Glioblastoma multiforme (GBM) is the most aggressive
and lethal primary brain tumor (1). Globally, it constitutes >50% of
all diagnosed glioma cases and 20% of all central nervous system
(CNS) tumors. In Europe and North America, the frequency of GBM is
3–5 cases per 100,000 individuals, mostly affecting non-Hispanic
males (2). Notably, the average
age of affected patients is 62 years. The tumor is characterized by
invasive growth, intensive angiogenesis and an unfavorable
prognosis. When following a modern protocol of complex treatment,
the median survival of patients is ~15 months (1,3). GBM
radiation resistance is attributed to plasticity and marked
proliferation abilities of cancer stem cells (CSCs), which are the
main catalyst of neoplastic processes. Irradiation and chemotherapy
are unable to eliminate CSCs effectively (4); thus, development of fundamentally
novel approaches to GBM treatment are required, and immunotherapy
(IT) has great potential.
IT has demonstrated a high level of efficiency in
treating hemoblastoses, melanomas, lung, prostate and bladder
cancer (5). IT effectively damages
CSCs, disrupts their interaction with local cellular
microsurroundings and the extracellular matrix (ECM), and controls
further actions of CSCs, such as inhibiting the interaction between
non-tumor cells and CSCs, thereby suppressing tumor progression and
prolonging the patients' life. However, unlike other cancer types,
GBM is protected from direct interaction with the immune system by
the blood-brain barrier (5,6). GBM
cells do not express numerous unique antigens, create
immunosuppressive microsurroundings, and express anti-inflammatory
cytokines and inhibitors of the immune checkpoint (6), thereby affecting the immune response.
The ability of these cells to suppress the immune system locally
classifies GBM as a ‘cold’ tumor that is almost completely
resistant to IT.
On the other hand, high radiation doses and
chemotherapy inevitably cause myelosuppression (7), followed by severe systemic
immunodeficiency (8) that is
intensified by glucocorticosteroids (9), which are used for preventing cerebral
edema during all stages of GBM treatment. A combination of local
and systemic immunosuppression limits the antitumor potential of
IT, and interferes with its ability to control CSCs (7–9).
Thus, a novel systemic approach to GBM treatment is required, that
combines classic methods of antitumor therapy, IT and innovative
biomedical technologies that would be aimed at overcoming local and
systemic immunosuppression.
The present study aimed to review systemic solutions
for the issues associated with immunosuppression that interfere
with the antitumor potential of cell-based IT used in patients with
GBM.
The majority of patients with GBM require surgery to
achieve brain decompression, minimize hydrocephalus and lower the
risk of fatal complications (10).
Using microsurgical equipment, modern neuro-navigation and
intra-operative neuro-monitoring increases the chances of effective
tumor removal, and minimizes the risk of damaging key functioning
areas of the brain (11). However,
GBM eradication via surgery is not possible due to brain tissue
infiltration with cancer cells; therefore, the main treatment focus
is chemoradiotherapy (12).
Traditionally, γ-radiation of 60 Gy is used,
including 2 Gy daily at 30 fractions for 6 weeks (13), together with chemotherapy, where
temozolomide (TMZ) is considered the gold standard for the
treatment of GBM. Treatment options may be extended by combining
γ-radiation with proton (14) or
boron neutron capture therapy (15), the use of hyperbaric oxygen
therapy, consuming a ketogenic diet or pharmacological support with
Olaparib (16), kynurenine pathway
metabolites (17) or other
inducers of genomic instability (18). Tumor-treating fields increase the
treatment effect (11,19) and improve the patient's
condition.
Current treatment options for GBM are relatively
ineffective. In the majority of cases, despite the efforts of
medical experts, tumor relapse occurs in 4–6 months following
removal (20). In the case of
recurrent GBM, patients with ~70 on the Karnofsky Performance Scale
and 0–1 on the Eastern Cooperative Oncology Group scale may be
reoperated (21). Further rounds
of irradiation are rarely used; however, brachytherapy and
radiosurgery are considered promising options (22). Chemotherapy may help extend a
patient's life, and TMZ or lomustine are often used with
bevacizumab (23). Treatment with
procarbazine, lomustine, vincristine and platinum-based medications
(24) remain limited. Best
supportive care is recommended for all patients with recurrent GBM,
as the associated prognosis is unfavorable, and the median survival
is ~15 months (25) due to the
coordinated combination of local factors and systemic
mechanisms.
Cancer cell plasticity is mostly due to epigenetic
damage. Notably, in 2016 the World Health Organization (25) selected the isocitrate dehydrogenase
(IDH)12 mutations as the main determinant of prognosis for patients
with GBM, as these mutations result in an excess of
2-hydroxyglutarate in cancer cells that causes hypermethylation of
the genome (31). This includes
hypermethylation of the promoter regions of O6-methylguanine-DNA
methyltransferase, which is responsible for the repair of damaged
DNA and the subsequent adaptation to radiation and chemotherapy.
The IDH-mutant GBM often affects patients of a young age,
developing from a diffuse or anaplastic astrocytoma and becoming
localized in the frontal cortex (32). The IDH-wild-type GBM constitutes up
to 90% of cases, develops de novo, and is localized in
parietal, occipital or temporal areas of the brain. The median
survival of patients with IDH-mutant GBM is 23 months, and for
those with IDH-wild-type GBM is 13 months.
CSCs acquire a number of new oncogenic properties by
interacting with neural stem and progenitor cells residing in the
subventricular zone (43–45) and other germinal centers of the
human brain. Activation of primitive self-organization mechanisms
of CSCs (46,47) allows them to actively interact with
other stem and differentiated cells (48), enabling the exchange of genes,
chromosomes and whole nuclei (49), thereby increasing the viability of
the tumor and its resistance to treatment.
TGF-β inhibits inflammation, triggers ECM repair,
increases the interaction between cancer cells and the ECM
(53), creates a niche for cancer
cells (54), hinders T cell
proliferation, suppresses antigen presentation by macrophages,
inhibits the expression of major histocompatibility complex (MHC)
class II antigens by dendritic cells (DCs), increases the synthesis
rate of atypical human leukocyte antigen molecules and activates M2
microglia (55) (Fig. 1). Subsequently, as a result of
inhibiting T cells via the production of programmed cell death
protein (PD)-1 and cytotoxic T lymphocyte-associated protein 4 by
the tumor, the exhausted phenotype of T lymphocytes is created to
interact with regulatory T cells, M2-macrophages and
myeloid-derived suppressor cells (56). Thus, CSCs are isolated from the
immune system, creating an optimum living environment and
increasing the production of kynurenine pathway metabolites in the
tumor (57,58); these metabolites act as negative
controls of local inflammation. However, the ability of GBM to
resist all existing types of treatment is not only due to local
factors, but also immunosuppressive mechanisms.
Key factors of systemic immunosuppression in GBM are
radiation and chemotherapy (59).
Following chemoradiation treatment in vitro in GBM cells,
these cells produce increased levels of IL-10, IL-6, prostaglandin
E2 and other immunosuppressive factors (60). Moreover, the supernatant obtained
during cell culturing directly suppressed the proliferation of
CD4+ and CD8+ T lymphocytes (61). After receiving 30 fractions of
radiation, lymphocytes circulating in the blood flow accumulated an
average radiation dose of 2.3 Gy, while the average amount of
СD4+ cells in the patients' body decreased by two-fold
following chemoradiation, and remained low for a year (62). As patients are recommended
radiation treatment (75 mg/m2/day) along with six
subsequent cycles of TMZ treatment (150–200 mg/m2/day),
subsequent adjuvant therapy with chemotherapeutic drugs leads to
the inhibition of immunopoiesis in the red bone marrow (63–65).
The use of corticosteroids (9,66,67)
that have an adverse effect on the survival of patients with GBM is
another factor of systemic immunosuppression.
Therefore, local immunosuppression and systemic
immunodeficiency create optimal conditions for CSCs to fulfill
their lethal potential, and a combination of these factors
decreases the survival rate of patients.
Despite notable advances in the IT of malignant
neoplasms, the application of IT in brain tumors has only been an
option in recent decades (5). For
>50 years, the brain was considered an immune-privileged organ
(68), due to its lack of a
lymphatic system and isolation from other tissues by the
blood-brain barrier. This notion was based on the research by
Medawar (69), who demonstrated
the potential allogeneic graft acceptance in a rodent brain, while
other grafts were rejected by the immune system. Furthermore,
immunocyte functions in the brain have previously been associated
with cells of resident microglia that originate from the yolk sac,
which maintain their population through proliferation without any
interaction with the immune system cells (70).
The presence of specific antigens in cancer cells is
a key element of successful IT (76). Since 2009, the National Cancer
Institute in the USA has regularly updated this list of antigens
(5,6), which include IL-13 receptor α2
(IL13Rα2) and HER2. IL13Rα2 is expressed by 60–80% of GBM cells, is
absent in healthy CNS cells, yet is often found in kidney cells.
HER2 is expressed by >80% of GBM cells, but is also present in
healthy tissues (77). The first
phase of a clinical trial that aimed to target these antigens
(78) demonstrated that the size
of the tumor lesion and the associated risk level may be reduced;
however, there was no significant increase in the survival rates of
patients. In this case, IT may not be successful, as it is not
specifically targeting CSCs, which have almost no specific
antigens. Therefore, further investigations into the molecular
targets are required to eliminate CSCs.
In order to specifically target CSCs, a number of
proteins have been targeted, including heat shock proteins
(79–81), telomerase reverse transcriptase
(82), Wilms' tumor protein
(83,84), glycoprotein 100 (85), tyrosinase-related protein 2
(86), ephrin type-A receptor 2
(87,88), A2B5 protein (89,90),
SOX family of transcription factors (91) and cytomegalovirus phosphoprotein 65
(92), among others. Alternative
methods include the delivery of specific antigens to CSCs, using
adenoviruses, lentiviruses, parvovirus and recombinant polioviruses
DNX-2401 and PVS-RIPO (93–95).
However, the relative treatment success associated with these
methods remains low. Selective elimination of CSCs in GBM is
currently not an option, and therefore requires the development of
novel and more systemic approaches.
CSCs are characterized by the heterogeneous nature
of molecular genetic landscapes (96) not only in cell clones, but also in
singular cells (97), and these
have previously been extracted from tumor biopsy samples. Numerous
immunocytochemical markers of CSCs (98–100) and metabolic pathway maps of
cancer cells have been described, and notable differences have been
demonstrated between patients with primary and recurrent GBM. In
addition, cancer cells that are immunopositive for the main CSC
marker, CD133 antigen (101), do
not always demonstrate properties of CSCs, while CD133−
cancer cells may demonstrate these (102). However, the development of a
systemic approach to CSCs should focus on the role of these cells
in the pathogenesis of GBM.
Proliferation is a key property of CSCs, and
intensive proliferation is accompanied by a notable increase in
cancer cell number, leading to an increased oxygen consumption and
hypoxia development (103).
Long-term hypoxia is detrimental for all cancer cells, including
CSCs (33,40). These cells can survive by
triggering angiogenesis and providing the corresponding tumor with
a blood supply, and this is only possible through interaction with
the ECM (48). Previous cell
proteome studies have demonstrated that increased synthesis of
proteins associated with the ECM-receptor interaction signaling
pathway is a key difference between CSCs and differentiated GBM
cells (53). Proteome profiles of
CSCs and normal stem cells are considered similar (104).
Thus, the optimum targets for managing CSCs may
include cell-surface receptors, such as integrin V, integrin β3 and
integrin β1, as well as other components of the ECM-receptor
interaction signaling pathway. Optimum targets may also include ECM
components secreted by CSCs, such as collagen type VI α1, laminin
β1, fibronectin 1 and tenascin. Targeting these elements may
inhibit the development of the CSC niche, and disrupt the
corresponding intercellular interactions mediated by these cells
(105). This method may be
possible using the technologies of adaptive and active IT.
Adaptive IT involves the extraction of the patient's
immune cells in order to activate them to tumor antigens, and
subsequently return them to the patient's body. An example of this
process involved the vaccination of the patient with the antigens
of destroyed cancer cells, along with Bacillus Calmette-Guérin
antigens (106). Furthermore,
subsequent extraction of T cells was carried out, and these were
additionally stimulated with IL-2 ex vivo, and injected into
the patient. In a previous study, vaccination with cancer cell
antigens was accompanied by injection of granulocyte-macrophage
colony-stimulating factor (GM-CSF), and T cells were subsequently
obtained from the blood, activated ex vivo with
Staphylococcus aureus antigens and returned to the patient
(107). The use of immunocytes
from patients with GBM may not be successful, as these immunocytes
have been exhausted by anti-inflammatory cytokines, inhibitors of
immune checkpoints and other reprogramming factors produced by GBM
cells, as well as by the systemic immune suppressing effect of
antitumor treatment. Thus, their cytotoxic effect is relatively low
(108).
A further example of adaptive IT involves
intravenous or intracranial injection of genetically modified T
cells equipped with chimeric antigen receptors (CAR), where the
areas of antigen-recognition domains, consisting of monoclonal
antibodies, are connected with the areas of intercellular signaling
domains of T lymphocytes (109).
A previous study using rat models and monovalent CAR-T cells
targeting IL-13Rα2, IDH1-R132H, HER2 or EGFR variant (v) III
demonstrated tumor regression, but the clinical application of
CAR-T cells (110) is yet to be
fully established.
Previous data highlighting 6 clinical trials using
CAR-T cells in patients with GBM that have reached phase I are
displayed in Table I. The
aforementioned trials are limited by the small number of
participants, the involvement of patients with recurrent and
refractory GBM who have received multiple chemotherapy cycles,
limited observation periods and treatment that does not target CSCs
(111). Additionally, the
combination of CAR-T cell therapy and immune checkpoint inhibitors
have been used in patients with first-time diagnoses and recurrent
GBM (112). Thus, this method has
no clear potential. Moreover, despite a wide range of side effects,
the life expectancy of patients with GBM who take part in clinical
trials exceeds the life expectancy of those who are treated
according to the standard protocol (113). The heterogeneous nature of GBM
cells predetermines the need for creating multivalent CAR-T cells
that are capable of eliminating cancer cells that express HER2,
ephrin type-A receptor 2, IL13Rα2 and other antigens (114). Multivalent CAR-T cells that have
the ability to inhibit key components of the ECM-receptor
interaction signaling pathway in CSCs may act as a potential
treatment option for managing CSCs.
Active IT stimulates the development of antitumor
immunity following injection of peptide vaccines, including tumor
cell vaccines (TCVs) and DC vaccines (DCV). An example of a peptide
vaccine is rindopepimut, that targets cancer cells expressing a
mutant peptide of EGFRvIII (115–117). In March 2016, the phase III ACT
IV trial was terminated as the overall survival rate of patients
was not increased (118). This
may be associated with a lower antigen load due to previous
surgery, influence of PD-ligand 1 (PD-L1) or the immunosuppressive
effect of radiation and chemotherapy. This peptide vaccine may
still be used in complex personalized treatment of patients with
GBM cells that express the EGFRvIII antigen.
The development of TCVs involves recruiting immune
cells into the blood stream and presenting them with specific
antigens, followed by stimulation with pro-inflammatory cytokines
that determine the main type of intercellular interactions in the
neoplastic lesion. Immunization is performed simultaneously with
injecting GM-CSF, and immunocytes are subsequently extracted from
the patient's body, activated with Staphylococcus
enterotoxin A antigens, multiplied in the culture medium with IL-2
and returned to the patient. Autologous or gene-modified allogeneic
cancer cells that are either treated with ultrasound or deactivated
with radiation, are used as antigens for TCV creation (6). One TCV has been developed with
genetically modified cancer cells producing GM-CSF (119). In addition, a vaccine from
radiation-treated autologous cancer cells with inactivated
allogeneic cancer cells producing GM-CSF has also been described
(55).
Previous data detailing 78 clinical trials for TCV
suggested that only 30 biomedical products passed phase I. These
studies were characterized by the small number of patients, lack of
a unified approach to selecting patients and complete disregard for
local immunosuppression and systemic immunodeficiency. Moreover,
only two studies targeted CSCs (NCT00846456 and NCT01171469).
Notably, the results of these clinical trials demonstrated that the
average life expectancy of patients that received TCV was
significantly increased, compared with the control group. However,
no biomedical product that is currently on trial can be
categorically classified as TCV, as a part of the immunocytes
returned to the patient following stimulation with cancer cell
antigens includes DCs, which present the antigens with MHC class I
and II molecules to T cells. This induces immune aggression in the
tumor tissue (122).
DCV is one of the most important elements of
adaptive tumor IT. These vaccines are created in a traditional
manner, for example, cancer cells are lysed, and incubated with
mononuclear CD45+ cells of the red bone marrow and the
corresponding cytokine mixture that contains IL1 or IL2, GM-CSF and
IFNγ (123). DCs attached to the
surface of the plate are washed, stimulated with pro-inflammatory
cytokines and returned to patients with GBM. DCs migrate from the
patient's blood flow into the lymphatic system, and subsequently
move into the regional lymph nodes, where they stimulate the
proliferation and differentiation of T lymphocytes, enabling an
antitumor immune response (124).
Solving issues associated with local
immunosuppression and systemic immunodeficiency is complex. The
increased synthesis of TGF-β by GBM cells is a key element of local
immunosuppression. Previous studies have focused on suppressing
TGF-β synthesis with pharmacological agents, such as trabedersen
(127) and galunisertib (128), but the use of TGF-β inhibitors
alone (129) for the management
of local immunosuppression was insufficient. Blockade of the
signaling axis colony stimulating factor 1 (CSF1)CSF1 receptor
(CSF1R; also known as macrophage colony-stimulating factor
receptor) is one of the most important ways of regulating local
immunosuppression. CSF1, through CSF1R, induces the differentiation
of hematopoietic stem cells (HSCs) and monocytes into
tumor-associated macrophages, which under the influence of TGF-β
are activated along the alternative M2 pathway and markedly
increase the synthesis of this cytokine (130). In this regard, certain prospects
may be associated with the combination of TGF-β inhibitors with
CSF1R antagonists (131) such as
pexidartinib, emactuzumab and cabiralizumab.
In addition, local immunosuppression is caused by
the production of PD-L1 and other molecules from cancer cells
(132–134), which inhibit receptor activation
on cytotoxic T lymphocytes (135)
allowing cancer cells to take control of the immune system.
Notably, following PD-L1 stimulation, resident tumor microglia with
M2 activation not only increase the levels of these ligands, but
also induce monocyte-derived macrophages that inhibit the immune
response and promote tumor growth (136). Nivolumab, an inhibitor of PD-1
receptors, is able to direct the tumor microglia to the
M1-phenotype and alter the microsurroundings of cancer cells
(137). However, the combination
of TGF-β antagonists with agents that prevent neutralization of T
lymphocytes remains inefficient (138). Only seven clinical trials of
immune checkpoint inhibitors in the complex treatment of
glioblastoma have been completed worldwide to date (Table III).
Bevacizumab is a monoclonal antibody that targets
vascular endothelial growth factor (139–141), and when used together with TMZ,
has been indicated to increase the average life expectancy of
patients with GBM from 6.5 to 9.5 months. Nivolumab is more
effective than bevacizumab (142), but a combination of these agents
allows the treatment of cerebral edema without corticosteroids
(6,143), which enhances the potential of
IT. However, a significant increase in the life expectancy of
patients is yet to be achieved, which highlights issues associated
with these treatment methods that are more profound. Nevertheless,
despite the small number of completed clinical trials (Table III), immune checkpoint inhibitors
are among the most promising drugs for immunochemotherapy of
GBM.
The results of previous studies have demonstrated
that patients with GBM with a high level of leukocytes exhibit an
improved prognosis following the use of antitumor vaccines
(144), inhibitors of immune
checkpoint (145–147) and other immunotherapeutic agents.
This indicates the impact of systemic immunodeficiency caused by
the damage to the bone marrow tissues following radiation and
chemotherapy. Systemic solutions involve transplantation of HSCs.
In cases of unimpaired hematopoiesis, the proportion of HSCs in the
blood of a healthy individual does not exceed 0.01%, and
stimulation with GM-CSF increases the number of HSCs in the blood
>100-fold, which allows HSC extraction for further use in the
reconstruction of the immune system (148). However, this task is difficult to
achieve. Following radiation and several courses of chemotherapy,
the bone marrow of patients becomes exhausted (132,133), and it is impossible to obtain an
adequate quantity of HSCs for transplantation. This issue may be
resolved with the transplantation of autologous HSCs obtained from
the patient, that have been cryopreserved prior to disease
occurrence, but only a number of patients have this option.
Restoring the immune system of a patient using
autologous HSCs may not be the only option, as the development of
novel antitumor immunity may counter high levels of tumor
aggression. In the entire existence of both mammals and humans,
infective agents have posed serious threats; however, life
expectancies were never long enough for cancer development. Thus,
evolution has led to the development of a number of mechanisms for
eliminating pathologically altered cells (such as inflammation,
apoptosis and autophagy), but only if their number is relatively
small and it does not interfere with cell or tissue homeostasis
(149).
HSCs are progenitors of the immune system, as well
as coordinators and stabilizers of regeneration processes.
Proliferation speed and the number of these cells decreases with
age, thus reducing the levels of immune protection and increasing
the number of pathologically altered cells in the body. Notable
increases in the number of cancer cells are observed during the
rapid invasive growth of GBM, resulting in the destruction of
neurons and glial cells, inflammation, cerebral ischemia and the
intensified production of chemo-attractants that stimulate the
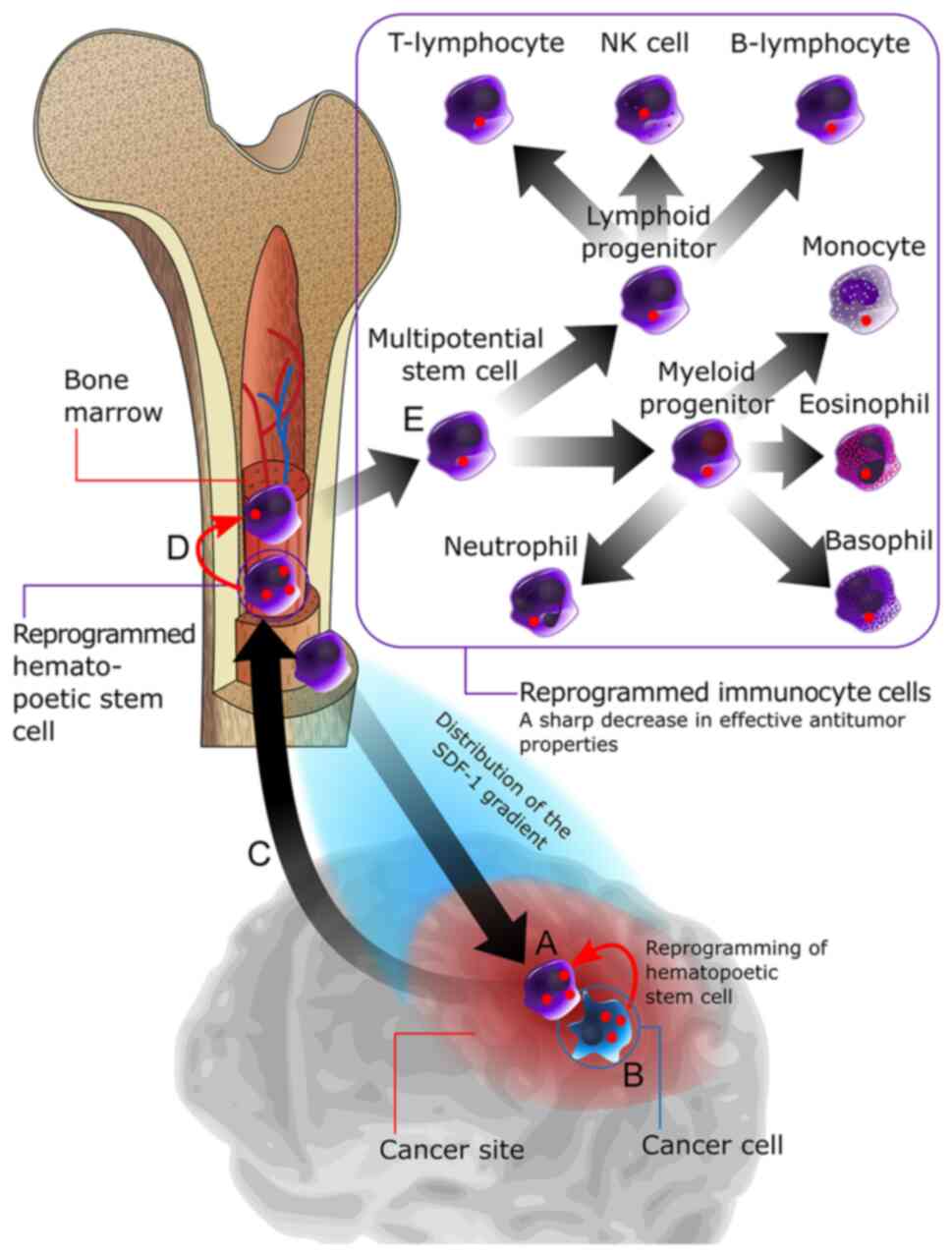
migration of HSCs into the brain (150).
The key mechanism underlying the recruitment of HSCs
into the tumor lesion is carried out by hypoxia-inducible factors a
group of ligands that activate the production of >80 cytokines
by damaged tissues. Key cytokines involved in this process include
monocyte chemoattractant protein-1 and stromal cell-derived
factor-1α, which interact with C-C motif chemokine 2 and C-X-C
chemokine receptor type 4 receptors on the HSC membrane, and induce
their migration to the tumor lesion (151). Results of a previous study
demonstrated that following 3 days of HSC injections into rats with
implanted C6 gliomas, the transplanted cells migrated into the
tumor and were visible in the blood vessels of glioma (152). In addition, following 4 and 5
days of the experiment, these cells penetrated areas of invasive
growth and necrotic segments of neoplastic tissue, where they
aggregated and interacted with cancer cells.
Briefly, HSCs adhere to cancer cells and interact
with them, which has been demonstrated by the aggregation of
fluorescent stain in cancer cells that is bound to the HSC
cytoplasmic proteins during this interaction (153). Results of a previous in
vitro study revealed that aggregation of the stain in the
cancer cells decreased the adhesion to the culture plate surface
and eliminated the interaction with the ECM, due to TGF-β
stimulation and reduced proliferation of GBM cells (154). This effect became more pronounced
when the cancer cell: HSC cell ratio reached 1:3, indicating the
antitumor potential of HSCs. In natural conditions, aggregation of
normal stem cells in a damaged area results in M2-polarization of
macrophages, suppression of antigen presentation, increased
production of IL-4 and other anti-inflammatory cytokines,
accompanied by the apoptosis of pathologically altered cells,
clearance of the dead cells and debris of the phlogogenic site, and
ECM remodeling (155). Thus,
disruption of the interaction between CSCs and the ECM induced by
HSCs is a powerful regulatory stimulus.
Furthermore, the exchange of the fluorescent stain
is not one-sided, indicating the transfer of information in both
directions. When HSCs interact with cancer cells, their cytoplasm
also displays fluorescent stain aggregation and the transfer of
specific proteins, accompanied by the epigenetic reprogramming of
HSCs (154). The ability of these
cells to effectively restore hematopoiesis and immunopoiesis
requires further investigation; however, a previous clinical trial
demonstrated that oncologic and autoimmune diseases cause
significant changes in the molecular phenotype of HSCs, impacting
treatment outcomes and the prognosis of patients (156).
Thus, whether GBM develops only as a result of
pathological transformation of neural stem cells in the human
brain, or arises as a result of the interaction of normal neural
stem cells with HSCs remain to be fully elucidated. During an in
vitro experiment, neural stem cells demonstrated a high
mobility to cancer cells of different lines, and this level of
migration was markedly increased between neural stem cells and GBM
cells, which may be attributed to their origin from the same
histogenetic source in the central nervous system Additionally, GBM
cells demonstrated a high mobility towards neural stem cells,
actively interacting with them and exchanging fluorescent stain.
HSCs are stem cells of a different origin, but their mobility
towards GBM cells is not as active as that of neural stem cells
(152) Notably, unlike stem cells
in germinal regions of the brain, HSCs play a key part in the
mechanisms underlying the memory retention of antigens, managing
the processes of immune tolerance and immunocyte activation
(157). Following the
aforementioned antitumor vaccination, HSCs may inactivate the
immune system, leading to ineffective methods for the treatment of
GBM (Fig. 2).
Collectively, the aforementioned research
demonstrated that transplantation of autologous HSCs to patients
with GBM is not a systemic approach to the issue of
immunodeficiency. Based on the hypothesis that epigenetic
reprogramming of HSCs occurs during their interaction with cancer
cells, IT may only be effective following complete regeneration or
replacement of the immune system, using allogeneic HSCs of a
healthy donor. The allogeneic transplantation of bone marrow should
become the basis for a novel approach to GBM treatment. Both the
duration and quality of remission for patients with leucosis
(158) and multiple myeloma
(159), who have received
allotransplantation of bone marrow, are significantly improved.
This is directly associated with the initiated graft-versus-tumor
effect. This approach requires extensive further investigation;
however, future treatment options should involve the use of
allogeneic HSCs.
Modern oncology exhibits a wide range of antitumor
products and methods. However, despite all recent scientific
advances, the survival rates of patients with GBM still remains
low. This issue requires novel systemic solutions, but novel
treatment options for patients with GBM should not be confined to
medication, radiation or a combination of both methods. GBM
treatment success is dependent on the use of modern surgical,
radiation and chemotherapy methods. Cytoreductive, cytotoxic and
cytostatic therapies are a first treatment stage that must involve
CSC management in order to be successful. At present, the only
method of managing CSCs is IT, but fulfilling the cytoregulatory
potential of IT is only possible following complete restoration of
the immune system functions.
HSC transplantation should be performed directly
after chemoradiation treatment. Autologous HSCs obtained from the
patient prior to disease development should be the first choice for
a graft. Therefore, cryopreservation of HSCs is required, to allow
potential cancer treatment before its occurrence. If this option is
unavailable, transplantation of allogeneic HSCs from a biologically
compatible donor may be considered; however, further experimental
studies are required.
Subsequent IT should be based on the use of
immunocytes from a healthy donor that are targeted against the key
proteins of CSCs, with a main focus on components of the
ECM-receptor interaction signaling pathway. Following HSC
transplantation and restoration of healthy immunopoiesis, TCVs are
required, based on CSC antigens, DCVs and multivalent CAR-T cells
targeting key elements of the ECM-receptor interaction signaling
pathway. Such IT should be used in combination with inhibitors of
the immune checkpoint, antagonists of TGF-β and antiangiogenic
agents.
The assumption that HSCs are the main tool of the
immune memory requires a healthy donor to receive a vaccine based
on key CSC antigens prior to obtaining HSCs. This would allow the
production of biomedical HSC-based products for personalized
proteome-based GBM therapy (Fig.
3). This method requires further experimental and clinical
investigation; however, the current review may provide a
theoretical basis for the development of novel GBM treatment
options.
Not applicable.
Funding: No funding was received.
Not applicable.
IB designed and wrote the review, and approved the
final version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Lukas RV, Wainwright DA, Ladomersky E,
Sachdev S, Sonabend AM and Stupp R: Newly diagnosed glioblastoma: A
review on clinical management. Oncology (Williston Park).
33:91–100. 2019.PubMed/NCBI
|
|
2
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gimple RC, Bhargava S, Dixit D and Rich
JN: Glioblastoma stem cells: Lessons from the tumor hierarchy in a
lethal cancer. Genes Dev. 33:591–609. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lathia JD, Mack SC, Mulkearns-Hubert EE,
Valentim CL and Rich JN: Cancer stem cells in glioblastoma. Genes
Dev. 29:1203–1217. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jackson CM, Choi J and Lim M: Mechanisms
of immunotherapy resistance: Lessons from glioblastoma. Nat
Immunol. 20:1100–1109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McGranahan T, Therkelsen KE, Ahmad S and
Nagpal S: Current state of immunotherapy for treatment of
glioblastoma. Curr Treat Options Oncol. 20:242019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chongsathidkiet P, Jackson C, Koyama S,
Loebel F, Cui X, Farber SH, Woroniecka K, Elsamadicy AA, Dechant
CA, Kemeny HR, et al: Sequestration of T cells in bone marrow in
the setting of glioblastoma and other intracranial tumors. Nat Med.
24:1459–1468. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Waziri A: Glioblastoma-derived mechanisms
of systemic immunosuppression. Neurosurg Clin N Am. 21:31–42. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pitter KL, Tamagno I, Alikhanyan K,
Hosni-Ahmed A, Pattwell SS, Donnola S, Dai C, Ozawa T, Chang M,
Chan TA, et al: Corticosteroids compromise survival in
glioblastoma. Brain. 139((Pt 5)): 1458–1471. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan AC, Ashley DM, López GY, Malinzak M,
Friedman HS and Khasraw M: Management of glioblastoma: State of the
art and future directions. CA Cancer J Clin. 70:299–312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weller M, Le Rhun E, Preusser M, Tonn JC
and Roth P: How we treat glioblastoma. ESMO Open. 4 (Suppl
2):e0005202019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nam JY and de Groot JF: Treatment of
glioblastoma. J Oncol Pract. 13:629–638. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scartoni D, Amelio D, Palumbo P,
Giacomelli I and Amichetti M: Proton therapy re-irradiation
preserves health-related quality of life in large recurrent
glioblastoma. J Cancer Res Clin Oncol. 146:1615–1622. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyatake SI, Wanibuchi M, Hu N and Ono K:
Boron neutron capture therapy for malignant brain tumors. J
Neurooncol. 149:1–11. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lesueur P, Lequesne J, Grellard JM, Dugué
A, Coquan E, Brachet PE, Geffrelot J, Kao W, Emery E, Berro DH, et
al: Phase I/IIa study of concomitant radiotherapy with olaparib and
temozolomide in unresectable or partially resectable glioblastoma:
OLA-TMZ-RTE-01 trial protocol. BMC Cancer. 19:1982019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bostian AC, Maddukuri L, Reed MR, Savenka
T, Hartman JH, Davis L, Pouncey DL, Miller GP and Eoff RL:
Kynurenine signaling increases DNA polymerase kappa expression and
promotes genomic instability in glioblastoma cells. Chem Res
Toxicol. 29:101–108. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Khotimchenko R, Bryukhovetskiy I,
Khotimchenko M and Khotimchenko Y: Bioactive compounds with
antiglioma activity from marine species. Biomedicines. 9:8862021.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anthony P, McArdle S and McHugh M: Tumor
treating fields: Adjuvant treatment for high-grade gliomas. Semin
Oncol Nurs. 34:454–464. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Di Nunno V, Franceschi E, Tosoni A, Di
Battista M, Gatto L, Lamperini C, Minichillo S, Mura A, Bartolini S
and Brandes AA: Treatment of recurrent glioblastoma:
State-of-the-art and future perspectives. Expert Rev Anticancer
Ther. 20:785–795. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kazmi F, Soon YY, Leong YH, Koh WY and
Vellayappan B: Re-irradiation for recurrent glioblastoma (GBM): A
systematic review and meta-analysis. J Neurooncol. 142:79–90. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gigliotti MJ, Hasan S, Karlovits SM,
Ranjan T and Wegner RE: Re-Irradiation with stereotactic
radiosurgery/radiotherapy for recurrent high-grade gliomas:
Improved survival in the modern Era. Stereotact Funct Neurosurg.
96:289–295. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weller M and Le Rhun E: How did lomustine
become standard of care in recurrent glioblastoma? Cancer Treat
Rev. 87:1020292020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmidt F, Fischer J, Herrlinger U, Dietz
K, Dichgans J and Weller M: PCV chemotherapy for recurrent
glioblastoma. Neurology. 66:587–589. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Soomro SH, Ting LR, Qing YY and Ren M:
Molecular biology of glioblastoma: Classification and mutational
locations. J Pak Med Assoc. 67:1410–1414. 2017.PubMed/NCBI
|
|
27
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jakovlevs A, Vanags A, Gardovskis J and
Strumfa I: Molecular classification of diffuse gliomas. Pol J
Pathol. 70:246–258. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhawe KM and Aghi MK: Microarray analysis
in glioblastomas. Methods Mol Biol. 1375:195–206. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qazi MA, Vora P, Venugopal C, Sidhu SS,
Moffat J, Swanton C and Singh SK: Intratumoral heterogeneity:
Pathways to treatment resistance and relapse in human glioblastoma.
Ann Oncol. 28:1448–1456. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Waitkus MS, Diplas BH and Yan H:
Biological role and therapeutic potential of IDH mutations in
cancer. Cancer Cell. 34:186–195. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Diplas BH, He X, Brosnan-Cashman JA, Liu
H, Chen LH, Wang Z, Moure CJ, Killela PJ, Loriaux DB, Lipp ES, et
al: The genomic landscape of TERT promoter wildtype-IDH wildtype
glioblastoma. Nat Commun. 9:20872018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Najafi M, Mortezaee K and Majidpoor J:
Cancer stem cell (CSC) resistance drivers. Life Sci.
234:1167812019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heng WS, Gosens R and Kruyt FAE: Lung
cancer stem cells: Origin, features, maintenance mechanisms and
therapeutic targeting. Biochem Pharmacol. 160:121–133. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Butti R, Gunasekaran VP, Kumar TVS,
Banerjee P and Kundu GC: Breast cancer stem cells: Biology and
therapeutic implications. Int J Biochem Cell Biol. 107:38–52. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ottevanger PB: Ovarian cancer stem cells
more questions than answers. Semin Cancer Biol. 44:67–71. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Munro MJ, Wickremesekera SK, Peng L, Tan
ST and Itinteang T: Cancer stem cells in colorectal cancer: A
review. J Clin Pathol. 71:110–116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sharifzad F, Ghavami S, Verdi J, Mardpour
S, Mollapour Sisakht M, Azizi Z, Taghikhani A, Łos MJ, Fakharian E,
Ebrahimi M and Hamidieh AA: Glioblastoma cancer stem cell biology:
Potential theranostic targets. Drug Resist Updat. 42:35–45. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:5821–5828.
2003.PubMed/NCBI
|
|
41
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Beier D, Schulz JB and Beier CP:
Chemoresistance of glioblastoma cancer stem cells-much more complex
than expected. Mol Cancer. 10:1282011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Altmann C, Keller S and Schmidt MHH: The
Role of SVZ stem cells in glioblastoma. Cancers (Basel).
11:4482019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Álvarez-Satta M, Moreno-Cugnon L and
Matheu A: Primary cilium and brain aging: Role in neural stem
cells, neurodegenerative diseases and glioblastoma. Ageing Res Rev.
52:53–63. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Álvarez-Satta M and Matheu A: Primary
cilium and glioblastoma. Ther Adv Med Oncol.
10:17588359188011692018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Qu K and Ortoleva P: Understanding stem
cell differentiation through self-organization theory. J Theor
Biol. 250:606–620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Beccari L, Moris N, Girgin M, Turner DA,
Baillie-Johnson P, Cossy AC, Lutolf MP, Duboule D and Arias AM:
Multi-axial self-organization properties of mouse embryonic stem
cells into gastruloids. Nature. 562:272–276. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Persano L, Rampazzo E, Basso G and Viola
G: Glioblastoma cancer stem cells: Role of the microenvironment and
therapeutic targeting. Biochem Pharmacol. 85:612–622. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Osswald M, Jung E, Sahm F, Solecki G,
Venkataramani V, Blaes J, Weil S, Horstmann H, Wiestler B, Syed M,
et al: Brain tumour cells interconnect to a functional and
resistant network. Nature. 528:93–98. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Taniguchi S, Elhance A, Van Duzer A, Kumar
S, Leitenberger JJ and Oshimori N: Tumor-initiating cells establish
an IL-33-TGF-beta niche signaling loop to promote cancer
progression. Science. 369:eaay18132020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Massagué J: TGFbeta in Cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Derynck R, Turley SJ and Akhurst RJ: TGFβ
biology in cancer progression and immunotherapy. Nat Rev Clin
Oncol. 18:9–34. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shevchenko V, Arnotskaya N, Pak O, Sharma
A, Sharma HS, Khotimchenko Y, Bryukhovetskiy A and Bryukhovetskiy
I: Molecular determinants of the interaction between glioblastoma
CD133+ cancer stem cells and the extracellular matrix.
Int Rev Neurobiol. 151:155–169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bryukhovetskiy I and Shevchenko V:
Molecular mechanisms of the effect of TGF-β1 on U87 human
glioblastoma cells. Oncol Lett. 12:1581–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fecci PE and Sampson JH: The current state
of immunotherapy for gliomas: An eye toward the future. J
Neurosurg. 131:657–666. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Choi BD, Maus MV, June CH and Sampson JH:
Immunotherapy for glioblastoma: Adoptive T-cell Strategies. Clin
Cancer Res. 25:2042–2048. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Takenaka MC, Gabriely G, Rothhammer V,
Mascanfroni ID, Wheeler MA, Chao CC, Gutiérrez-Vázquez C, Kenison
J, Tjon EC, Barroso A, et al: Control of tumor-associated
macrophages and T cells in glioblastoma via AHR and CD39. Nat
Neurosci. 22:729–740. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dehhaghi M, Kazemi Shariat Panahi H, Heng
B and Guillemin GJ: The gut microbiota, kynurenine pathway, and
immune system interaction in the development of brain cancer. Front
Cell Dev Biol. 8:5628122020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Authier A, Farrand KJ, Broadley KW,
Ancelet LR, Hunn MK, Stone S, McConnell MJ and Hermans IF: Enhanced
immunosuppression by therapy-exposed glioblastoma multiforme tumor
cells. Int J Cancer. 136:2566–2578. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yovino S, Kleinberg L, Grossman SA,
Narayanan M and Ford E: The etiology of treatment-related
lymphopenia in patients with malignant gliomas: Modeling radiation
dose to circulating lymphocytes explains clinical observations and
suggests methods of modifying the impact of radiation on immune
cells. Cancer Invest. 31:140–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Grossman SA, Ye X, Lesser G, Sloan A,
Carraway H, Desideri S and Piantadosi S; NABTT CNS Consortium, :
Immunosuppression in patients with high-grade gliomas treated with
radiation and temozolomide. Clin Cancer Res. 17:5473–5480. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kim WJ, Dho YS, Ock CY, Kim JW, Choi SH,
Lee ST, Kim IH, Kim TM and Park CK: Clinical observation of
lymphopenia in patients with newly diagnosed glioblastoma. J
Neurooncol. 143:321–328. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sampson JH, Aldape KD, Archer GE, Coan A,
Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE,
McLendon RE, et al: Greater chemotherapy-induced lymphopenia
enhances tumor-specific immune responses that eliminate
EGFRvIII-expressing tumor cells in patients with glioblastoma.
Neuro Oncol. 13:324–333. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Byun HK, Kim N, Yoon HI, Kang SG, Kim SH,
Cho J, Baek JG, Chang JH and Suh CO: Clinical predictors of
radiation-induced lymphopenia in patients receiving chemoradiation
for glioblastoma: Clinical usefulness of intensity-modulated
radiotherapy in the immuno-oncology era. Radiat Oncol. 14:512019.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gwin WR III, Disis ML and Ruiz-Garcia E:
Immuno-oncology in the era of personalized medicine. Adv Exp Med
Biol. 1168:117–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Alkhalili K, Zenonos G and
Fernandez-Miranda JC: Do corticosteroids compromise survival in
glioblastoma? Neurosurgery. 79:N15–N16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Klement RJ and Champ CE: Corticosteroids
compromise survival in glioblastoma in part through their elevation
of blood glucose levels. Brain. 140:e162017.PubMed/NCBI
|
|
68
|
Sampson JH, Gunn MD, Fecci PE and Ashley
DM: Brain immunology and immunotherapy in brain tumours. Nat Rev
Cancer. 20:12–25. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Medawar PB: Immunity to homologous grafted
skin; The fate of skin homografts transplanted to the brain, to
subcutaneous tissue, and to the anterior chamber of the eye. Br J
Exp Pathol. 29:58–69. 1948.PubMed/NCBI
|
|
70
|
Zhou Q, Wang Y and Ma W: The progress of
immunotherapy for glioblastoma. Hum Vaccin Immunother.
11:2654–2658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
da Fonseca AC, Amaral R, Garcia C, Geraldo
LH, Matias D and Lima FR: Microglia in Cancer: For good or for bad?
Adv Exp Med Biol. 949:245–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bryukhovetskiy I, Manzhulo I, Mischenko P,
Milkina E, Dyuizen I, Bryukhovetskiy A and Khotimchenko Y: Cancer
stem cells and microglia in the processes of glioblastoma
multiforme invasive growth. Oncol Lett. 12:1721–1728. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Herz J, Louveau A, Da Mesquita S and
Kipnis J: Morphological and functional analysis of CNS-associated
lymphatics. Methods Mol Biol. 1846:141–151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Louveau A and Nau JY: Nervous and
lymphatic system communicate with each other. ‘Zyka virus’ unravels
its mystery. Rev Med Suisse. 11:1462–1463. 2015.(In French).
PubMed/NCBI
|
|
75
|
Lim M, Xia Y, Bettegowda C and Weller M:
Current state of immunotherapy for glioblastoma. Nat Rev Clin
Oncol. 15:422–442. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sarah C: Immunotherapy: CAR T cells in
glioblastoma. Nat Rev Drug Discov. 16:6022017. View Article : Google Scholar
|
|
77
|
Li L, Zhu X, Qian Y, Yuan X, Ding Y, Hu D,
He X and Wu Y: Chimeric antigen receptor T-cell therapy in
glioblastoma: Current and future. Front Immunol. 11:5942712020.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ahmed N, Brawley V, Hegde M, Bielamowicz
K, Kalra M, Landi D, Robertson C, Gray TL, Diouf O, Wakefield A, et
al: HER2-specific chimeric antigen receptor-modified virus-specific
T cells for progressive glioblastoma: A phase 1 dose-escalation
trial. JAMA Oncol. 3:1094–1101. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lettini G, Lepore S, Crispo F, Sisinni L,
Esposito F and Landriscina M: Heat shock proteins in cancer stem
cell maintenance: A potential therapeutic target? Histol
Histopathol. 35:25–37. 2020.PubMed/NCBI
|
|
80
|
Iglesia RP, Fernandes CFL, Coelho BP,
Prado MB, Melo Escobar MI, Almeida GHDR and Lopes MH: Heat Shock
Proteins in Glioblastoma Biology: Where Do We Stand? Int J Mol Sci.
20:57942019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ammendola M, Currò G, Memeo R, Curto LS,
Luposella M, Zuccalà V, Pessaux P, Navarra G, Gadaleta CD and
Ranieri G: Targeting stem cells with hyperthermia: Translational
relevance in cancer patients. Oncol. 98:755–762. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zanetti M: A second chance for telomerase
reverse transcriptase in anticancer immunotherapy. Nat Rev Clin
Oncol. 14:115–128. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Oji Y, Hashimoto N, Tsuboi A, Murakami Y,
Iwai M, Kagawa N, Chiba Y, Izumoto S, Elisseeva O, Ichinohasama R,
et al: Association of WT1 IgG antibody against WT1 peptide with
prolonged survival in glioblastoma multiforme patients vaccinated
with WT1 peptide. Int J Cancer. 139:1391–1401. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kijima N, Hosen N, Kagawa N, Hashimoto N,
Kinoshita M, Oji Y, Sugiyama H and Yoshimine T: Wilms' tumor 1 is
involved in tumorigenicity of glioblastoma by regulating cell
proliferation and apoptosis. Anticancer Res. 34:61–67.
2014.PubMed/NCBI
|
|
85
|
Saikali S, Avril T, Collet B, Hamlat A,
Bansard JY, Drenou B, Guegan Y and Quillien V: Expression of nine
tumour antigens in a series of human glioblastoma multiforme::
Interest of EGFRvIII, IL-13Ralpha2, gp100 and TRP-2 for
immunotherapy. J Neurooncol. 81:139–148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu G, Khong HT, Wheeler CJ, Yu JS, Black
KL and Ying H: Molecular and functional analysis of
tyrosinase-related protein (TRP)-2 as a cytotoxic T lymphocyte
target in patients with malignant glioma. J Immunother. 26:301–312.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Affinito A, Quintavalle C, Esposito CL,
Roscigno G, Giordano C, Nuzzo S, Ricci-Vitiani L, Scognamiglio I,
Minic Z, Pallini R, et al: Targeting ephrin receptor tyrosine
kinase A2 with a selective aptamer for glioblastoma stem cells. Mol
Ther Nucleic Acids. 20:176–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Qazi MA, Vora P, Venugopal C, Adams J,
Singh M, Hu A, Gorelik M, Subapanditha MK, Savage N, Yang J, et al:
Cotargeting ephrin receptor tyrosine kinases A2 and A3 in cancer
stem cells reduces growth of recurrent glioblastoma. Cancer Res.
78:5023–5037. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Tchoghandjian A, Baeza N, Colin C, Cayre
M, Metellus P, Beclin C, Ouafik L and Figarella-Branger D: A2B5
cells from human glioblastoma have cancer stem cell properties.
Brain Pathol. 20:211–221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Baeza-Kallee N, Bergès R, Soubéran A,
Colin C, Denicolaï E, Appay R, Tchoghandjian A and
Figarella-Branger D: Glycolipids recognized by A2B5 antibody
promote proliferation, migration, and clonogenicity in glioblastoma
cells. Cancers (Basel). 11:12672019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
De la Rocha AM, Sampron N, Alonso MM and
Matheu A: Role of SOX family of transcription factors in central
nervous system tumors. Am J Cancer Res. 4:312–324. 2014.PubMed/NCBI
|
|
92
|
Weathers SP, Penas-Prado M, Pei BL, Ling
X, Kassab C, Banerjee P, Bdiwi M, Shaim H, Alsuliman A, Shanley M,
et al: Glioblastoma-mediated immune dysfunction limits CMV-specific
T cells and therapeutic responses: Results from a phase I/II trial.
Clin Cancer Res. 26:3565–3577. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
De Haan P, Van Diemen FR and Toscano MG:
Viral gene delivery vectors: The next generation medicines for
immune-related diseases. Hum Vaccin Immunother. 17:14–21. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang JL, Scheitler KM, Wenger NM and Elder
JB: Viral therapies for glioblastoma and high-grade gliomas in
adults: A systematic review. Neurosurg Focus. 50:E22021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Desjardins A, Gromeier M, Herndon JE II,
Beaubier N, Bolognesi DP, Friedman AH, Friedman HS, McSherry F,
Muscat AM, Nair S, et al: Recurrent Glioblastoma treated with
Recombinant Poliovirus. N Engl J Med. 379:150–161. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Bhaduri A, Di Lullo E, Jung D, Müller S,
Crouch EE, Espinosa CS, Ozawa T, Alvarado B, Spatazza J, Cadwell
CR, et al: Outer Radial Glia-like cancer stem cells contribute to
heterogeneity of glioblastoma. Cell Stem Cell. 26:48–63.e6. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Patel AP, Tirosh I, Trombetta JJ, Shalek
AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT,
Martuza RL, et al: Single-cell RNA-seq highlights intratumoral
heterogeneity in primary glioblastoma. Science. 344:1396–1401.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xu HS, Qin XL, Zong HL, He XG and Cao L:
Cancer stem cell markers in glioblastoma-an update. Eur Rev Med
Pharmacol Sci. 21:3207–3211. 2017.PubMed/NCBI
|
|
99
|
Ludwig K and Kornblum HI: Molecular
markers in glioma. J Neurooncol. 134:505–512. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Pointer KB, Clark PA, Zorniak M, Alrfaei
BM and Kuo JS: Glioblastoma cancer stem cells: Biomarker and
therapeutic advances. Neurochem Int. 71:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Ahmed SI, Javed G, Laghari AA, Bareeqa SB,
Farrukh S, Zahid S, Samar SS and Aziz K: CD133 expression in
glioblastoma multiforme: A literature review. Cureus.
10:e34392018.PubMed/NCBI
|
|
102
|
Beier CP and Beier D: CD133 negative
cancer stem cells in glioblastoma. Front Biosci (Elite Ed).
3:701–710. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
103
|
Colwell N, Larion M, Giles AJ, Seldomridge
AN, Sizdahkhani S, Gilbert MR and Park DM: Hypoxia in the
glioblastoma microenvironment: Shaping the phenotype of cancer
stem-like cells. Neuro Oncol. 19:887–896. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bryukhovetskiy A, Shevchenko V, Kovalev S,
Chekhonin V, Baklaushev V, Bryukhovetskiy I and Zhukova M: To the
novel paradigm of proteome-based cell therapy of tumors: Through
comparative proteome mapping of tumor stem cells and
tissue-specific stem cells of humans. Cell Transplant. 23 (Suppl
1):S151–S170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bryukhovetskiy I, Shevchenko V, Arnotskaya
N, Kushnir T, Pak O, Victor Z, Zaitsev S, Khotimchenko Y,
Bryukhovetskiy A, Sharma A and Sharma HS: Transforming growth
factor-β mimics the key proteome properties of CD133-differentiated
and CD133+ cancer stem cells in glioblastoma. Int Rev
Neurobiol. 151:219–242. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Holladay FP, Heitz-Turner T, Bayer WL and
Wood GW: Autologous tumor cell vaccination combined with adoptive
cellular immunotherapy in patients with grade III/IV astrocytoma. J
Neurooncol. 27:179–189. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wood GW, Holladay FP, Turner T, Wang YY
and Chiga M: A pilot study of autologous cancer cell vaccination
and cellular immunotherapy using anti-CD3 stimulated lymphocytes in
patients with recurrent grade III/IV astrocytoma. J Neurooncol.
48:113–120. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Mitchell DA, Sayour EJ, Reap E,
Schmittling R, DeLeon G, Norberg P, Desjardins A, Friedman AH,
Friedman HS, Archer G and Sampson JH: Severe adverse immunologic
reaction in a patient with glioblastoma receiving autologous
dendritic cell vaccines combined with GM-CSF and dose-intensified
temozolomide. Cancer Immunol Res. 3:320–325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Brown CE, Alizadeh D, Starr R, Weng L,
Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J,
Simpson J, et al: Regression of glioblastoma after chimeric antigen
receptor T-cell therapy. N Engl J Med. 375:2561–2569. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Babar Khan M, Chakraborty S and Boockvar
JA: Use of chimeric antigen receptor T cells as a potential
therapeutic for glioblastoma. Neurosurgery. 80:N33–N34. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Land CA, Musich PR, Haydar D, Krenciute G
and Xie Q: Chimeric antigen receptor T-cell therapy in
glioblastoma: Charging the T cells to fight. J Transl Med.
18:4282020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Shen SH, Woroniecka K, Barbour AB, Fecci
PE, Sanchez-Perez L and Sampson JH: CAR T cells and checkpoint
inhibition for the treatment of glioblastoma. Expert Opin Biol
Ther. 20:579–591. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Salinas RD, Durgin JS and O'Rourke DM:
Potential of glioblastoma-targeted chimeric antigen receptor (CAR)
T-cell therapy. CNS Drugs. 34:127–145. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Bielamowicz K, Fousek K, Byrd TT, Samaha
H, Mukherjee M, Aware N, Wu MF, Orange JS, Sumazin P, Man TK, et
al: Trivalent CAR T cells overcome interpatient antigenic
variability in glioblastoma. Neuro Oncol. 20:506–518. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Neagu MR and Reardon DA: Rindopepimut
vaccine and bevacizumab combination therapy: Improving survival
rates in relapsed glioblastoma patients? Immunotherapy. 7:603–606.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Weller M, Butowski N, Tran DD, Recht LD,
Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al:
Rindopepimut with temozolomide for patients with newly diagnosed,
EGFRvIII-expressing glioblastoma (ACT IV): A randomised,
double-blind, international phase 3 trial. Lancet Oncol.
18:1373–1385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Elsamadicy AA, Chongsathidkiet P, Desai R,
Woroniecka K, Farber SH, Fecci PE and Sampson JH: Prospect of
rindopepimut in the treatment of glioblastoma. Expert Opin Biol
Ther. 17:507–513. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Gerstner ER: ACT IV: The final act for
rindopepimut? Lancet Oncol. 18:1294–1296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Parney IF, Chang LJ, Farr-Jones MA, Hao C,
Smylie M and Petruk KC: Technical hurdles in a pilot clinical trial
of combined B7-2 and GM-CSF immunogene therapy for glioblastomas
and melanomas. J Neurooncol. 78:71–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zaitsev S, Sharma HS, Sharma A, Manzhulo
I, Polevshchikov A, Kudriavtsev I, Khotimchenko Y, Pak O,
Bryukhovetskiy A and Bryukhovetskiy I: Pro-inflammatory
modification of cancer cells microsurroundings increases the
survival rates for rats with low differentiated malignant glioma of
brain. Int Rev Neurobiol. 151:253–279. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC,
Due-Tønnesen P, Suso EM, Sæbøe-Larssen S, Sandberg C, Brinchmann
JE, Helseth E, et al: Therapeutic vaccination against autologous
cancer stem cells with mRNA-transfected dendritic cells in patients
with glioblastoma. Cancer Immunol Immunother. 62:1499–509. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Thomas AA, Fisher JL, Ernstoff MS and
Fadul CE: Vaccine-based immunotherapy for glioblastoma. CNS Oncol.
2:331–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Cuoco JA, Benko MJ, Busch CM, Rogers CM,
Prickett JT and Marvin EA: Vaccine-Based immunotherapeutics for the
treatment of glioblastoma: Advances, challenges, and future
perspectives. World Neurosurg. 120:302–315. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lee YS and Radford KJ: The role of
dendritic cells in cancer. Int Rev Cell Mol Biol. 348:123–178.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Reardon DA and Mitchell DA: The
development of dendritic cell vaccine-based immunotherapies for
glioblastoma. Semin Immunopathol. 39:225–239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Chang CN, Huang YC, Yang DM, Kikuta K, Wei
KJ, Kubota T and Yang WK: A phase I/II clinical trial investigating
the adverse and therapeutic effects of a postoperative autologous
dendritic cell tumor vaccine in patients with malignant glioma. J
Clin Neurosci. 18:1048–1054. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bogdahn U, Hau P, Stockhammer G,
Venkataramana NK, Mahapatra AK, Suri A, Balasubramaniam A, Nair S,
Oliushine V, Parfenov V, et al: Targeted therapy for high-grade
glioma with the TGF-β2 inhibitor trabedersen: Results of a
randomized and controlled phase IIb study. Neuro Oncol. 13:132–142.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wick A, Desjardins A, Suarez C, Forsyth P,
Gueorguieva I, Burkholder T, Cleverly AL, Estrem ST, Wang S, Lahn
MM, et al: Phase 1b/2a study of galunisertib, a small molecule
inhibitor of transforming growth factor-beta receptor I, in
combination with standard temozolomide-based radiochemotherapy in
patients with newly diagnosed malignant glioma. Invest New Drugs.
38:1570–1579. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Birch JL, Coull BJ, Spender LC, Watt C,
Willison A, Syed N, Chalmers AJ, Hossain-Ibrahim MK and Inman GJ:
Multifaceted transforming growth factor-beta (TGFbeta) signalling
in glioblastoma. Cell Signal. 72:1096382020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Cannarile MA, Weisser M, Jacob W, Jegg AM,
Ries CH and Rüttinger D: Colony-stimulating factor 1 receptor
(CSF1R) inhibitors in cancer therapy. J Immunother Cancer.
5:532017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Akkari L, Bowman RL, Tessier J, Klemm F,
Handgraaf SM, de Groot M, Quail DF, Tillard L, Gadiot J, Huse JT,
et al: Dynamic changes in glioma macrophage populations after
radiotherapy reveal CSF-1R inhibition as a strategy to overcome
resistance. Sci Transl Med. 12:eaaw78432020. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wherry EJ: T cell exhaustion. Nat Immunol.
12:492–499. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ando M, Ito M, Srirat T, Kondo T and
Yoshimura A: Memory T cell, exhaustion, and tumor immunity. Immunol
Med. 43:1–9. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Mohme M, Schliffke S, Maire CL, Rünger A,
Glau L, Mende KC, Matschke J, Gehbauer C, Akyüz N, Zapf S, et al:
Immunophenotyping of newly diagnosed and recurrent glioblastoma
defines distinct immune exhaustion profiles in peripheral and
tumor-infiltrating lymphocytes. Clin Cancer Res. 24:4187–4200.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Litak J, Mazurek M, Grochowski C,
Kamieniak P and Roliński J: PD-L1/PD-1 axis in glioblastoma
multiforme. Int J Mol Sci. 20:53472019. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Bloch O, Crane CA, Kaur R, Safaee M,
Rutkowski MJ and Parsa AT: Gliomas promote immunosuppression
through induction of B7-H1 expression in tumor-associated
macrophages. Clin Cancer Res. 19:3165–3175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle
R, López-Janeiro A, Porciuncula A, Idoate MA, Inogés S, de Andrea
C, López-Diaz de Cerio A, Tejada S, et al: Neoadjuvant nivolumab
modifies the tumor immune microenvironment in resectable
glioblastoma. Nat Med. 25:470–476. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Wang X, Guo G, Guan H, Yu Y, Lu J and Yu
J: Challenges and potential of PD-1/PD-L1 checkpoint blockade
immunotherapy for glioblastoma. J Exp Clin Cancer Res. 38:872019.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Prionisti I, Bühler LH, Walker PR and
Jolivet RB: Harnessing Microglia and macrophages for the treatment
of glioblastoma. Front Pharmacol. 10:5062019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Di Tacchio M, Macas J, Weissenberger J,
Sommer K, Bähr O, Steinbach JP, Senft C, Seifert V, Glas M,
Herrlinger U, et al: Tumor vessel normalization, immunostimulatory
reprogramming, and improved survival in glioblastoma with combined
inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol Res.
7:1910–1927. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kim MM, Umemura Y and Leung D: Bevacizumab
and glioblastoma: Past, present, and future directions. Cancer J.
24:180–186. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Reardon DA, Brandes AA, Omuro A,
Mulholland P, Lim M, Wick A, Baehring J, Ahluwalia MS, Roth P, Bähr
O, et al: Effect of nivolumab vs. bevacizumab in patients with
recurrent glioblastoma: The CheckMate 143 Phase 3 randomized
clinical trial. JAMA Oncol. 6:1003–1010. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Chen C, Zuo W, Yang P and Zhang Y:
Anti-PD-1, anti-VEGF, and temozolomide therapy in a patient with
recurrent glioblastoma: A case report. J Int Med Res.
48:3000605209513952020.PubMed/NCBI
|
|
144
|
Kong Z, Wang Y and Ma W: Vaccination in
the immunotherapy of glioblastoma. Hum Vaccin Immunother.
14:255–268. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
De Felice F, Pranno N, Marampon F, Musio
D, Salducci M, Polimeni A and Tombolini V: Immune check-point in
glioblastoma multiforme. Crit Rev Oncol Hematol. 138:60–69. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Di Giacomo AM, Valente M, Covre A,
Danielli R and Maio M: Immunotherapy targeting immune
check-point(s) in brain metastases. Cytokine Growth Factor Rev.
36:33–38. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
De Felice F, Musio D, Cassese R, Gravina
GL and Tombolini V: New approaches in glioblastoma multiforme: The
potential role of immune-check point inhibitors. Curr Cancer Drug
Targets. 17:282–289. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Yang M, Oh IY, Mahanty A, Jin WL and Yoo
JS: Immunotherapy for glioblastoma: Current state, challenges, and
future perspectives. Cancers (Basel). 12:23342020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Blumenthal DT, Gorlia T, Gilbert MR, Kim
MM, Burt Nabors L, Mason WP, Hegi ME, Zhang P, Golfinopoulos V,
Perry JR, et al: Is more better? The impact of extended adjuvant
temozolomide in newly diagnosed glioblastoma: A secondary analysis
of EORTC and NRG Oncology/RTOG. Neuro Oncol. 19:1119–1126. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Bryukhovetskiy I, Bryukhovetskiy A,
Khotimchenko Y and Mischenko P: Novel cellular and post-genomic
technologies in the treatment of glioblastoma multiforme (Review).
Oncol Rep. 35:639–648. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Chen Z and Hambardzumyan D: Immune
microenvironment in glioblastoma subtypes. Front Immunol.
9:10042018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Bryukhovetskiy IS, Dyuizen IV, Shevchenko
VE, Bryukhovetskiy AS, Mischenko PV, Milkina EV and Khotimchenko
YS: Hematopoietic stem cells as a tool for the treatment of
glioblastoma multiforme. Mol Med Rep. 14:4511–4520. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Bryukhovetskiy IS, Mischenko PV, Tolok EV,
Zaitcev SV, Khotimchenko YS and Bryukhovetskiy AS: Directional
migration of adult hematopoeitic progenitors to C6 glioma in
vitro. Oncol Lett. 9:1839–1844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Milkina E, Ponomarenko A, Korneyko M,
Lyakhova I, Zayats Y, Zaitsev S, Mischenko P, Eliseikina M,
Khotimchenko Y, Shevchenko V, et al: Interaction of hematopoietic
CD34+ CD45+ stem cells and cancer cells
stimulated by TGF-β1 in a model of glioblastoma in vitro.
Oncol Rep. 40:2595–2607. 2018.PubMed/NCBI
|
|
155
|
Calvi LM and Link DC: The hematopoietic
stem cell niche in homeostasis and disease. Blood. 126:2443–2451.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Bryukhovetskiy AS, Grivtsova LY and Sharma
HS: Is the ALS a motor neuron disease or a hematopoietic stem cell
disease? Prog Brain Res. 258:381–396. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
de Laval B, Maurizio J, Kandalla PK,
Brisou G, Simonnet L, Huber C, Gimenez G, Matcovitch-Natan O,
Reinhardt S, David E, et al: C/EBPβ-dependent epigenetic memory
induces trained immunity in hematopoietic stem cells. Cell Stem
Cell. 26:657–674.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Yanada M, Takami A, Mizuno S, Mori J, Chou
T, Usuki K, Uchiyama H, Amano I, Fujii S, Miyamoto T, et al:
Autologous hematopoietic cell transplantation for acute myeloid
leukemia in adults: 25 years of experience in Japan. Int J Hematol.
111:93–102. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Arora S, Majhail NS and Liu H:
Hematopoietic progenitor cell mobilization for autologous stem cell
transplantation in multiple myeloma in contemporary Era. Clin
Lymphoma Myeloma Leuk. 19:200–205. 2019. View Article : Google Scholar : PubMed/NCBI
|