Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide and the third leading cause of
cancer-related mortality (1). In
recent years, the incidence of HCC in western countries has
increased markedly (2). HCC has a
number of notable epidemiological features, including marked
variations between geographical regions, racial and ethnic groups,
and genders. Previous studies revealed that men have a higher
prevalence of HCC than women; the ratio of affected men to affected
women varies between 2:1 and 4:1 (3).
In high-risk Chinese populations, the male:female ratio of HCC
patients is ~2.65:1 (2). HCC
frequently occurs within an established background of chronic liver
disease and cirrhosis. Major causes of cirrhosis in patients with
HCC include hepatitis B virus (HBV) and hepatitis C virus (HCV),
alcoholic liver disease and, possibly, non-alcoholic
steatohepatitis (3,4). China has a high incidence of HCC, and
the newly diagnosed HCC patients in China account for ~55% of the
newly diagnosed HCC patients globally each year (5). HBV infection is one major risk factor
for HCC occurrence; ≥75% cases of HCC are associated with HBV
infection in China (6). However, not
all individuals in HBV-infected populations develop HCC over their
lifetime. This phenomenon indicates that an individual's genetic
background is important in HBV-related hepatocarcinogenesis
(3).
To date, the predominant strategy employed to
investigate HCC-associated genes has been the candidate gene-based
case-control association study. The candidate genes have included
GSTT1, GSTM1, UGT1A7, CYPs,
NAT2, HFE, MTHFR, TGFβ, TNFα and
MnSOD (7–11). By reviewing these studies, we observed
numerous inconsistent results, and the associations reported were
based on strong evidence. As the molecular mechanisms of most
complex diseases, including HCC, are still unknown, the candidate
strategy has inherent limitations; in fact, it is a process of
validation of the equivocal hypothesis and the conclusions of such
studies should also be drawn cautiously. Based on the success of
the human genome project, the HapMap Project, and the availability
of high throughput gene chips, the whole genome-wide association
study (GWAS) has become the most powerful approach to search for
and map the susceptibility genes of complex diseases (12,13). Since
2005, GWAS has achieved great success in identifying susceptibility
genes in complex diseases, including diabetes (14), cancer (15,16) and
systemic lupus erythematosus (17).
Recently, a GWAS conducted in China reported 1p36.22 as a novel
susceptibility locus for HCC (18).
Distinct clinical characteristics of HBV infection
have been reported in different geographical regions of the world,
and increasing evidence indicates an association with the genetic
diversity of infected patients (19,20).
However, such data are largely lacking in Qidong, China, where
chronic HBV infection is highly endemic (21). In the present study, a pilot two-stage
GWAS was conducted to search for susceptibility loci for HCC. In
order to maximize the statistical power to identify associations,
homogeneity of the samples was ensured by selecting participants
from the male Chinese Han population with HBV surface antigen
(HBsAg) seropositivity, and a high proportion of HCC cases with a
family history of HCC was included.
Materials and methods
Participants
The present analysis used data and stored samples
from a prospective cohort in Qidong, Jiangsu, China. The enrollment
of the study cohort has been previously described (22–24).
Briefly, male HCC patients with HBsAg seropositivity were recruited
from inpatients at the Qidong Liver Cancer Institute (Qidong,
China), between August 1, 2006 and July 31, 2008. HCC was diagnosed
by histopathological biopsy, or by elevated α-fetoprotein (AFP)
levels and distinct changes on imaging (ultrasonography, computed
tomography and magnetic resonance imaging), according to the 2004
Barcelona guidelines (25). Blood
samples were collected at the time of HCC diagnosis. All male
control subjects were selected from a cohort consisting of HBsAg
carriers, established by Qidong Liver Cancer Institute in 1996. The
absence of HCC in the controls was verified by assessing AFP levels
and ultrasonography when the blood samples were collected in 2006.
All samples were HCV seronegative. According to the tenets of the
Declaration of Helsinki (26), the
study was approved by the ethics committees of Qidong County Liver
Cancer Institute, (Qidong, China) and Fudan University (Shanghai,
China). All participants provided written informed consent,
including consent for genetic studies. Data do not contain any
information that may lead to the identification of the
patients.
Genotyping
DNA was extracted from peripheral blood mononuclear
cells using SHENamp Blood DNA Kit according to the manufacturer's
instructions (Shengyou Biotechnology Co., Ltd., Hangzhou, China).
In phase 1, a total of 500,447 single-nucleotide polymorphisms
(SNPs) were genotyped using an Affymetrix GeneChip Mapping 500k
Array Set (Affymetrix, Santa Clara, CA, USA) in 50 cases and 50
controls. In phase 2, a total of 1,152 SNPs, selected from phase 1,
were genotyped in 282 cases and 278 controls using an Illumina
GoldenGate genotyping assay (Illumina, San Diego, CA, USA) at
Shanghai Biochip Co., Ltd. (Shanghai, China). The quality control,
genotyping and data analyses were performed according to the
protocols of the respective manufacturers. For phases 1 and 2, a
DNA sample was deemed to have failed if it generated genotypes at
<93% of loci. A SNP was deemed to have failed if <90% of DNA
samples generated a genotype at the locus. In phase 2, 8 duplicate
samples were genotyped to ensure quality of genotyping. For all
1,152 SNPs, >99.8% concordant results were obtained. The
genotyping results of 11 SNPs were also validated in 41 samples
using the iPLEX Gold assay on the MassARRAY® platform (Sequenom,
Inc., San Diego, CA, USA) and >98.4% concordant results were
obtained.
Statistical analysis
The statistical analysis of the GeneChip data was
performed using Genotyping Console Software, version 4.0
(Affymetrix, Santa Clara, CA, USA) and BeadStudio Genotyping
Module, version 3.2 (Illumina, San Diego, CA, USA). Combined
analysis was conducted using Stata software version 10.0
(StataCorp, College Station, TX, USA). For each SNP, allele
P-value, Cochran-Armitage trend P-value, odds ratio (ORs) and 95%
confidence interval (CI) were calculated. The prior probability of
HCC in control subjects was assigned at 0.01, and false-positive
report probability (FPRP) was used to assess the reliability of the
associations (27). FPRP<0.20 was
set as the significance threshold for the associations.
Results
Two stage GWA study identified 9
susceptibility loci
In phase 1, the age distribution of 50 cases [mean
age (± SD) at diagnosis, 52.3±7.9 years] and 50 controls (mean age
at diagnosis, 54.88±8.68 years) was matched. Of the 50 cases, 11
had one or more first-degree relatives affected by HCC (mean age at
diagnosis, 54.1±8.1 years). All 100 samples had a call rate of
>96%. The call rate is the percentage of successful genotype
calls per passing SNP. Exclusion criteria for SNPs included the
following: i) overall frequency of SNP <90%; ii) minor allele
frequency (MAF) <0.05; and iii) P-value of Hardy-Weinberg
equilibrium (HWE) <0.001. Altogether, 279,757 eligible SNPs
passed quality control and were included in further analyses. For
each SNP, allele P-values, genotype P-values and Cochran-Armitage
trend P-values were calculated. The minimum P-value was designated
as minP. For autosome, min P = min (allele_P, genotype_P, P_trend);
for sex chromosome, min P =min (allele_P, allele_exact_P). There
were 355 SNPs on autosomes and 2 SNPs on the X chromosome with
minP<1×10−3. A total of 26 SNPs were identified with
minP<1×10−4 (Table I).
One SNP (rs2212522) demonstrated a significant association with HCC
(Pallele=5.23×10−8; ORallele=4.96;
95% CI, 2.72–9.03).
 | Table I.Twenty-six SNPs with
minP<1×10−4 in phase 1. |
Table I.
Twenty-six SNPs with
minP<1×10−4 in phase 1.
|
|
| Cases, n | Controls, n |
|
|
|---|
|
|
|
|
|
|
|
|---|
| dbSNP
rsIDa | Associated
gene(s) | A | B | A | B | minP-value |
ORallele |
|---|
| rs2212522 | L3MBTL4 | 68 | 32 | 30 | 70 |
7.65×10−8 | 0.2016807 |
| rs4713039 | NO145 | 44 | 56 | 77 | 23 |
1.81×10−6 | 4.2608696 |
| rs4539982 | TBL1XR1 | 79 | 15 | 49 | 45 |
2.68×10−6 | 0.2067511 |
| rs9877175 | TBL1XR1 | 18 | 80 | 49 | 51 |
5.25×10−6 | 4.2701525 |
| rs4277177 | TMEM16F | 51 | 45 | 62 | 36 |
8.52×10−6 | 1.5196078 |
| rs8031646 | ARRDC4 | 72 | 26 | 42 | 56 |
1.40×10−5 | 0.2708333 |
| rs11057529 | FAM101A | 17 | 83 | 45 | 55 |
1.40×10−5 | 3.9946524 |
| rs7069096 | LYZL1 | 36 | 62 | 67 | 33 |
2.03×10−5 | 3.4966332 |
| rs946351 | NO145 | 51 | 49 | 22 | 78 |
2.05×10−5 | 0.2709904 |
| rs12044483 | FLJ32784,
UBXD3 | 39 | 61 | 69 | 31 |
2.08×10−5 | 3.4813896 |
| rs10926832 | PLD5 | 32 | 66 | 61 | 39 |
2.16×10−5 | 3.2259615 |
| rs1334125 | PRMT6 | 34 | 66 | 64 | 36 |
2.19×10−5 | 3.4509804 |
| rs12580388 |
TMEM132D | 80 | 20 | 53 | 47 |
3.17×10−5 | 0.2819149 |
| rs6910232 | NO145 | 64 | 34 | 35 | 63 |
3.43×10−5 | 0.2951389 |
| rs7854810 |
ENST00000380100 | 97 | 3 | 76 | 22 |
3.80×10−5 | 0.1068416 |
| rs12034802 | PRMT6 | 60 | 30 | 37 | 63 |
4.42×10−5 | 0.2936508 |
| rs7870157 |
ENST00000387810 | 4 | 96 | 24 | 76 |
4.59×10−5 | 7.5789474 |
| rs1883165 |
FLJ32784 | 65 | 31 | 35 | 57 |
4.60×10−5 | 0.2928475 |
| rs2292723 |
TMEM132D | 11 | 89 | 32 | 66 |
5.63×10−5 | 3.9228651 |
| rs10735541 | TLE4 | 32 | 68 | 13 | 87 |
5.93×10−5 | 0.3175287 |
| rs1543940 | LOC440337,
FAM86A | 13 | 79 | 36 | 54 |
7.63×10−5 | 4.0512821 |
| rs9571852 | NBEA | 14 | 86 | 38 | 62 |
7.89×10−5 | 3.7649773 |
| rs9949516 | L3MBTL4 | 31 | 63 | 60 | 38 |
8.92×10−5 | 3.2088285 |
| rs6828409 | EREG,
EPGN | 75 | 25 | 47 | 51 |
9.16×10−5 | 0.3071895 |
| rs7105477 | AP2A2 | 82 | 8 | 68 | 32 |
9.56×10−5 | 0.2073171 |
| rs4417097 | PRMT6,
AMY1C | 39 | 53 | 69 | 29 |
9.76×10−5 | 3.2334218 |
In phase 2, 282 HCC cases and 278 controls were
recruited from Qidong. Of the HCC cases, 61 (21%) had a family
history of HCC in a first or second-degree relative. The age
distributions were 51.4±10.0 and 53.7±10.6 years in cases and
controls, respectively. The selected 1,152 SNPs included 4
categories: i) SNPs whose minP in phase 1 was <1×10−3
or SNPs well clustered in chromosome with minP<0.05 (n=398); ii)
predicted deleterious non-synonymous SNPs (nsSNPs) (n=430) of the
nearest genes; iii) SNPs possibly associated with copy number
variation (CNV) (n=315); and iv) SNPs located at 8q24 which had
been reported to be associated with other types of cancers (n=9:
rs13254738, rs6983561, rs16901979, rs13281615, rs10505447,
rs10808556, rs6983267, rs7000448 and rs1447295) (28–33).
All 560 samples had a call rate of >93%. Of the
selected 1,152 SNPs, 598 passed the quality control following the
exclusion procedure (MAF <0.05, HWE disequilibrium P<0.001
and call frequency <0.95). There were 35 SNPs with unadjusted
Pallele<0.05. Of these, 8 SNPs whose minor allele
number was ≤3 were further excluded, leaving 27 SNPs with
unadjusted Pallele<0.05. The most significant signal
in phase 1, rs2212522, was replicated in phase 2 with
Pallele<0.05 and combined
Pallele=7.91×10−5. Out of 27 SNPs, 20 were
genotyped in phases 1 and 2, and 7 SNPs were genotyped only in
phase 2. Combined analysis of the 20 SNPs revealed that there were
15 SNPs with combined Pallele<0.05. Thus, there were
22 SNPs with combined allele P-value or with phase 2 allele P-value
of <0.05 (Table II). As all
controls were men with HBsAg seropositivity, a prior probability of
0.01 for HCC occurrence was assigned and FPRP was calculated
(34). The results revealed that 9 of
the 22 SNPs were associated with HCC (FPRP<0.20) (Table III). CNV was validated by genotyping
315 high density SNPs using the Illumina GoldenGate platform and
quantitative polymerase chain reaction (qPCR) analysis; this
identified a number of CNVs in HCC, reported in other studies
(35,36). No associations were identified between
the 9 SNPs in 8q24 and HCC.
| Table II.Twenty-two SNPs with allele P<0.05
in phase 2 or combined allele P<0.05. |
Table II.
Twenty-two SNPs with allele P<0.05
in phase 2 or combined allele P<0.05.
|
|
|
|
| Allele P-value |
|---|
|
|
|
|
|
|
|---|
| dbSNP
rsIDa | Associated
gene | Chromosome | Position | Phase 1 | Phase 2 | Combined |
|---|
| rs2120243 | VEPH1 | 3q24–25 | 158630262 |
1.37×10−4 |
3.23×10−4 |
2.00×10−6 |
| rs1350171 | FZD4 | 11q14.2 |
86322037 |
1.68×10−4 |
9.16×10−4 |
7.47×10−6 |
| rs1048338 | FZD4 | 11q14.2 |
86310465 |
1.57×10−4 |
1.44×10−3 |
1.11×10−5 |
| rs2212522 | L3MBTL4 | 18p11.31 |
5890773 |
5.23×10−8 |
4.81×10−2 |
7.91×10−5 |
| rs7116140 | FZD4 | 11q14.2 |
86313003 |
2.14×10−4 |
1.20×10−2 |
1.76×10−4 |
| rs4480667 | PCDH9 | 13q14.3 |
66825714 |
6.60×10−1 |
1.92×10−4 |
3.02×10−4 |
| rs4417097 | PRMT6 | 1p13.3 | 106601576 |
8.78×10−5 |
2.79×10−2 |
4.27×10−4 |
| rs9893681 | LHX1 | 17q12 |
32362128 |
4.47×10−4 |
2.82×10−2 |
9.31×10−4 |
| rs4767254 | TBX3 | 12q24.1 | 113682775 |
6.13×10−4 |
3.22×10−2 |
1.18×10−3 |
| rs132024 | PHF21B | 22q13.31 |
43804508 |
5.77×10−4 |
4.65×10−2 |
2.20×10−3 |
| rs4561519 | KIF2B | 17q22 |
49256702 |
1.90×10−1 |
9.26×10−3 |
3.61×10−3 |
| rs729565 | CNTNAP2 | 7q35 | 146668586 |
3.70×10−1 |
1.02×10−2 |
6.49×10−3 |
| rs4726849 | CNTNAP2 | 7q35 | 146704681 |
3.40×10−1 |
3.23×10−2 |
1.90×10−2 |
| rs10500181 | CNTNAP2 | 7q35 | 146704487 |
3.60×10−1 |
3.75×10−2 |
2.30×10−2 |
| rs12532315 | CNTNAP2 | 7q35 | 146730215 |
5.40×10−1 |
4.95×10−2 |
4.04×10−2 |
| rs3818605 | PTPRA | 20p12 |
2788773 |
|
6.12×10−3 |
|
| rs9898643 | KIF2B | 17q22 |
49254648 |
|
7.49×10−3 |
|
| rs3806523 | SH3BP4 | 2q37.1 | 235523908 |
|
1.33×10−2 |
|
| rs1155569 | RPL38 | 17q23 |
61082421 |
|
2.54×10−2 |
|
| rs1057090 | MCPH1 | 8p23.1 |
6466450 |
|
2.80×10−2 |
|
| rs17138848 | COMMD10 | 5q23.1 | 115447202 |
|
4.22×10−2 |
|
| rs730819 | PTPRA | 20p12 |
2793130 |
|
4.28×10−2 |
|
| Table III.Nine SNPs with combined allele
P<0.05 and FPRP <0.20. |
Table III.
Nine SNPs with combined allele
P<0.05 and FPRP <0.20.
|
|
| Allele P-value |
|
|
|
|---|
|
|
|
|
|
|
|
|---|
| dbSNP
rsIDa | Associated
gene | Phase 1 | Phase 2 | Combined |
ORallele | 95% CI | FPRP |
|---|
| rs2120243 | VEPH1 |
1.37×10−4 |
3.32×10−4 |
2.00×10−6 | 1.76 | 1.39–2.22 | <0.001 |
| rs1350171 | FZD4 |
1.68×10−4 |
9.28×10−4 |
6.48×10−6 | 1.66 | 1.33–2.07 |
0.014 |
| rs1048338 | FZD4 |
1.58×10−4 |
1.44×10−3 |
1.11×10−5 | 1.64 | 1.31–2.04 |
0.046 |
| rs2212522 | L3MBTL4 |
5.23×10−8 |
4.81×10−2 |
7.91×10−5 | 1.57 | 1.25–1.97 |
0.019 |
| rs7116140 | FZD4 |
2.14×10−4 |
1.20×10−2 |
1.76×10−4 | 1.51 | 1.22–1.88 |
0.039 |
| rs4480667 | PCDH9 |
6.60×10−1 |
1.92×10−4 |
3.02×10−4 | 1.52 | 1.21–1.90 |
0.037 |
| rs4417097 | PRMT6 |
8.78×10−5 |
2.78×10−2 |
4.27×10−4 | 1.48 | 1.19–1.85 |
0.057 |
| rs9893681 | LHX1 |
4.46×10−4 |
2.82×10−2 |
9.31×10−4 | 1.65 | 1.22–2.21 |
0.159 |
| rs4561519 | KIF2B |
1.90×10−1 |
9.26×10−3 |
3.61×10−3 | 1.52 | 1.14–2.02 |
0.176 |
Genes associated with the 9
susceptibility loci
rs2120243 (C>A) maps within the fourth intron of
ventricular zone expressed PH domain homolog 1 [VEPH1; HUGO
Gene Nomenclature Committee (HGNC) ID: 25735]. VEPH1 maps at
3q24–25 and covers 273.88 kb.
Frizzled homolog 4 (FZD4; HGNC ID: 4042) is a
member of the frizzled gene family, maps at 11q14.2 and covers 9.73
kb. The majority of frizzled receptors are coupled to the β-catenin
signaling pathway. It has been reported FZD4 is associated
with numerous types of cancer (37).
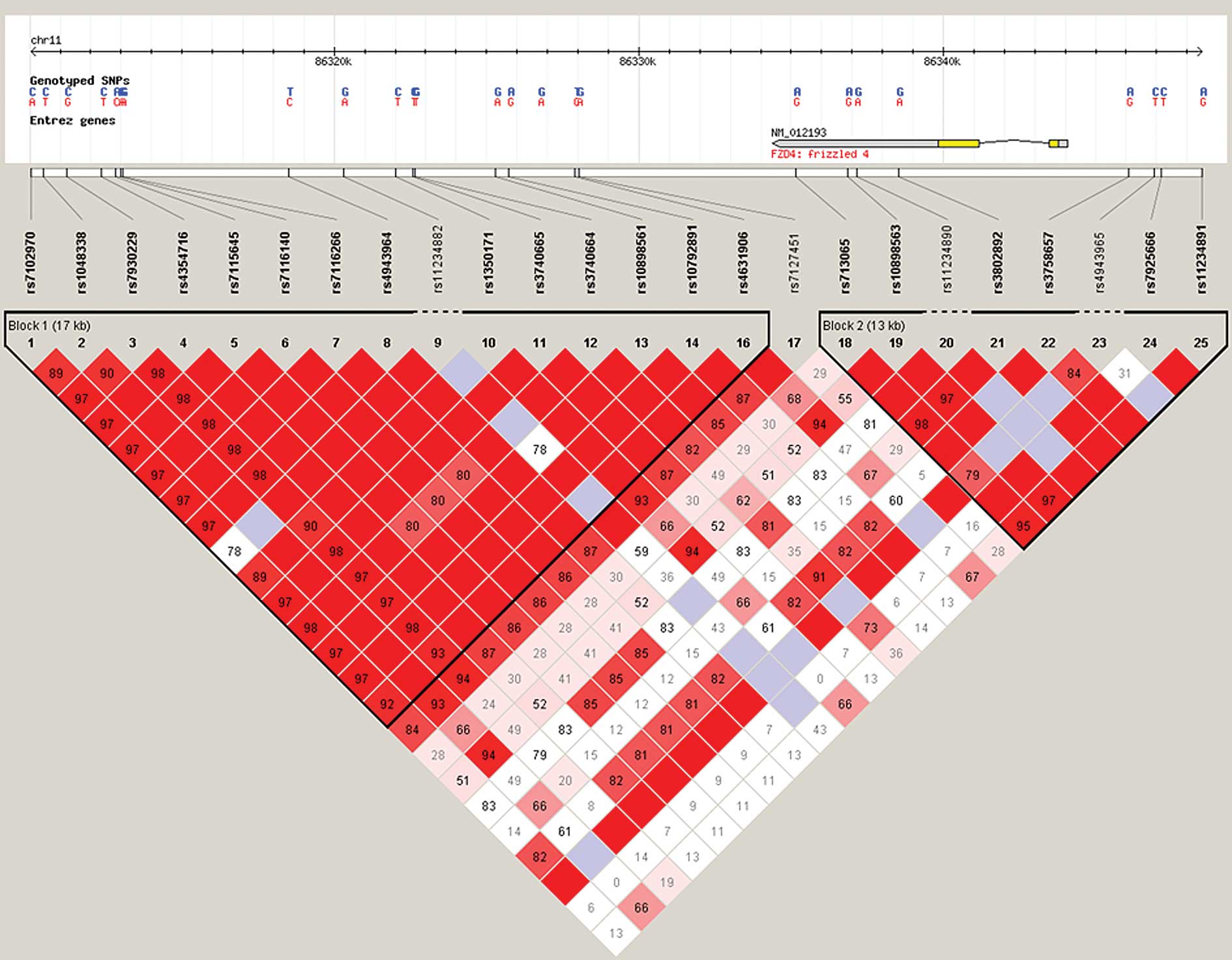
In the present study, 3 SNPs (rs1048338, rs7116140 and rs1350171)
were identified in the downstream 12–23 kb of FZD4.
Haploview version 4.1 1 (The Broad Institute) revealed that the 3
SNPs are in one strong linkage disequilibrium (LD) block (Fig. 1). Using the haplotype analysis
software PLINK (pngu.mgh.harvard.edu/purcell/plink/), it was
demonstrated that the haplotype exhibited significantly different
distributions between cases and controls (P<0.01).
rs4480667 is located in the 125 kb upstream of
protocadherin 9 (PCDH9; HGNC ID: 8661) and maps in a 24 kb
block (38). PCDH9, which
belongs to the protocadherin gene family, maps at 13q14.3-q21.1 and
covers 927.62 kb. It has been reported that CNV of PCDH9 may
be associated with glioblastoma as a tumor suppressor gene (TSG)
(39). Patch 1.0 revealed that allele
A of rs4480667 (A>G) created a new binding site for the
transcription factors C-Ets-1 (HGNC ID: 3488) and Elf-1 (HGNC ID:
3316). These two transcription factors have been reported to be
associated with several cancer types, including lung cancer and HCC
(40).
rs9893681 maps at the upstream 6.4 kb of the LIM
homeobox 1 (LHX1) gene. LHX1 (HGNC ID: 6593) maps at
17q12 and covers 7.15 kb. It has been reported the CNV of
LHX1 is associated with gastric cancer (41). Patch 1.0 indicated that allele A of
rs9893681 (T>A) created a new binding site for transcription
factor Fushi tarazu (FTZ gene). Liu et al (42) reported that FTZ regulated the
ubiquitin E3 ligase complex factor Speckle-type POZ protein (SPOP;
HGNC ID: 11254), which mediated degradation of the Jun-kinase
phosphatase, thereby inducing tumor necrosis factor/Eiger-dependent
apoptosis (42). The human homolog of
FTZ, nuclear receptor subfamily 5 group A member 2
(NR5A2; HGNC ID: 7984) has been shown to play critical roles
in various cancer types (43,44).
rs4417097 is located in a region of gene desert, the
nearest gene being protein arginine methyltransferase 6
(PRMT6; HGNC ID: 18241). rs4417097 maps in the upstream
799kb of PRMT6. A previous GWAS reported that PRMT6
gene is involved in acquired immune deficiency syndrome (45). Another study reported that
thrombospondin-1 was a transcriptional repression target of PRMT6,
and suggested that neutralizing the activity of PRMT6 could inhibit
tumor progression (46).
rs4561519 is a nsSNP of kinesin family member 2B
(KIF2B; HGNC ID: 29443). KIF2B has been proposed to
participate in the process of microtubule-based movement. Three
protein function prediction software packages: SIFT (sift.jcvi.org/), Polyphen (genetics.bwh.harvard.edu/pph2/) and SNPs3D (snps3d.org/), predicted that rs4561519 is deleterious
to the protein's function (47,48).
L(3)mbt-like 4
(L3MBTL4) maps on chromosome 18, at 18p11.3. It covers
460.53 kb, from 6405235 to 5944705 [National Center for
Biotechnology Information (NCBI) 36, March 2006; ncbi.nlm.nih.gov/IEB/Research/Acembly/av.cgi?c=geneid&org=9606&l=91133],
on the reverse strand. rs2212522 maps to 53.9 kb downstream of
L3MBTL4. Functionally, this gene has been proposed to
participate in various processes, including cell adhesion, platelet
activation and regulation of transcription. The protein is also
predicted to have molecular functions (transcription factor
activity and zinc ion binding) and to localize to various
compartments (integrin complex, cytoplasm, extracellular space and
the nucleus). L3MBT has been reported to be a TSG in
Drosophila (49). In addition, one
study reported that L3MBTL4 was associated with HCC
(50).
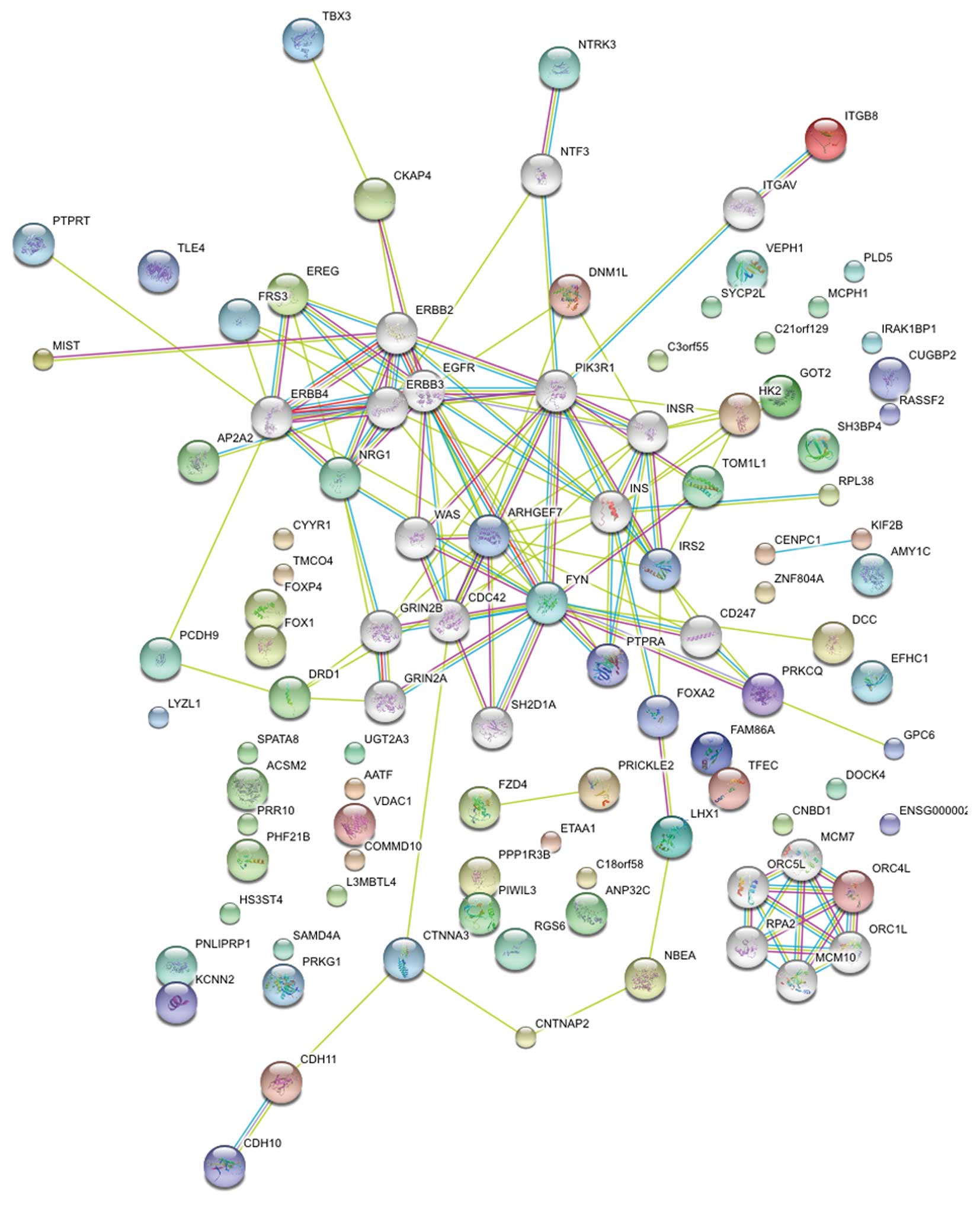
Gene gene interactions
In order to find the gene-gene interactions, we
selected all SNPs with combined Pallele<0.05 and SNPs
with Pallele<0.05 in phase 2 and searched for the
nearest genes using the NCBI Map Viewer (www.ncbi.nlm.nih.gov/mapview/). A total of 109 SNPs
associated with 84 genes were identified. Protein-protein
interaction networks were then explored using STRING software
(string-db.org/); 80 proteins were recognized and
analyzed. By adding 20 protein notes automatically with STRING,
many crucial proteins were identified, including EGFR, ERBBs, FYN,
CDC42, PIK3R1, ARHGEF, INS, INSR, CDC42 and PRKCQ (Fig. 2). These genes involved a number of
important signaling pathways involved in carcinogenesis, such as
transforming growth factor β, insulin/phosphoinositide 3 kinase
(PI3K) and Wnt/β-catenin.
Discussion
The success of GWAS relies on much of the risk of
common diseases being due to common genetic variants; however,
evidence for this hypothesis is inconclusive, and the possibility
that some complex diseases are due to certain rare variants with
high genetic risk cannot be excluded. Therefore, the present study
investigated both common variants and relatively rare variants of
the selected nsSNPs, with possibly deleterious effects on protein
function, to perform a two-stage GWAS.
Chen et al (33) genotyped over 350,000 genome-wide
autosomal SNPs by using Illumina Human 610-Quad BeadChips in over
6,000 Han Chinese samples from ten provinces, showing that, in the
Han Chinese population, geographic matching is a good proxy for
genetic matching (51). In phase 1 of
the present study, 100 participants were enrolled from Qidong,
China. Therefore, we considered it unnecessary to conduct a
population stratification analysis. In order to increase the
statistical power to search for genetic risk factors, potential
confounding factors (gender, ethnicity, HBV status and age) were
controlled by selecting only male HCC patients (including familial
HCC patients) and male controls with HBsAg seropositivity from
Qidong, where there is a high incidence of HCC. Due to the
relatively small sample size (660 samples) of this GWAS, the
associations did not reach previously established statistical
criteria for GWAS (P<5×10−7) (52). In addition, certain researchers
consider the Bonferroni adjustment too strict and not applicable to
small studies (53). In the current
study, by using the FPRP criterion (FPRP<0.20), 9 SNPs were
identified to be associated with HCC. The majority of these SNPs
were located in a gene region, including intron, promoter and
coding regions. The strongest statistical evidence for an
association was found in rs2120243, which maps within the fourth
intron of VEPH1. According to Haploview, rs2120243 is in a
25 kb block including 19 SNPs. Teleman et al (54) identified the Drosophila melted protein
as a modulator of the insulin/PI3K signaling pathway; VEPH1 in
Homo sapiens and Drosophila melted protein are homologous
proteins. One study has reported VEPH1 to be a
cancer-associated gene in breast cancer (55). By using the online functional protein
network software STRING, VEPH1 was found to be connected to
ACVR1, TGFBR1, AKT1, SRC, FRAP1
and TP53 (56). Patch 1.0
software, which predicts changes in the binding sites of
transcription factors, revealed that allele A of rs2120243 creates
a new binding site for transcription factor retinoid X receptor α
(RXR-α) (57). It has been reported
that RXR-α is associated with HCC (58). In addition, a recent GWAS conducted in
China reported a susceptibility locus KIF1B on 1p36.22
(rs17401966) for HBV-related HCC (18). The current GWAS discovery analysis did
not reveal a consistent result for the association between
rs17401966 and the development of HBV-related HCC. However, one
nsSNP (rs4561519) of the KIF2B gene was associated with HCC
in this study. Three protein function prediction softwares (SIFT,
Polyphen and SNPs3D) all predicted that rs4561519 was deleterious
to the protein's function. KIF1B and KIF2B belong to
the kinesin family, which may indicate that kinesin families are
associated with HCC. We further investigated the protein expression
of KIF2B in chronic HBV carriers by immunohistochemistry and
western blot analysis in another unpublished study. Significantly
increased expression of KIF2B was detected in adjacent
non-tumor liver tissues compared with that of paired HCC tissues
(data not shown). The exact functional consequences of rs17401966
remain unknown. Further studies will be needed to finely map and
identify the causative polymorphism and to clarify which genes
drive the genetic association. Using STRING network analysis, many
cancer-related genes were identified, and these were involved in
various important signaling pathways associated with HCC that have
been validated by other studies (59,60).
Previous studies have clearly demonstrated the
existence of a subpopulation structure among the Chinese Han
population along the north-south axis. In the present study, all
cases and controls were residents of Qidong. Therefore, the
findings should be free of adverse effects of population
stratification. The sample size in this pilot two-stage GWAS was
relatively small and had relatively limited statistical power to
detect risk alleles with relatively low allele frequency
(MAF<0.1) or genetic power (OR<1.2). Due to these
limitations, the results must be interpreted and conclusions made
with caution. At present, we are collecting more samples and intend
to investigate certain proposed SNPs and genotypes in larger
studies to replicate and validate the associations. The risk
alleles and related gene functions must also be studied
further.
In conclusion, by conducting two-stage GWAS and
mining bioinformatic data, the present study has identified a
number of potential susceptibility loci for HCC in male Chinese
individuals with HBsAg seropositivity. These findings offer
valuable clues in the study of hepatocarcinogenesis and may have
potential clinical value. The associations and molecular mechanisms
of HCC merit further research.
Acknowledgements
The authors would like to thank the clinical
investigators involved in the present study. This study was
supported by the China Ministry of Health (no. W201202), National
Natural Science Foundation (no. 81302056), Natural Science
Foundation of Jiangsu Province (no. BK2012225) and Foundation of
Jiangsu Province (no. WS056).
References
|
1
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: From genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shariff MI, Cox IJ, Gomaa AI, Khan SA,
Gedroyc W and Taylor-Robinson SD: Hepatocellular carcinoma: Current
trends in worldwide epidemiology, risk factors, diagnosis and
therapeutics. Expert Rev Gastroenterol Hepatol. 3:353–367. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trinchet JC, Alperovitch A, Bedossa P,
Degos F, Hainaut P and Beers BV: Epidemiology, prevention,
screening and diagnosis of hepatocellular carcinoma. Bull Cancer.
96:35–43. 2009.(In French). PubMed/NCBI
|
|
5
|
Poon D, Anderson BO, Chen LT, Tanaka K,
Lau WY, Van Cutsem E, Singh H, Chow WC, Ooi LL, Chow P, et al:
Asian Oncology Summit: Management of hepatocellular carcinoma in
Asia: Consensus statement from the Asian Oncology Summit 2009.
Lancet Oncol. 10:1111–1118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yeh FS, Yu MC, Mo CC, Luo S, Tong MJ and
Henderson BE: Hepatitis B virus, aflatoxins, and hepatocellular
carcinoma in southern Guangxi, China. Cancer Res. 49:2506–2509.
1989.PubMed/NCBI
|
|
7
|
Long XD, Ma Y, Wei YP and Deng ZL: The
polymorphisms of GSTM1, GSTT1, HYL1*2, and XRCC1, and aflatoxin
B1-related hepatocellular carcinoma in Guangxi population, China.
Hepatol Res. 36:48–55. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Imaizumi T, Higaki Y, Hara M, Sakamoto T,
Horita M, Mizuta T, Eguchi Y, Yasutake T, Ozaki I, Yamamoto K, et
al: Interaction between cytochrome P450 1A2 genetic polymorphism
and cigarette smoking on the risk of hepatocellular carcinoma in a
Japanese population. Carcinogenesis. 30:1729–1734. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nahon P, Sutton A, Rufat P, Ziol M, Thabut
G, Schischmanoff PO, Vidaud D, Charnaux N, Couvert P, Ganne-Carrie
N, et al: Liver iron, HFE gene mutations, and hepatocellular
carcinoma occurrence in patients with cirrhosis. Gastroenterology.
134:102–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Migita K, Miyazoe S, Maeda Y, Daikoku M,
Abiru S, Ueki T, Yano K, Nagaoka S, Matsumoto T, Nakao K, et al:
Cytokine gene polymorphisms in Japanese patients with hepatitis B
virus infection - association between TGF-beta1 polymorphisms and
hepatocellular carcinoma. J Hepatol. 42:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ognjanovic S, Yuan JM, Chaptman AK, Fan Y
and Yu MC: Genetic polymorphisms in the cytokine genes and risk of
hepatocellular carcinoma in low-risk non-Asians of USA.
Carcinogenesis. 30:758–762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirschhorn JN and Daly MJ: Genome-wide
association studies for common diseases and complex traits. Nat Rev
Genet. 6:95–108. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Bakker PI, Yelensky R, Pe'er I, Gabriel
SB, Daly MJ and Altshuler D: Efficiency and power in genetic
association studies. Nat Genet. 37:1217–1223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Florez JC, Manning AK, Dupuis J, McAteer
J, Irenze K, Gianniny L, Mirel DB, Fox CS, Cupples LA and Meigs JB:
A 100K genome-wide association scan for diabetes and related traits
in the Framingham Heart Study: Replication and integration with
other genome-wide datasets. Diabetes. 56:3063–3074. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rothman N, Garcia-Closas M, Chatterjee N,
Malats N, Wu X, Figueroa JD, Real FX, Van Den Berg D, Matullo G,
Baris D, et al: A multi-stage genome-wide association study of
bladder cancer identifies multiple susceptibility loci. Nat Genet.
42(11): 978–84. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu C, Miao X, Huang L, Che X, Jiang G, Yu
D, Yang X, Cao G, Hu Z, Zhou Y, et al: Genome-wide association
study identifies five loci associated with susceptibility to
pancreatic cancer in Chinese populations. Nat Genet. 44:62–66.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu
Z, Xu JH, Cai ZM, Huang W, Zhao GP, et al: Genome-wide association
study in a Chinese Han population identifies nine new
susceptibility loci for systemic lupus erythematosus. Nat Genet.
41:1234–1237. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kurbanov F, Tanaka Y and Mizokami M:
Geographical and genetic diversity of the human hepatitis B virus.
Hepatol Res. 40:14–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen CJ and Chen DS: Interaction of
hepatitis B virus, chemical carcinogen, and genetic susceptibility:
Multistage hepatocarcinogenesis with multifactorial etiology.
Hepatology. 36:1046–1049. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ming L, Thorgeirsson SS, Gail MH, Lu P,
Harris CC, Wang N, Shao Y, Wu Z, Liu G, Wang X, et al: Dominant
role of hepatitis B virus and cofactor role of aflatoxin in
hepatocarcinogenesis in Qidong, China. Hepatology. 36:1214–1220.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Zhai Y, Hu Z, Wu C, Qian J, Jia
W, Ma F, Huang W, Yu L, Yue W, et al: Genome-wide association study
identifies 1p36.22 as a new susceptibility locus for hepatocellular
carcinoma in chronic hepatitis B virus carriers. Nat Genet.
42:755–758. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qu LS, Liu TT, Jin F, Guo YM, Chen TY, Ni
ZP and Shen XZ: Combined pre-S deletion and core promoter mutations
related to hepatocellular carcinoma: A nested case-control study in
China. Hepatol Res. 41:54–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qu L, Kuai X, Liu T, Chen T, Ni Z and Shen
X: Pre-S deletion and complex mutations of hepatitis B virus
related to young age hepatocellular carcinoma in Qidong, China.
PLoS One. 8:e595832013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qu LS, Liu JX, Liu TT, Shen XZ, Chen TY,
Ni ZP and Lu CH: Association of hepatitis B virus pre-S deletions
with the development of hepatocellular carcinoma in Qidong, China.
PLoS One. 9:e982572014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Llovet JM, Fuster J and Bruix J:
Barcelona-Clínic Liver Cancer Group: The Barcelona approach:
Diagnosis, staging, and treatment of hepatocellular carcinoma.
Liver Transpl. 10:S115–S120. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
World Medical Association Declaration of
Helsinki: Ethical principles for medical research involving human
subjects. JAMA. 284:3043–3045. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Al Olama AA, Kote-Jarai Z, Giles GG, Guy
M, Morrison J, Severi G, Leongamornlert DA, Tymrakiewicz M, Jhavar
S, Saunders E, et al: Multiple loci on 8q24 associated with
prostate cancer susceptibility. Nat Genet. 41:1058–1060. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kiemeney LA, Thorlacius S, Sulem P, Geller
F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson
JT, Sigurdsson A, et al: Sequence variant on 8q24 confers
susceptibility to urinary bladder cancer. Nat Genet. 40:1307–1312.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fletcher O, Johnson N, Gibson L, Coupland
B, Fraser A, Leonard A, dos Santos Silva I, Ashworth A, Houlston R
and Peto J: Association of genetic variants at 8q24 with breast
cancer risk. Cancer Epidemiol Biomarkers Prev. 17:702–705. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beebe-Dimmer JL, Levin AM, Ray AM, Zuhlke
KA, Machiela MJ, Halstead-Nussloch BA, Johnson GR, Cooney KA and
Douglas JA: Chromosome 8q24 markers: Risk of early-onset and
familial prostate cancer. Int J Cancer. 122:2876–2879. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cheng I, Plummer SJ, Jorgenson E, Liu X,
Rybicki BA, Casey G and Witte JS: 8q24 and prostate cancer:
Association with advanced disease and meta-analysis. Eur J Hum
Genet. 16:496–505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yeager M, Orr N, Hayes RB, Jacobs KB,
Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee
N, et al: Genome-wide association study of prostate cancer
identifies a second risk locus at 8q24. Nat Genet. 39:645–649.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen CF, Hsu EC, Lin KT, Tu PH, Chang HW,
Lin CH, Chen YJ, Gu DL, Lin CH, Wu JY, et al: Overlapping
high-resolution copy number alterations in cancer genomes
identified putative cancer genes in hepatocellular carcinoma.
Hepatology. 52:1690–1701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z,
Zhao Y, Ge C, Zhao F, Chen T, et al: Genome-wide copy number
analyses identified novel cancer genes in hepatocellular carcinoma.
Hepatology. 54:1227–1236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wacholder S, Chanock S, Garcia-Closas M,
El Ghormli L and Rothman N: Assessing the probability that a
positive report is false: An approach for molecular epidemiology
studies. J Natl Cancer Inst. 96:434–442. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Blakely TA, Bates MN, Baker MG and Tobias
M: Hepatitis B carriage explains the excess rate of hepatocellular
carcinoma for Maori, Pacific Island and Asian people compared to
Europeans in New Zealand. Int J Epidemiol. 28:204–210. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Katoh M: Networking of WNT, FGF, Notch,
BMP, and Hedgehog signaling pathways during carcinogenesis. Stem
Cell Rev. 3:30–38. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Purcell S, Neale B, Todd-Brown K, Thomas
L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ,
et al: PLINK: A tool set for whole-genome association and
population-based linkage analyses. Am J Hum Genet. 81:559–575.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
de Tayrac M, Etcheverry A, Aubry M,
Saïkali S, Hamlat A, Quillien V, Le Treut A, Galibert MD and Mosser
J: Integrative genome-wide analysis reveals a robust genomic
glioblastoma signature associated with copy number driving changes
in gene expression. Genes Chromosomes Cancer. 48:55–68. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mamori S and Tajiri H: Ets-1 is increased
in anticancer drug-containing media and hypoxic cultures, similar
to TACE. Scand J Gastroenterol. 44:507–508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Varis A, Wolf M, Monni O, Vakkari ML,
Kokkola A, Moskaluk C, Frierson H Jr, Powell SM, Knuutila S,
Kallioniemi A, et al: Targets of gene amplification and
overexpression at 17q in gastric cancer. Cancer Res. 62:2625–2629.
2002.PubMed/NCBI
|
|
42
|
Liu J, Ghanim M, Xue L, Brown CD, Iossifov
I, Angeletti C, Hua S, Nègre N, Ludwig M, Stricker T, et al:
Analysis of Drosophila segmentation network identifies a JNK
pathway factor overexpressed in kidney cancer. Science.
323:1218–1222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xing Y, Shi S, Le L, Lee CA, Silver-Morse
L and Li WX: Evidence for transgenerational transmission of
epigenetic tumor susceptibility in Drosophila. PLoS Genet.
3:1598–1606. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hahn SA, Schutte M, Hoque AT, Moskaluk CA,
da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban
RH, et al: DPC4, a candidate tumor suppressor gene at human
chromosome 18q21.1. Science. 271:350–353. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Le Clerc S, Limou S, Coulonges C,
Carpentier W, Dina C, Taing L, Delaneau O, Labib T, Sladek R,
Deveau C, et al: ANRS Genomic Group: Genomewide association study
of a rapid progression cohort identifies new susceptibility alleles
for AIDS (ANRS Genomewide Association Study 03). J Infect Dis.
200:1194–1201. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
46
|
Michaud-Levesque J and Richard S:
Thrombospondin-1 is a transcriptional repression target of PRMT6. J
Biol Chem. 284:21338–21346. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jegga AG, Gowrisankar S, Chen J and Aronow
BJ: PolyDoms: A whole genome database for the identification of
non-synonymous coding SNPs with the potential to impact disease.
Nucleic Acids Res. 35(Database): D700–D706. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yue P and Moult J: Identification and
analysis of deleterious human SNPs. J Mol Biol. 356:1263–1274.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bonasio R, Lecona E and Reinberg D: MBT
domain proteins in development and disease. Semin Cell Dev Biol.
21:221–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ip WK, Lai PB, Wong NL, Sy SM, Beheshti B,
Squire JA and Wong N: Identification of PEG10 as a progression
related biomarker for hepatocellular carcinoma. Cancer Lett.
250:284–291. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen J, Zheng H, Bei JX, et al: Genetic
structure of the Han Chinese population revealed by genome-wide SNP
variation. Am J Hum Genet. 85:775–785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wellcome Trust Case Control Consortium:
Genome-wide association study of 14,000 cases of seven common
diseases and 3,000 shared controls. Nature. 447:661–678. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Duggal P, Gillanders EM, Holmes TN and
Bailey-Wilson JE: Establishing an adjusted p-value threshold to
control the family-wide type 1 error in genome wide association
studies. BMC Genomics. 9:5162008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Teleman AA, Chen YW and Cohen SM:
Drosophila Melted modulates FOXO and TOR activity. Dev Cell.
9:271–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sjöblom T, Jones S, Wood LD, et al: The
consensus coding sequences of human breast and colorectal cancers.
Science. 314:268–274. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jensen LJ, Kuhn M, Stark M, Chaffron S,
Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et
al: STRING 8 - a global view on proteins and their functional
interactions in 630 organisms. Nucleic Acids Res. 37(Database):
D412–D416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Matys V, Fricke E, Geffers R, Gössling E,
Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis
OV, et al: TRANSFAC: Transcriptional regulation, from patterns to
profiles. Nucleic Acids Res. 31:374–378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Matsushima-Nishiwaki R, Shidoji Y,
Nishiwaki S, Yamada T, Moriwaki H and Muto Y: Aberrant metabolism
of retinoid X receptor proteins in human hepatocellular carcinoma.
Mol Cell Endocrinol. 121:179–190. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zender L, Villanueva A, Tovar V, Sia D,
Chiang DY and Llovet JM: Cancer gene discovery in hepatocellular
carcinoma. J Hepatol. 52:921–929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
van Hengel J, D'Hooge P, Hooghe B, Wu X,
Libbrecht L, De Vos R, Quondamatteo F, Klempt M, Brakebusch C and
van Roy F: Continuous cell injury promotes hepatic tumorigenesis in
cdc42-deficient mouse liver. Gastroenterology. 134:781–792. 2008.
View Article : Google Scholar : PubMed/NCBI
|