Introduction
The inflammatory response is the body's natural
response to invasion by an infectious agents or tissue damage.
However, prolonged inflammation may activate cell proliferation and
induce deregulation of cell death in affected tissues (1). Thus, chronic inflammation is now
recognized as a predisposing cause of many types of cancer,
including liver and gastric (2).
Proinflammatory cytokines, including interleukin-1β (IL-1β),
interleukin-6 (IL-6) and tumor necrosis factor (TNF-α), are
produced by stimulated inflammatory cells and epithelial cells
during mucosal inflammation (3).
IL-1β is a potent inflammatory mediator that has a significant role
in initiating and amplifying the inflammatory response against
Helicobacter pylori infection (4). The present authors and others have
previously demonstrated that IL-1β induced by H. pylori
infection promotes gastric carcinogenesis (5,6).
Furthermore, the significant role of IL-1β in increasing the risk
of gastric inflammation and cancer has been demonstrated by using a
transgenic model overexpressing human IL-1β protein (7). IL-1β is known to activate the nuclear
factor κB (NF-κB) signaling pathway and elicit IL-6 production
(8). IL-1β induced by H.
pylori infection led to upregulation of inducible nitric oxide
synthase (iNOS) and overproduction of nitric oxide (NO) (9). Accumulating evidence demonstrates that
overproduction of NO may induce irreversible mucosal DNA damage,
which has been proposed to be involved in the initiation and
promotion of gastric tumor growth (10).
Promoter CpG island methylation is frequently
present in non-neoplastic gastric mucosa with H.
pylori-induced gastritis (11,12).
E-cadherin (E-cad) is a tumor-suppressor gene and its promoter
hypermethylation has been observed to have a significant role in
gastric carcinogenesis (11,13,14).
Therefore, inflammation has been suggested to be an inducer of
aberrant DNA methylation and a crucial driving force in the
development of gastric cancer. This is evidenced by the frequent
detection of E-cad methylation in non-neoplastic gastric mucosa
with H. pylori infection (15).
It has been demonstrated that IL-1β may induce DNA
methylation in vitro (16–18). This
methylation-dependent gene silencing mechanism is via IL-1β
activation of NO production (16,18). Thus,
the present study used mice in which the interleukin 1 receptor
type 1 (IL-1R1) was deleted in order to characterize the specific
role of IL-1β in H. pylori-induced DNA methylation.
Previously the present authors demonstrated that H.
pylori infection induced severe gastritis with enhanced
IL-1β production is likely a prerequisite mechanism for DNA
methylation (5). However, whether
H. pylori-induced DNA methylation is IL-1β-dependent, or
whether this phenomenon is the result of a complex interaction due
to the host immune response to H. pylori infection,
irrespective of IL-1β, remains to be elucidated.
The present study aimed to elucidate the biological
activity of IL-1β in the initiation of gastric inflammation, and
additionally investigated the specific role of IL-1β in linking
inflammation with DNA methylation by using
IL-1R1-homozygous-knockout (IL-1R1−/−)
mice.
Materials and methods
Animals
IL-1R1−/− mice from a C57BL/6
background were purchased from the Jackson Laboratory (Bar Harbor,
ME, USA). The IL-1R1−/− mice and their
C57BL/6 wild-type cohorts (WT) were bred and housed in the Animal
Unit at the University of Hong Kong (Hong Kong, China). The mice
were kept in individually ventilated cages (Tecniplast, West
Chester, PA, USA) in an air-conditioned room at 23±1°C with a 12/12
h light/dark cycle with access ad libitum to water and food
(Ralston Purina Co., Chicago, IL, USA). Male WT and
IL-1R1−/− mice (4–6 weeks old), weighing
20–25 g were randomly split into groups (n=4-8 for each time
point). All experimental protocols were approved by the Hong Kong
Department of Health and the Committee on the Use of Live Animals
for Teaching and Research of The University of Hong Kong (Hong
Kong, China; approval no., 1327–06).
Intraperitoneal challenges
To elucidate the role of IL-1β in induction of DNA
methylation in vivo, WT and IL-1R1−/−
mice (n=8 in each group) were treated with intraperitoneal
injection of recombinant mouse IL-1β (5 µg/kg/day; R&D Systems,
Inc., Minneapolis, MN, USA) on day 0 and day 2. Controls for the
two strains (n=4) were treated with an identical volume of
phosphate-buffered saline (PBS; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) solution. The dose of IL-1β was
selected based on dose response studies to induce an inflammatory
response (19) and preliminary
experiments where it was observed that 5 µg/kg/day of IL-1β caused
measurable effects.
Preparation of specimens
Mice were sacrificed using anesthesia at 1 day, 3
days, 1 week and 2 weeks subsequent to injection. Blood was taken
from the tail vein 0, 1, 4 and 8 h following IL-1β injection and
subsequent to sacrifice to investigate the serum levels of IL-1β,
IL-6 and NO. The stomach was resected and snap-frozen for
additional analysis.
Measurement of cytokines
The blood levels of proinflammatory cytokines IL-1β
and IL-6 were determined using the mouse IL-1β and IL-6 Quantikine®
Colorimetric Sandwich enzyme-linked immunosorbent assay (ELISA)
(R&D Systems, Inc.) according to the manufacturer's protocol.
Absorbance was measured at 450 nm by a microplate reader (µQaunt™
Microplate Spectrophotometer; Bio-Tek Instruments, Inc., Winooski,
VT, USA). Each measurement was repeated in triplicate, and the mean
value was recorded (in pg/ml).
Methylation analysis
Genomic DNA was extracted from frozen mucosa
specimens using the DNeasy Blood & Tissue kit (Qiagen GmbH,
Hilden, Germany). The promoter methylation status of E-cad was
assessed by restriction enzyme digestion followed by polymerase
chain reaction (PCR) as previously described (20). Briefly, 500 ng of genomic DNA was
digested with restriction enzymes HpaII (40 units/ml) and
MspI (50 units/ml; both Invitrogen; Thermo Fisher
Scientific) at 37°C overnight in a total volume of 20 µl.
HpaII and MspI cut DNA at the CCGG sequence; however,
HpaII does not cut when the internal cytosine is methylated.
A parallel assay using heat inactivated MspI was performed
for each DNA sample as the negative control (20). DNA samples extracted from the mouse
tail was methylated in vitro with CpG methylase
(M.SssI; Zymo Research, Irvine, CA, USA) was used as a
positive control. Following digestion, 1 µl of each digest
underwent PCR amplification. The PCR reaction was performed using
AmpliTaq Gold® DNA Polymerase (Invitrogen; Thermo Fisher
Scientific, Inc.) at 95°C for 2 min, followed by 35 cycles of
denaturation at 94°C for 45 s, annealing at 57°C for 45 sec and
elongation at 72°C for 45 sec, followed by a final extension step
of 5 min at 72°C. The PCR amplification was performed using the
GeneAmp® 9700 PCR System (Applied Biosystems®; Thermo Fisher
Scientific, Inc.). PCR products and a 50 bp DNA ladder (Invitrogen;
Thermo Fisher Scientific, Inc.) were subjected to electrophoresis
on a 2% agarose gel, stained with ethidium bromide (Invitrogen;
Thermo Fisher Scientific, Inc.) and visualized using the Gel Doc XR
System (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Methylation-positive PCR fragments were purified and confirmed by
sequencing using the TA-cloning method (Invitrogen; Thermo Fisher
Scientific, Inc.) as previously described (21). This experiment was repeated in
triplicate. Gene-specific PCR primers are listed in Table I (synthesised by Invitrogen; Thermo
Fisher Scientific, Inc.).
 | Table I.Primer sequences (5′ to 3′) used for
methylation-specific PCR and RT-PCR analysis. |
Table I.
Primer sequences (5′ to 3′) used for
methylation-specific PCR and RT-PCR analysis.
| Gene name | Forward | Reverse |
|---|
|
E-cadherina |
TCCAGGAACCTCCGTGATGA |
CCGGTGTCCCTATTGACAG |
|
E-cadherinb |
GCGTCCCCAGCCAATCAG |
GCAGACGCCGAGCAAACAC |
|
Interleukin-1βa |
CAGGATGAGGACATGAGCACC |
CTCTGCAGACTCAAACTCCAC |
|
Interleukin-6a |
GATGATGCACTTGCAGAA |
GTACTCCAGAAGACCAGAGG |
| Inducible nitric
oxide synthasea |
CAGCTGGGCTGTACAAACCTT |
CATTGGAAGTGAAGCGTTTCG |
|
Glyceraldehyde-3-Phosphate
Dehydrogenasea |
GACATCAAGAAGGTGGTGAAGC |
GTCCACCACCCTGTTGCTGTAG |
Reverse transcription
(RT)-quantitative (q)PCR analysis of gene expression
Total RNA was extracted from snap-frozen mucosa
specimens using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Total
RNA (2 µg) was reverse transcribed using SuperScript® First-Strand
Synthesis system (Invitrogen; Thermo Fisher Scientific, Inc.) at
65°C for 10 min, followed by 50°C for 30 min and 85°C for 5 min
using the Opticon 2 Real-Time PCR system (Bio-Rad Laboratories,
Inc.). A control reaction without reverse transcriptase was
included to identify any DNA contamination in the samples. RT-qPCR
amplification of IL-1β, IL-6, E-cad and iNOS was performed in the
Opticon 2 Real-Time PCR system (Bio-Rad Laboratories, Inc.) using
SYBR Green (Bio-Rad Laboratories, Inc.). Gene expression levels
were normalized using glyceraldehyde-3-phosphate dehydrogenase as
an internal control gene, and compared with the data of untreated
mice using the ∆∆Cq method (22). The
experiment was repeated in three times. Gene-specific PCR primers
are listed in Table I (synthesised by
Invitrogen; Thermo Fisher Scientific, Inc.).
Nitric oxide assay
Production of NO in mouse serum was measured as
nitrate, a more stable end product of NO, based on the Griess
reaction method (23) using a Total
Nitric Oxide Assay kit (Enzo Life Sciences, Inc., Farmingdale, NY,
USA). The optical density was measured at 550 nm using a microplate
reader (µQaunt Microplate Spectrophotometer). All standards and
samples were measured in triplicate.
Measurement of total DNA
methyltransferase (DNMT) activity
Nuclear protein extract was obtained from the frozen
gastric mucosa samples according to the manufacturer's protocol
using the EpiQuik™ Nuclear Extraction kit (Epigentek, Farmingdale,
NY, USA). Protein concentrations were determined using the Bradford
method (Bio-Rad Laboratories, Inc.) (24). The total DNMT activity was measured
using the EpiQuik™ DNA methyltransferase activity assay kit
(Epigentek). Absorbance was determined using a microplate
spectrophotometer (µQaunt Microplate Spectrophotometer) at 450 nm,
and DNMT activity (OD/h/mg) was calculated according to the
manufacturer's protocol.
Statistical analysis
All data are expressed as the mean ± standard error.
The statistical significance between the two experimental groups
was analyzed using the student's t-test. All statistical analysis
was performed using GraphPad Prism version 5.0 (GraphPad Software,
Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of IL-1β on induction of
inflammatory cytokines
To investigate the impact of peripheral IL-1β
challenge on serum levels of IL-1β and IL-6, ELISAs were performed
on blood collected at 0, 1, 4 and 8 h subsequent to IL-1β
injection, and 1 day, 3 days, 1 week and 2 weeks following
sacrifice. Control mice injected with PBS solution demonstrated an
undetectable serum level of IL-1β and IL-6 at all the time points.
As shown in Fig. 1A, the IL-1β level
increased significantly between 1 h and 8 h post-injection in
wild-type (WT) mice (23±2.6 pg/ml at 1 h, 133±4.8 pg/ml at 4 h,
74±5.2 pg/ml at 8 h; all P<0.005) when compared with
IL-1R1−/− mice. Following a peak at 4 h, the
levels of IL-1β progressively decreased to an undetectable level at
1-day post-injection in WT mice. In IL-1R1−/−
mice, a low level of IL-1β was observed only at 1 h post-injection
(9.3±1.4 pg/ml), and the IL-1β level became undetectable even
following a second dose of IL-1β on day 2. By contrast, in WT mice,
following a second dose of IL-1β, serum levels of IL-1β remained
detectable at 3 days, 1 week and 2 weeks post-injection (30±2.3
pg/ml, 18±1.2 pg/ml and 13±2.3 pg/ml, respectively; all P<0.05)
when compared with IL-1R1−/− mice.
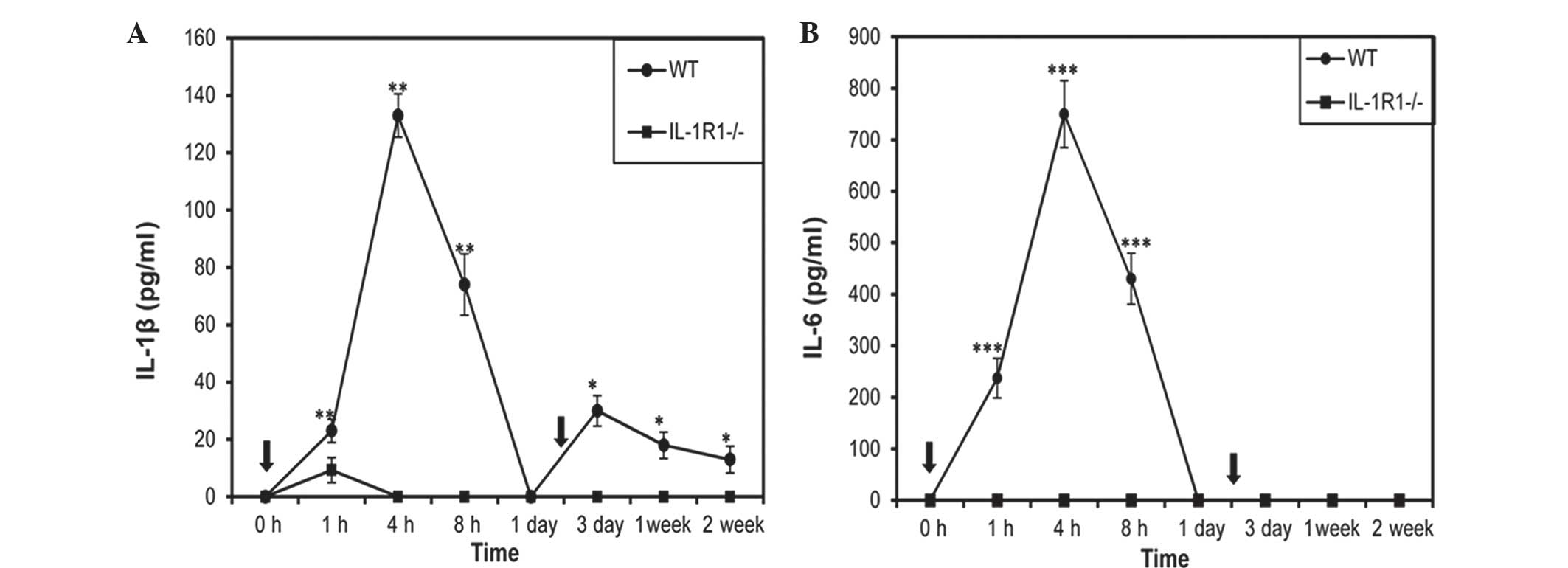 | Figure 1.Effect of peripheral IL-1β challenge
on serum levels of (A) IL-1β and (B) IL-6 in WT and
IL-1R1−/− mice. Mice were injected with IL-1β
(5 µg/kg/day) on day 0 and day 2 (indicated by arrows). Blood was
collected and measured by enzyme-linked immunosorbent assay at 0,
1, 4, 8 h subsequent to IL-1β injection, and after sacrifice at 1
day, 3 days, 1 week and 2 weeks. Data are presented as the mean ±
standard error. Significant differences are shown by *P<0.05,
**P<0.005, ***P<0.0005 vs. control mice without injection.
IL, interleukin; WT, wild-type; IL-1R1, interleukin 1 receptor type
1. |
The serum level of IL-6 was assessed to confirm that
the induced IL-1β was due to peripheral inflammatory cytokine IL-1β
challenge and not injection. IL-6 levels increased significantly at
1 h post-injection and reached a peak at 4 h, and subsequently
became undetectable at 1-day post-injection (237±11.5 pg/ml at 1 h,
750±24.7 pg/ml at 4 h, 430±5.6 pg/ml at 8 h; all P<0.0005) in WT
mice when compared with IL-1R1−/− mice
(Fig. 1B). Unlike IL-1β, the IL-6
level was not detectable at day 3, 1 week or 2 weeks, even
following a second dose of IL-1β on day 2. In injected
IL-1R1−/− mice and control mice treated with
PBS solution, no IL-6 induction was observed, and the IL-6 level
was undetectable at all the time points analyzed (Fig. 1B).
Induction of promoter methylation of
E-cad by IL-1β injection
The promoter methylation status of E-cad was
assessed in mice gastric mucosae collected at 1 day, 3 days, 1 week
and 2 weeks post-injection of IL-1β. Methylation of E-cad was
absent in the injected WT and IL-1R1−/− mice
(n=8 in each group) at 1-day post-injection. Following a second
dose of IL-1β on day 2, promoter methylation of E-cad was observed
in 3/8 (37.5%) WT mice at the 3 day and 1 week time points, and 1/8
(12.5%) WT mice at 2-weeks post-injection (Fig. 2). However, methylation of E-cad was
not detected in IL-1R1−/− mice (n=8) and
PBS-treated control mice (n=4) at all time points.
Effect of IL-1β treatment on mRNA
expression of proinflammatory genes, E-cad and iNOS
A significant change in IL-1β messenger (m)RNA
expression in the gastric mucosa was observed on day 3 in treated
WT mice when compared with IL-1R1−/− mice or
controls treated with PBS (all P<0.001). Otherwise, the mRNA
expression was comparable between treated WT and
IL-1R1−/− mice or controls at all other time
points measured (data not shown). IL-1β treatment did not induce a
statistically significant difference in mRNA expression of IL-6 in
WT and IL-1R1−/− mouse gastric mucosae, and
expression of IL-6 was almost undetectable in the control group
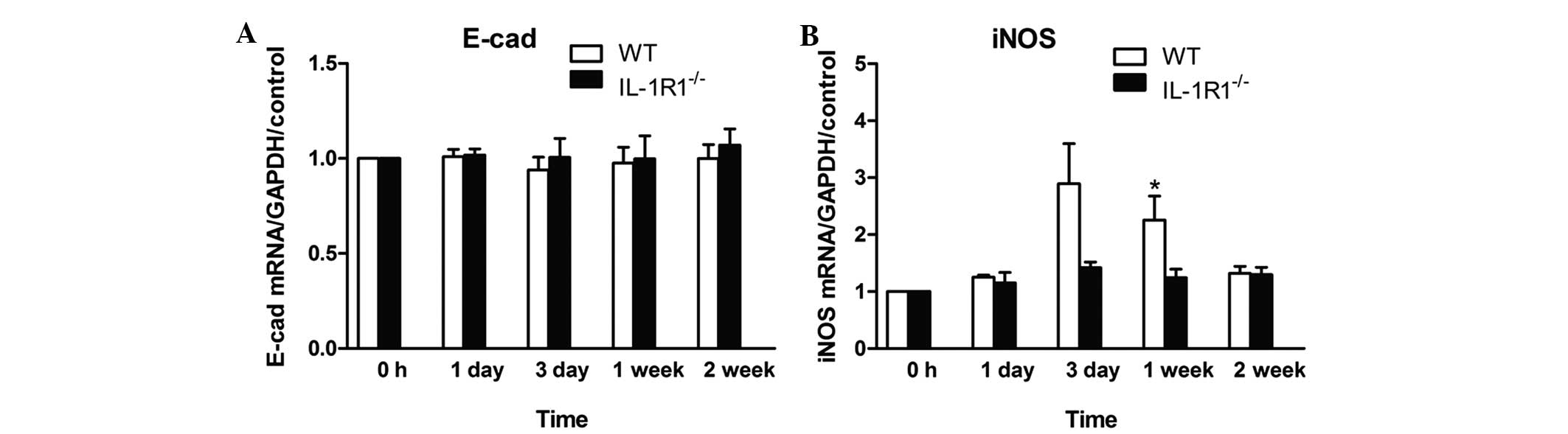
(data not shown). A slight decrease in the mRNA expression of E-cad
was observed between the IL-1β-treated WT and
IL-1R1−/− mice, as well as in the control
mice treated with PBS solution (Fig.
3A). However, IL-1β treatment induced a higher expression of
iNOS in WT mice when compared with IL-1R1−/−
mice. The expression of iNOS was comparable between treated WT mice
and IL-1R1−/− mice on day 3 (P=0.058). A
statistically significant difference in iNOS expression was
observed at 1-week post-infection in WT mice (P=0.0411; Fig. 3B).
Effect of IL-1β treatment on NO
production
The serum NO levels obtained in the various groups
of mice at different time points are illustrated in Fig. 4. The serum level of NO was
significantly increased in IL-1β-injected WT mice when compared
with IL-1R1−/− mice at 4 h (195.33±22.33 µM;
P<0.001), 8 h (138.34±16.24 µM; P<0.005), 3 days (182.60±32.2
µM; P<0.001) and 1 week (127.34±12.14 µM; P<0.005),
respectively. IL-1β injection did not affect the NO level in
IL-1R1−/− mice; the nitrate concentrations
measured at all three time points were comparable to the basal
physiological levels measured in the control mice (Fig. 4).
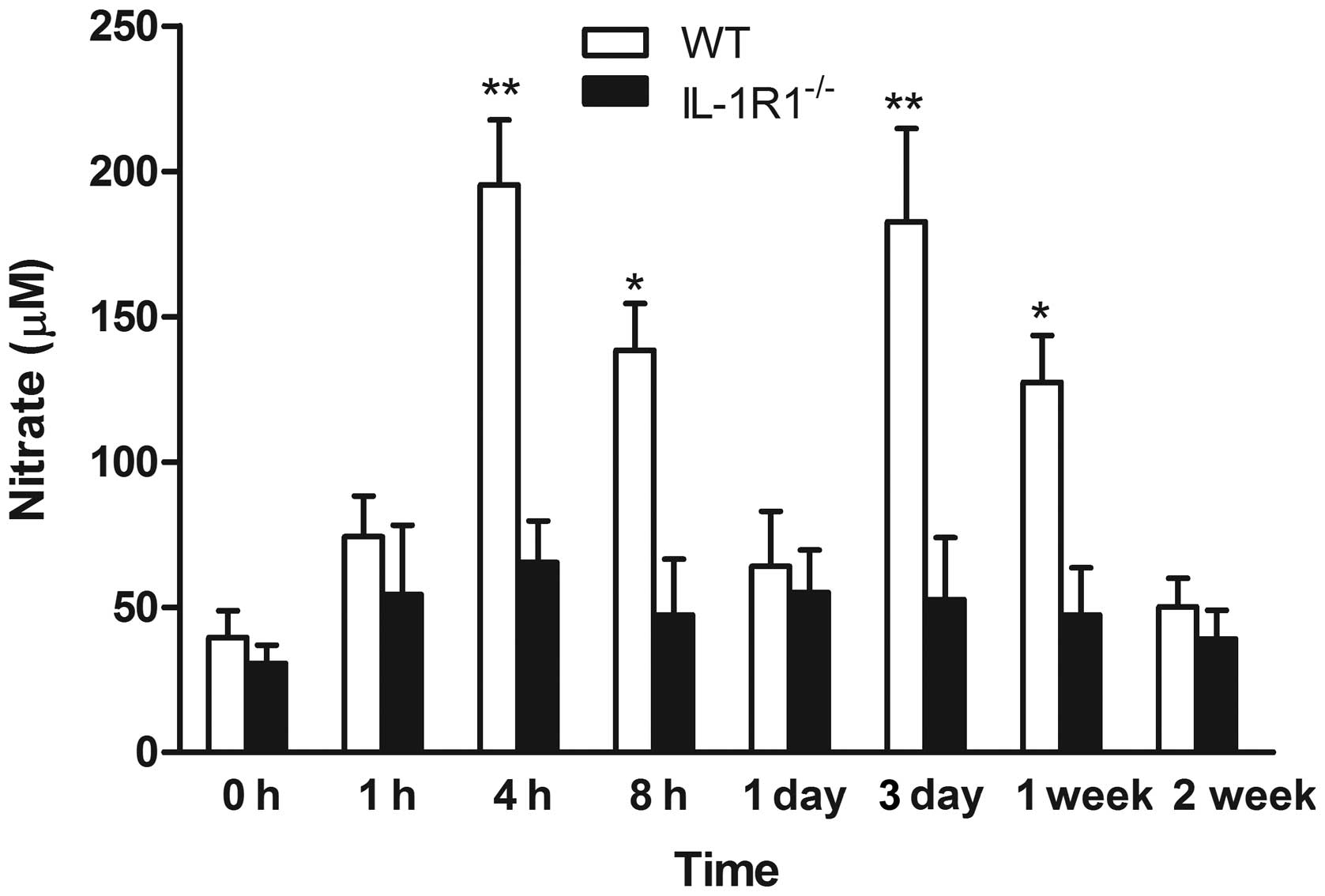 | Figure 4.Effect of peripheral IL-1β challenge
on serum levels of NO production. WT and
IL-1R1−/− mice were treated with IL-1β (5
µg/kg/day) on day 0 and day 2. NO was measured as nitrate in mouse
serum 0, 1, 4 and 8 h subsequent to IL-1β injection, and following
sacrifice at 1 day, 3 days, 1 week and 2 weeks. Data are presented
as the mean ± standard error. Significant differences are shown by
*P<0.005, **P<0.001 vs. control mice without injection. IL,
interleukin; NO, nitric oxide; WT, wild-type; IL-1R1, interleukin 1
receptor type 1. |
Discussion
Proinflammatory cytokine IL-1β has a significant
role in gastric carcinogenesis (16,17,25). In
the present study, IL-1R1−/− mice, which are
unresponsive to IL-1β, were used to fully elucidate the specific
role of IL-1β in linking gastric inflammation to DNA methylation
induction. The results indicated that peripheral IL-1β challenge
may induce proinflammatory cytokine release. This is evidenced by
the inducible serum IL-1β and IL-6 in injected WT mice when
compared with IL-1R1−/− mice lacking
functional receptors to IL-1β, or control mice without IL-1β
injection (these mice did not have inducible production of serum
IL-1β and IL-6). These findings were in agreement with previous
studies that reported that IL-1β is a significant mediator in
initiation of the immune response (26,27). The
present study additionally assayed the serum levels of IL-6 to
confirm that the increase in IL-1β was due to induction instead of
exogenous injection. A previous study additionally demonstrated
that peripheral challenge of IL-1β was able to induce IL-6 release
(28). However, the serum levels of
IL-6 were very low and dissipated quickly, suggesting that the
immune response initiated by IL-1β treatment may not be a potent
stimulator for IL-6 production (28).
The authors of the present study previously
demonstrated that H. pylori infection induced IL-1β
production and E-cad methylation in in vitro and in
vivo experiments. This methylation induction effect could be
reversed following the administration of IL-1β antagonist,
interleukin-1 receptor antagonist (5,17,18). These findings suggest the potential
mechanistic basis of H. pylori in induction of epigenetic
changes for gastric diseases. In the present study, having
demonstrated that IL-1β injection was able to initiate an immune
response evidenced by release of proinflammatory cytokines, the
methylation status of E-cad was assessed. Methylation of E-cad was
detected in IL-1β injected WT mice on day 3, following a second
dose of IL-1β. This finding was supported by a significant
upregulation of NO detected on day 3. The need for a second dose of
IL-1β may be associated with the short half-life of the IL-1β
protein, which inadequately elicited a strong immune response
following a single dose (29).
Disappearance of E-cad methylation was detected 2 weeks
post-infection, which was consistent with the plateau of IL-1β.
This is additionally in agreement with previous reports that severe
inflammation is required for initiation and maintenance of
methylation induction (5,30). Mechanistically, the present study
observed the absence of E-cad methylation in
IL-1R1−/− mice and controls, suggesting that
E-cad methylation may be mediated through an IL-1β-dependent
signaling pathway. To the best of our knowledge, no other study has
demonstrated that IL-1β injection may induce promoter methylation
of E-cad in vivo. Furthermore, the results of the present
study suggest that inflammatory cytokines may directly trigger
epigenetic changes.
The results of the present study additionally
support the mechanism of H. pylori-induced epigenetic
changes and gastric carcinogenesis via IL-1β activation of iNOS and
NO production, as proposed in our previous studies (5,17,18). Excessive NO production causes tissue
damage with subsequent cellular genome injuries (10). Previous studies have demonstrated that
NO may activate expression of DNMTs, which are the final effector
of DNA methylation (16,18,31).
However, no significant changes in total DNMT activity were
detected in the present study (data not shown). These findings are
consistent with previous studies, suggesting that mRNA expression
of DNMTs was not increased in non-cancerous gastric mucosa in
humans and mice (5,32). It is likely that aberrant DNA
methylation results from an imbalance between various DNMTs
(32–34). However, the present study provides an
improved insight into the role of NO in the IL-1β activated
methylation-dependent silencing pathway that may link inflammation
with gastric carcinogenesis.
Previous studies have reported that
IL-1R1−/− mice demonstrate attenuated
infiltration of inflammatory cells in the stomach with H.
pylori infection compared with WT mice (5,6). Among the
infiltrating inflammatory cells in H. pylori-infected
gastric mucosa, the IL-1R1−/− mice
demonstrated a significantly reduced number of macrophages and
neutrophils compared with the WT mice. Based on the absence of
E-cad methylation in IL-1R1−/− mice, it may
be speculated that IL-1β directly induces epigenetic deregulation
and thus promotes gastric carcinogenesis. This is supported by the
observation that IL-1R1−/− mice demonstrate
reduced susceptibility to gastric carcinogenesis (5,6). Although
significant progress has been made with regard to our understanding
of the mechanistic link between inflammation and gastric cancer
development, more comprehensive characterization of the role of
inflammation in the initiation and maintenance of epigenetic
changes is required.
In conclusion, the current study demonstrated that
IL-1β may directly trigger epigenetic alterations that lead to
suppression of gene expression. The results provide a significant
mechanistic link between inflammation and gastric cancer
development. These findings also highlight the importance of
developing treatment strategies that target persistent inflammatory
responses to achieve the prevention and eradication of disease.
References
|
1
|
Mantovani A, Marchesi F, Portal C,
Allavena P and Sica A: Linking inflammation reactions to cancer:
Novel targets for therapeutic strategies. Adv Exp Med Biol.
610:112–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
March CJ, Mosley B, Larsen A, et al:
Cloning, sequence and expression of two distinct human
interleukin-1 complementary DNAs. Nature. 315:641–647. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Noach LA, Bosma NB, Jansen J, et al:
Mucosal tumor necrosis factor-α, interleukin-1 β, and interleukin-8
production in patients with Helicobacter pylori infection.
Scand J Gastroenterol. 29:425–429. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang FY, Chan AO, Lo RC, et al:
Characterization of interleukin-1β in Helicobacter
pylori-induced gastric inflammation and DNA methylation in
interleukin-1 receptor type 1 knockout (IL-1R1(−/-)) mice. Eur J
Cancer. 49:2760–2770. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shigematsu Y, Niwa T, Rehnberg E, et al:
Interleukin-1β induced by Helicobacter pylori infection
enhances mouse gastric carcinogenesis. Cancer Lett. 340:141–147.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tu S, Bhagat G, Cui G, et al:
Overexpression of interleukin-1β induces gastric inflammation and
cancer and mobilizes myeloid-derived suppressor cells in mice.
Cancer Cell. 14:408–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kohase M, May LT, Tamm I, Vilcek J and
Sehgal PB: A cytokine network in human diploid fibroblasts:
Interactions of β-interferons, tumor necrosis factor,
platelet-derived growth factor, and interleukin-1. Mol Cell Biol.
7:273–280. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jaiswal M, LaRusso NF and Gores GJ: Nitric
oxide in gastrointestinal epithelial cell carcinogenesis: Linking
inflammation to oncogenesis. Am J Physiol Gastrointest Liver
Physiol. 281:G626–G634. 2001.PubMed/NCBI
|
|
10
|
Wink DA and Mitchell JB: Nitric oxide and
cancer: An introduction. Free Radic Biol Med. 34:951–954. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chan AO, Lam SK, Wong BC, et al: Promoter
methylation of E-cadherin gene in gastric mucosa associated with
Helicobacter pylori infection and in gastric cancer. Gut.
52:502–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park SY, Yoo EJ, Cho NY, Kim N and Kang
GH: Comparison of CpG island hypermethylation and repetitive DNA
hypomethylation in premalignant stages of gastric cancer,
stratified for Helicobacter pylori infection. J Pathol.
219:410–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maekita T, Nakazawa K, Mihara M, et al:
High levels of aberrant DNA methylation in Helicobacter
pylori-infected gastric mucosae and its possible association
with gastric cancer risk. Clin Cancer Res. 12:989–995. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choi IS and Wu TT: Epigenetic alterations
in gastric carcinogenesis. Cell Res. 15:247–254. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chan AO, Peng JZ, Lam SK, et al:
Eradication of Helicobacter pylori infection reverses
E-cadherin promoter hypermethylation. Gut. 55:463–468. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hmadcha A, Bedoya FJ, Sobrino F and
Pintado E: Methylation-dependent gene silencing induced by
interleukin 1β via nitric oxide production. J Exp Med.
190:1595–1604. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qian X, Huang C, Cho CH, Hui WM, Rashid A
and Chan AO: E-cadherin promoter hypermethylation induced by
interleukin-1β treatment or H. pylori infection in human
gastric cancer cell lines. Cancer Lett. 263:107–113. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang FY, Chan AO, Rashid A, et al:
Helicobacter pylori induces promoter methylation of
E-cadherin via interleukin-1β activation of nitric oxide production
in gastric cancer cells. Cancer. 118:4969–4980. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kwon KH, Murakami A, Hayashi R and
Ohigashi H: Interleukin-1β targets interleukin-6 in progressing
dextran sulfate sodium-induced experimental colitis. Biochem
Biophys Res Commun. 337:647–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang H, Ushijima T, Nagao M, Sugimura T
and Ohgaki H: β-catenin mutations in liver tumors induced by
2-amino-3,4-dimethylimidazo[4,5-f]quinoline in CDF1 mice. Cancer
Lett. 198:29–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamada H, Vijayachandra K, Penner C and
Glick A: Increased sensitivity of transforming growth factor (TGF)
β 1 null cells to alkylating agents reveals a novel link between
TGFβ signaling and O(6)-methylguanine methyltransferase promoter
hypermethylation. J Biol Chem. 276:19052–19058. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak and Schmittgen: Analysis of relative
gene expression data using real-time quantitative PCR and the
2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chan AO, Chu KM, Huang C, et al:
Association between Helicobacter pylori infection and
interleukin 1β polymorphism predispose to CpG island methylation in
gastric cancer. Gut. 56:595–597. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Griess P: Comments on the discussion of HH
Weselsky and Benedict ‘On some Azo Compounds’. Ber Deutsch Chem
Ges. 12:426–428. 1879. View Article : Google Scholar
|
|
25
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Epstein NJ, Warme BA, Spanogle J, et al:
Interleukin-1 modulates periprosthetic tissue formation in an
intramedullary model of particle-induced inflammation. J Orthop
Res. 23:501–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
El-Omar EM: The importance of interleukin
1β in Helicobacter pylori associated disease. Gut.
48:743–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Skelly DT, Hennessy E, Dansereau MA and
Cunningham C: A systematic analysis of the peripheral and CNS
effects of systemic LPS, IL-1β, TNF-β and IL-6 challenges in
C57BL/6 mice. PloS One. 8:e691232013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Klapproth J, Castell J, Geiger T, Andus T
and Heinrich PC: Fate and biological action of human recombinant
interleukin 1 β in the rat in vivo. Eur J Immunol.
19:1485–1490. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hur K, Niwa T, Toyoda T, et al:
Insufficient role of cell proliferation in aberrant DNA methylation
induction and involvement of specific types of inflammation.
Carcinogenesis. 32:35–41. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Attwood J and Richardson B: Relative
quantitation of DNA methyltransferase mRNA by real-time RT-PCR
assay. Methods Mol Biol. 287:273–283. 2004.PubMed/NCBI
|
|
32
|
Kanai Y and Hirohashi S: Alterations of
DNA methylation associated with abnormalities of DNA
methyltransferases in human cancers during transition from a
precancerous to a malignant state. Carcinogenesis. 28:2434–2442.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nakajima T, Yamashita S, Maekita T, et al:
The presence of a methylation fingerprint of Helicobacter
pylori infection in human gastric mucosae. Int J Cancer.
124:905–910. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Niwa T, Tsukamoto T, Toyoda T, et al:
Inflammatory processes triggered by Helicobacter pylori
infection cause aberrant DNA methylation in gastric epithelial
cells. Cancer Res. 70:1430–1440. 2010. View Article : Google Scholar : PubMed/NCBI
|