation of the intestinal
barrier by nutrients: The role of tight junctions. Anim Sci J.
91:e133572020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou C, Li L, Li T, Sun L, Yin J, Guan H,
Wang L, Zhu H, Xu P, Fan X, et al: SCFAs induce autophagy in
intestinal epithelial cells and relieve colitis by stabilizing
HIF-1α. J Mol Med (Berl). 98:1189–1202. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Jia J, Gong X, Zhao Y, Yang Z, Ji K, Luan
T, Zang B and Li G: Autophagy enhancing contributes to the organ
protective effect of alpha-lipoic acid in septic rats. Front
Immunol. 10:14912019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Tang C, Livingston MJ, Liu Z and Dong Z:
Autophagy in kidney homeostasis and disease. Nat Rev Nephrol.
16:489–508. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Foerster EG, Mukherjee T, Cabral-Fernandes
L, Rocha JDB, Girardin SE and Philpott DJ: How autophagy controls
the intestinal epithelial barrier. Autophagy. 18:86–103. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang SL, Shao BZ, Zhao SB, Chang X, Wang
P, Miao CY, Li ZS and Bai Y: Intestinal autophagy links
psychosocial stress with gut microbiota to promote inflammatory
bowel disease. Cell Death Dis. 10:3912019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mizushima N and Yoshimori T: How to
interpret LC3 immunoblotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|