Introduction
Hepatocellular carcinoma (HCC) is the most common
type of primary liver cancer worldwide (1–3), which
is prone to metastasis, recurrence and poor prognosis (4–6). Since
HCC is an insidious tumor, most patients are diagnosed with liver
cancer at an advanced stage and only receive systemic therapies
(7). A variety of targeted drugs
have been studied and used in clinical trials, such as sorafenib,
lenvatinib and regorafenib (8–10).
However, resistance to these drugs can emerge after several months
of treatment (11). Therefore, it
is crucial to investigate the molecular mechanisms underlying HCC
occurrence and development for the treatment of HCC. Overexpression
and abnormal activation of epidermal growth factor receptor (EGFR)
is considered an important factor leading to tumorigenesis and
development of cancer, such as non-small cell lung cancer, breast
cancer and HCC (12–14). Downstream signaling pathways of EGFR
can promote biological effects, such as proliferation, migration,
angiogenesis and inhibition of apoptosis in tumor cells (15–17).
Epithelial-mesenchymal transition (EMT) is an important step in the
process of tumorigenesis and development (18,19),
which involves multiple signaling pathways that regulate gene
expression by modulating major transcription factors, thereby
promoting cell invasion and metastasis (20–23).
However, the molecular mechanism underlying EGFR-mediated EMT in
HCC is currently unclear.
Glycogen synthase kinase-3β (GSK-3β) is a
ubiquitously expressed serine/threonine protein kinase, that is
involved in the regulation of various key cellular processes,
including cell proliferation, cell survival and cell signaling
(24). GSK-3β is a key downstream
component of the PI3K/Akt pathway, and its activity can be
inhibited by the Akt-mediated phosphorylation of GSK-3β at Ser9
(25). In addition, GSK-3β can be
regulated by Wnt to participate in the EMT process (26). Snail is a zinc finger transcription
factor that regulates cellular EMT process by inhibiting E-cadherin
transcription (27). However,
whether EGFR is involved in cell EMT via the Akt/GSK-3β/Snail
pathway in HCC is currently unknown.
The present study aimed to explore the expression
and function of EGFR in HCC and to analyze the molecular mechanism
of EGFR-mediated EMT in HepG2 cells.
Materials and methods
Patients and HCC tissue specimens
A total of 40 patients who had received curative
resection or biopsies for HCC between January 2021 and January 2023
at The First Affiliated Hospital of Bengbu Medical College (Bengbu,
China) were enrolled in the present study. The First Affiliated
Hospital of Bengbu Medical College is the teaching hospital of
Anhui University of Science & Technology. The morphology of
tissue samples from 40 patients was observed and analyzed by light
microscopy, some of which did not meet the research criteria. For
example, the adjacent tissue was not paracancerous, too little
tissue was obtained or the tissue cells were not obvious in
immunohistochemical staining. Therefore, 20 pairs of cancer tissues
and adjacent tissues (2 cm away from cancerous tissues) were
selected, and were stained by immunohistochemistry and analyzed.
All clinical specimens were collected from patients after they gave
written informed consent in accordance with a protocol approved by
the Ethics Committee of Anhui University of Science &
Technology (Huainan, China). Detailed clinicopathological
characteristics of the patients are provided in Table SI. Tumors were staged according to
the AJCC Cancer Staging Manual (8th edition) (28).
Immunohistochemistry
Clinical specimens were fixed with 4%
paraformaldehyde (cat. no. BL539A; Biosharp Life Sciences) at room
temperature for 24 h, embedded in paraffin and sectioned into 4-µm
slices. Sections were deparaffinized in xylene three times (5 min
each time) and were rehydrated in a descending ethanol series.
After deparaffinization and hydrated, tissue sections were
incubated in sodium citrate antigen retrieval solution (pH 6.0;
cat. no. KGIHC001; Nanjing KeyGen Biotech Co., Ltd.) at 100°C for
30 min. After natural cooling, the slides were washed three times
with PBS. Subsequently, 3% H2O2 was added to
the sections and they were blocked with 10% goat serum (cat. no.
SP-9000; Universal SP Kit; OriGene Technologies, Inc.) at room
temperature for 10 min. The slides were then incubated with
anti-EGFR (1:50; cat. no. 4267; Cell Signaling Technology, Inc.)
and anti-phosphorylated (p)-EGFR (1:200; cat. no. 3777; Cell
Signaling Technology, Inc.) at 4°C overnight to detect specific.
Biotinylated goat anti-rabbit IgG (cat. no. SP-9000; 1:100;
Universal SP Kit; OriGene Technologies, Inc.) was then added and
incubated at room temperature for 10 min, followed by incubation
with HRP-conjugated streptavidin at room temperature for 10 min.
Diaminobenzidine chromogenic solution was added and incubated for 5
min at room temperature. The stained tissue sections were
counterstained with hematoxylin, dehydrated in a graded ethanol
series and permeabilized in xylene for 5 min. Finally, the slices
were sealed and examined under a light microscope.
Cell culture
The HepG2 liver cancer cell line (HB-8065) was
purchased from American Type Culture Collection, and was cultured
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% fetal bovine serum (Biological Industries; Sartorius
AG) at 37°C with 5% CO2 to maintain a constant cell
growth environment. The Huh7 liver cancer cell line (BFN60800691)
and HHL-5 normal liver cell line (BFN6072012687) were purchased
from BLUEFBIO, and were cultured in DMEM (Biosharp Life Sciences)
containing 10% fetal bovine serum at 37°C with 5% CO2 to
maintain a constant cell growth environment. To ensure their
identity and purity, the cells were identified using the short
tandem repeat method.
Lentivirus transduction
To knockdown the target gene EGFR (NM_005228.3), a
short hairpin (sh)EGFR lentivirus vector, which was constructed and
validated by sequencing performed by Sangon Biotech Co., Ltd., was
used. The shRNA sequences used in the present study are listed as
follows: pLVE3753,
5′-CCGGCCTCCAGAGGATGTTCAATAACTCGAGTTATTGAACATCCTCTGGAGGTTTTTTG-3′;
pLVE3754,
5′-CCGGGCTGGATGATAGACGCAGATACTCGAGTATCTGCGTCTATCATCCAGCTTTTTTG-3′;
pLVE3755,
5′-CCGGGCCACAAAGCAGTGAATTTATCTCGAGATAAATTCACTGCTTTGTGGCTTTTTTG-3′;
and negative control (NC) pLVT1, scrambled sequence:
5′-CCGGGTTCTCCGAACGTGTCACGTACTCGAGTACGTGACACGTTCGGAGAACTTTTTTG-3′.
The shEGFR was synthesized and cloned into the pMAGIC1.1 vector
(Addgene, Inc.) to construct the pMAGIC1.1-shEGFR plasmid.
Subsequently, pMAGIC1.1-shEGFR (100 µg), pCMV-dR8.9 (65 µg) and
pCMV–VSV-G (35 µg) (both Addgene, Inc.) were transfected into 293T
cells (The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences) using CalPhos™ Mammalian Transfection Kit
(cat. no. 631312; Clontech Laboratories, Inc.) for 4 h at 37°C,
after which, the medium was replaced with fresh medium. After 72 h,
the 293T cell supernatant was collected. Lentivirus particles were
obtained by purification, and the virus titers were determined.
HepG2 cells (5×104 cells/well) were seeded in 6-well plates. Based
on a multiplicity of infection value of 20, the appropriate volume
of the virus was added to the cell culture medium for infection
after culturing for 24 h at 37°C. After 24 h infection at 37°C, the
medium was replaced with fresh medium. The selection of stable cell
lines was performed with puromycin (cat. no. HY-B1743A;
MedChemExpress) at 2 µg/ml 48 h and 1 µg/ml puromycin was used for
maintenance. Western blotting was used to screen the most effective
cells for subsequent experiments.
Western blotting
Adherent HepG2 cells treated with or without 100
ng/ml EGF (cat. no. GMP-10605-HNAE; Sino Biological, Inc.) or 100
ng/ml EGF + 3 µM MK-2206 (cat. no. HY-108232; MedChemExpress) at
37°C for 24 h were washed with PBS and lysed with RIPA lysis buffer
(Nanjing KeyGen Biotech Co., Ltd.) containing protease inhibitors.
Subsequently, proteins (25 µg/lane), quantified using the BCA
method, were separated by SDS-PAGE on 10% gels and were then
transferred to PVDF membranes, before being blocked with 5% skim
milk for 1 h at room temperature. The membranes were then incubated
with the following primary antibodies at 4°C overnight to detect
specific proteins: EGFR (cat. no. 4267), p-EGFR (Tyr1068) (cat. no.
3777), Akt (cat. no. 9272), GSK-3β (cat. no. 9315), p-GSK-3β (Ser9)
(cat. no. 5558), E-cadherin (cat. no. 3195), Vimentin (cat. no.
5741), Snail (cat. no. 3879), β-actin (cat. no. 4970) (all 1:1,000;
Cell Signaling Technology, Inc.), p-Akt (Ser473) (cat. no. 4060;
1:2,000; Cell Signaling Technology, Inc.), MMP2 (cat. no. ab92536),
MMP9 (cat. no. ab76003) (both 1:500; Abcam). An HRP-conjugated
anti-rabbit IgG secondary antibody (cat. no. 7074; 1:3,000; Cell
Signaling Technology, Inc.) was then applied at room temperature
for 1 h. An automatic gel imaging analysis system (JS-M6P; Shanghai
Peiqing Science & Technology Co., Ltd.) was used to analyze the
target proteins and the gray values of the bands were
semi-quantified with ImageJ V1.8.0 software (National Institutes of
Health).
Immunocytochemistry
HHL-5, HepG2 and Huh7 cells (2.5×104 cells/well)
were seeded in 24-well plates containing cover slips and the cells
were fixed with 4% paraformaldehyde for 10 min at room temperature
after adherence. Endogenous peroxidase was blocked with 3%
H2O2 for 15 min at room temperature and 10%
goat serum (cat. no. SP-9000; Universal SP Kit; OriGene
Technologies, Inc.) was applied for 10–15 min at room temperature.
Subsequently, anti-EGFR (cat. no. 4267; 1:50; Cell Signaling
Technology, Inc.) was added and incubated overnight at 4°C, after
which, a biotinylated goat anti-rabbit IgG secondary antibody
polymer (cat. no. SP-9000; 1:100; Universal SP Kit; OriGene
Technologies, Inc.) was added and incubated at room temperature for
10–15 min. HRP-conjugated streptavidin working solution was then
added and incubated at room temperature for 10–15 min. Finally,
diaminobenzidine chromogenic solution was added and incubated at
room temperature for 5 min, cells were counterstained with
hematoxylin and were observed under a light microscope.
Indirect immunofluorescence
HHL-5, HepG2 and Huh7 cells (2.5×104 cells/well)
were seeded in 24-well plates containing cover slips, and the cells
were fixed with 4% paraformaldehyde for 10 min at room temperature
after adherence. After cells were blocked with 10% goat serum (cat.
no. SP-9000; Universal SP Kit; OriGene Technologies, Inc.) for
10–15 min at room temperature, the following primary antibodies
were added and incubated overnight at 4°C: EGFR (cat. no. 4267;
1:50; Cell Signaling Technology, Inc.), E-cadherin (cat. no. 3195;
1:1,600; Cell Signaling Technology, Inc.) and Vimentin (cat. no.
5741; 1:200; Cell Signaling Technology, Inc.). Subsequently, an
Alexa Fluor® 488-labeled secondary anti-rabbit IgG
antibody (1:250; cat. no. 4412; Cell Signaling Technology, Inc.)
was added and incubated at 37°C for 30 min. After DAPI
counterstaining, the slides were sealed and observed under a
fluorescence microscope, and images were captured.
EdU proliferation assay
HepG2 cells (5×104 cells/well) were seeded in
24-well plates and treated with or without 100 ng/ml EGF for 24 h
at 37°C or 100 ng/ml EGF + 3 µM MK-2206 for 24 h at 37°C before
being replaced with medium containing EdU (10 µM; EdU-594 cell
proliferation detection kit; Beyotime Institute of Biotechnology).
The medium was removed after continued incubation for 2 h, and the
cells were fixed with 4% paraformaldehyde for 15 min at room
temperature and washed three times with PBS. Triton X-100 was used
for permeabilization for 15 min at room temperature, and the slides
were washed three times with PBS. Click reaction solution was then
added and incubated for 30 min at room temperature in the dark.
Hoechst-33342 staining was performed for 10 min at room
temperature. The cells were then observed under an inverted
fluorescence microscope, images were captured and the cell
proliferation rate was calculated according to the red fluorescence
ratio.
Colony formation assay
HepG2 cells were seeded in 6-well plates (1,000
cells/well), treated with 100 ng/ml EGF for 2 weeks at 37°C and the
medium was discarded after 2 weeks. After washing, the samples were
fixed with 4% paraformaldehyde for 15 min at room temperature and
further stained with 0.1% crystal violet for 10 min at room
temperature. Cell proliferation was determined by visually counting
the number of cell colonies in the sample. Each colony was 0.3–1 mm
in size.
CCK-8 assay
HepG2 cells (4×103 cells/well) were inoculated into
96-well plates, and after treatment with 100 ng/ml EGF for 1, 2, 3
and 4 days at 37°C, 10 µl CCK-8 solution (Beyotime Institute of
Biotechnology) was added and incubated at 37°C for 2 h. Absorbance
was measured at 450 nm and cell viability was calculated using
GraphPad Prism 8 software (Dotmatics).
Wound healing assay
HepG2 cells (2.5×105 cells/well) were seeded in a
12-well plate. After 24 h, cells grew to ~100% confluence and a
10-µl pipette tip was used to generate a scratch. After washing
three times with PBS, the medium was replaced with serum-free
medium containing 100 ng/ml EGF for 48 h at 37°C or 100 ng/ml EGF +
3 µM MK-2206 for 48 h at 37°C. Images were captured under a light
microscope at 0 and 48 h after scratching. Wound healing rate (%)
was calculated as follows: (0 h wound area −48 h wound area)/0 h
wound area ×100.
Transwell migration assay
HepG2 cells were pretreated with 100 ng/ml EGF for 6
h at 37°C. A total of 3×105 cells/ml resuspended in 100 µl
serum-free RPMI-1640 medium were added to the upper chamber of a
Transwell system (pore size, 8 µm), and 600 µl RPMI-1640 medium
containing 10% fetal bovine serum was added to the lower chamber.
After 48 h at 37°C, non-migrating cells in the upper chamber were
wiped away and the cells were fixed with 4% paraformaldehyde for 10
min at room temperature, stained with 0.5% crystal violet for 10
min at room temperature and observed under a light microscope, and
images were captured. The number of migratory cells was
recorded.
Statistical analysis
All experiments were repeated at least three times
and measured in three independent experiments. Data are presented
as the mean ± SD and GraphPad Prism 8 was used for statistical
analysis. Differences between two groups were compared using a
paired Student's t-test, and differences between three or more
groups were compared using one-way ANOVA followed by Bonferroni's
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
EGFR is highly expressed in HCC
tissues and cells
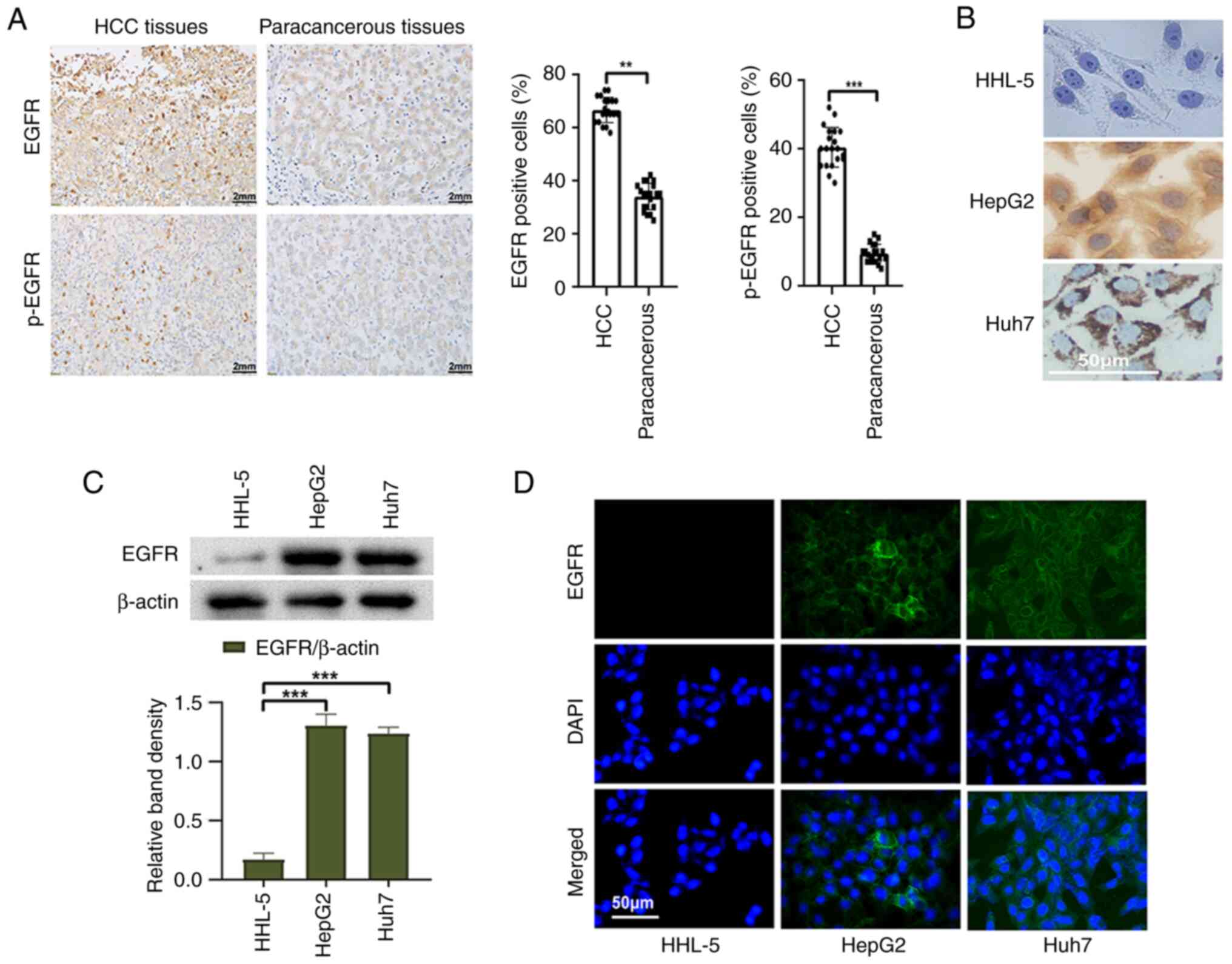
To detect the expression of EGFR in HCC tissues and
cells, a series of experiments were conducted. First, 20 HCC
tissues and corresponding paracancerous tissues were selected for
immunohistochemical staining. The positive rates of EGFR and p-EGFR
in HCC tissues were significantly higher than those in
paracancerous tissues (Fig. 1A).
Furthermore, the expression of EGFR in liver cancer cells and
normal liver cells was examined using western blot analysis. The
results revealed a significantly higher expression of EGFR in liver
cancer cells compared with those in normal liver cells (Fig. 1C). Immunocytochemistry and
immunofluorescence analyses provided additional confirmation of
these results (Fig. 1B and D).
These results serve as evidence for the high expression of EGFR in
liver cancer cells and tissues.
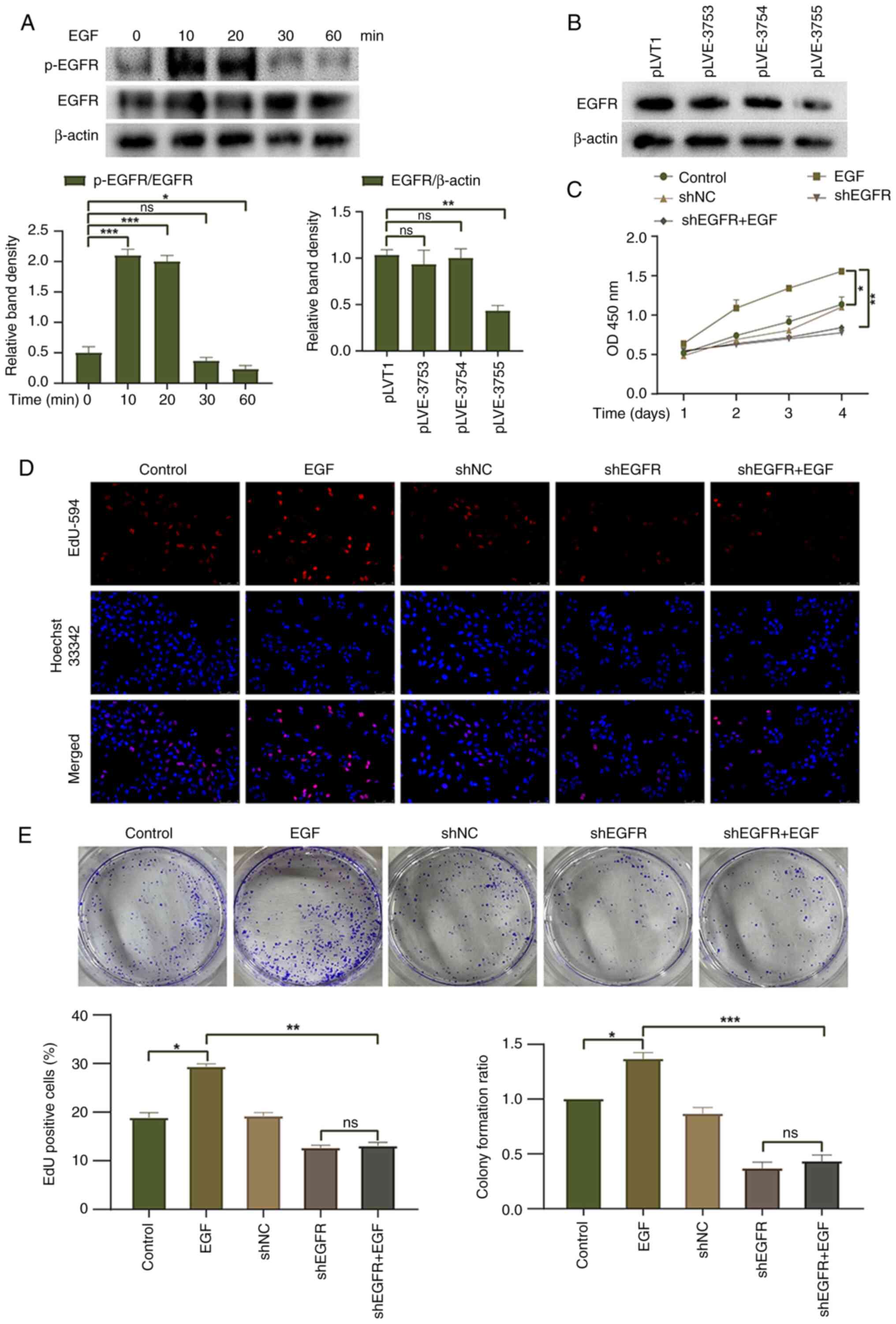
Knockdown of EGFR reduces HepG2 cell
proliferation
To investigate the biological effects of EGFR on
HepG2 liver cancer cells, EGFR activation was induced in HepG2
cells following treatment with EGF (100 ng/ml). The peak level of
EGFR phosphorylation was reached 10 min after EGF treatment and
gradually decreased thereafter (Fig.
2A). Furthermore, an EGFR knockdown lentivirus was transduced
into HepG2 cells. Western blotting showed that pLVE-3755 had the
best knockdown effect on EGFR and was thus used for subsequent
experiments (Fig. 2B). CCK-8, EdU
and colony formation assays were used to evaluate the proliferation
and colony-forming ability of HepG2 cells, respectively. The EGF
group exhibited increased cell proliferation and clonogenic ability
compared with the control group, suggesting that EGFR activation
promotes cell proliferation (Fig.
2C-E). EGF treatment did not enhance the cell proliferation or
clonogenic ability of HepG2 cells with EGFR knockdown, and there
was no significant difference observed between the shEGFR group and
the shEGFR + EGF group. To further validate the role of EGFR
activation in HepG2 cells, the EGF group was compared with the
shEGFR + EGF group. The findings revealed that EGFR knockdown had a
significant inhibitory effect on the proliferation and clonogenic
ability of HepG2 cells. These results indicated that EGFR
activation may promote the proliferation of HepG2 cells, whereas
downregulation of EGFR expression exerts an inhibitory effect on
the proliferation of HepG2 cells.
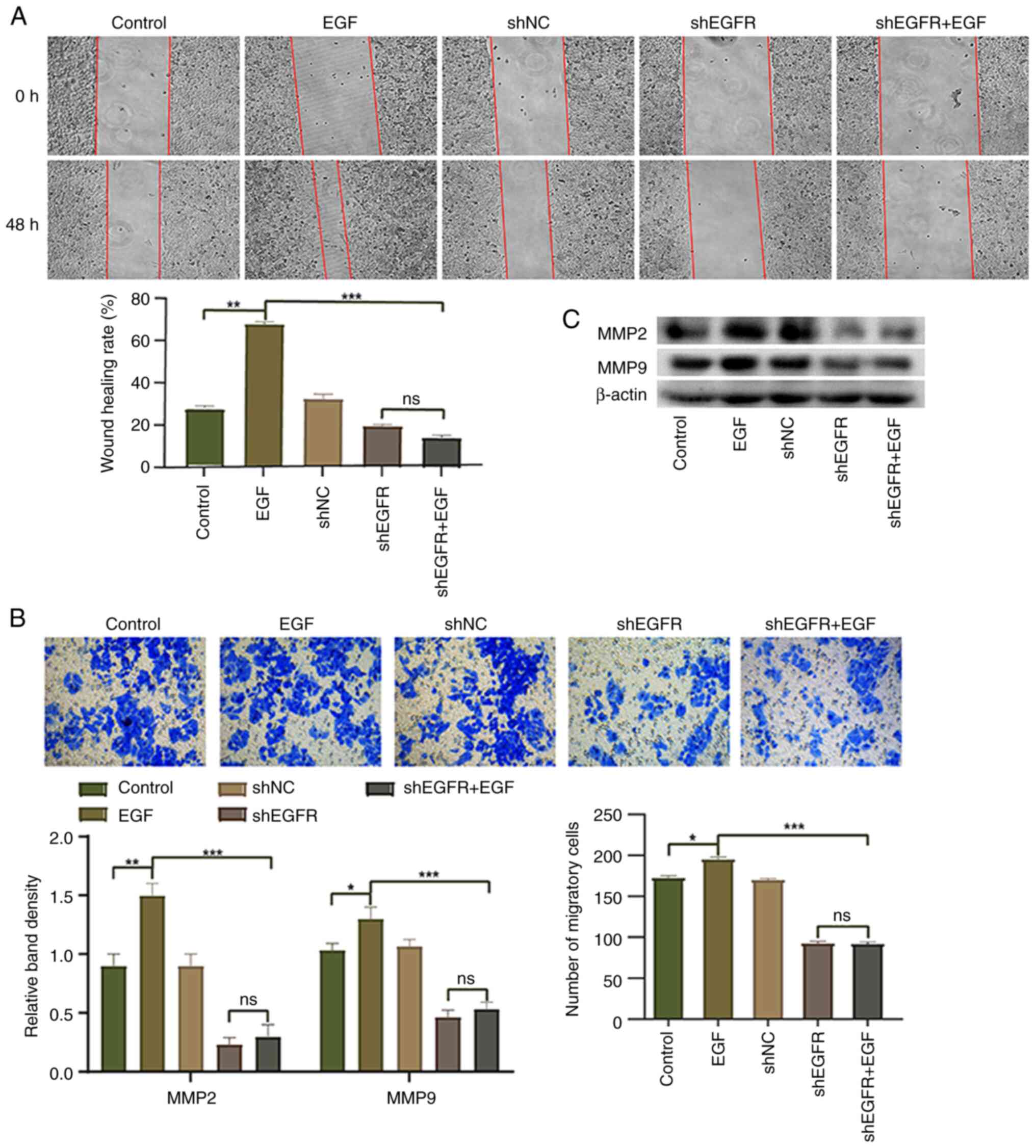
Knockdown of EGFR reduces HepG2 cell
migration
EGFR has been reported to serve an important role in
cell migration and invasion (29).
Therefore, the present study conducted wound healing and Transwell
assays to investigate the effect of EGFR knockdown on the migration
of HepG2 cells. Compared with in the control group, the migration
of HepG2 cells in the EGF group was significantly upregulated,
whereas the migration of HepG2 cells in the shEGFR + EGF group was
significantly inhibited (Fig. 3A and
B). Western blotting was performed to detect the expression
levels of MMP9 and MMP2, and the expression levels of MMP9 and MMP2
were increased in the EGF group compared with the control group,
whereas the expression levels of these proteins were significantly
decreased in the shEGFR + EGF group (Fig. 3C). These results suggested that EGFR
activation induced by EGF may increase cell migration, whereas
downregulation of EGFR expression can decrease the migration of
HepG2 cells.
Knockdown of EGFR partially reverses
HepG2 cell EMT
To investigate whether EGFR regulates the EMT
process of HepG2 cells, the present study analyzed the expression
of EMT markers in HepG2 cells. The results of western blotting and
indirect immunofluorescence showed that compared with the control
group, the expression levels of E-cadherin were significantly
decreased, whereas those of vimentin were significantly increased
in the EGF group, which is a typical feature of the EMT process
(Fig. 4). Compared with those in
the EGF group, the expression levels of E-cadherin were
significantly increased and those of vimentin were significantly
decreased in the shEGFR + EGF group. These results indicated that
EGFR may promote the EMT process of HepG2 cells, thereby promoting
HepG2 cell migration and invasion.
EGFR mediates HepG2 cell EMT through
the Akt/GSK-3β/Snail signaling pathway
The Akt/GSK-3β/Snail pathway has been reported to be
involved in the EMT process (30);
however, it is unclear whether EGFR mediates the EMT of HepG2 cells
through this pathway. The present study conducted western blot
analysis and revealed that compared with the control group, the
phosphorylation of Akt and GSK-3β, and the expression levels of
Snail were significantly increased in the EGF group, whereas the
expression levels of AKT and GSK-3β were not significantly altered
(Fig. 5C). By contrast, the
phosphorylation of Akt and GSK-3β, and the expression levels of
Snail were significantly decreased in the shEGFR + EGF group
compared with the EGF group. Additionally, following treatment with
the Akt inhibitor MK-2206, compared with the EGF group, the
phosphorylation of Akt and GSK-3β, and the expression levels of
Snail and vimentin were decreased, whereas the expression levels of
E-cadherin were increased (Fig.
5D). Moreover, the proliferation and migration of HepG2 cells
were significantly decreased in response to MK-2206 treatment
compared with those in the EGF group (Fig. 5A and B). These results indicated
that EGFR regulates the EMT process of HepG2 cells by modulating
the Akt/GSK-3β/Snail pathway, affecting cell proliferation,
migration and EMT marker expression.
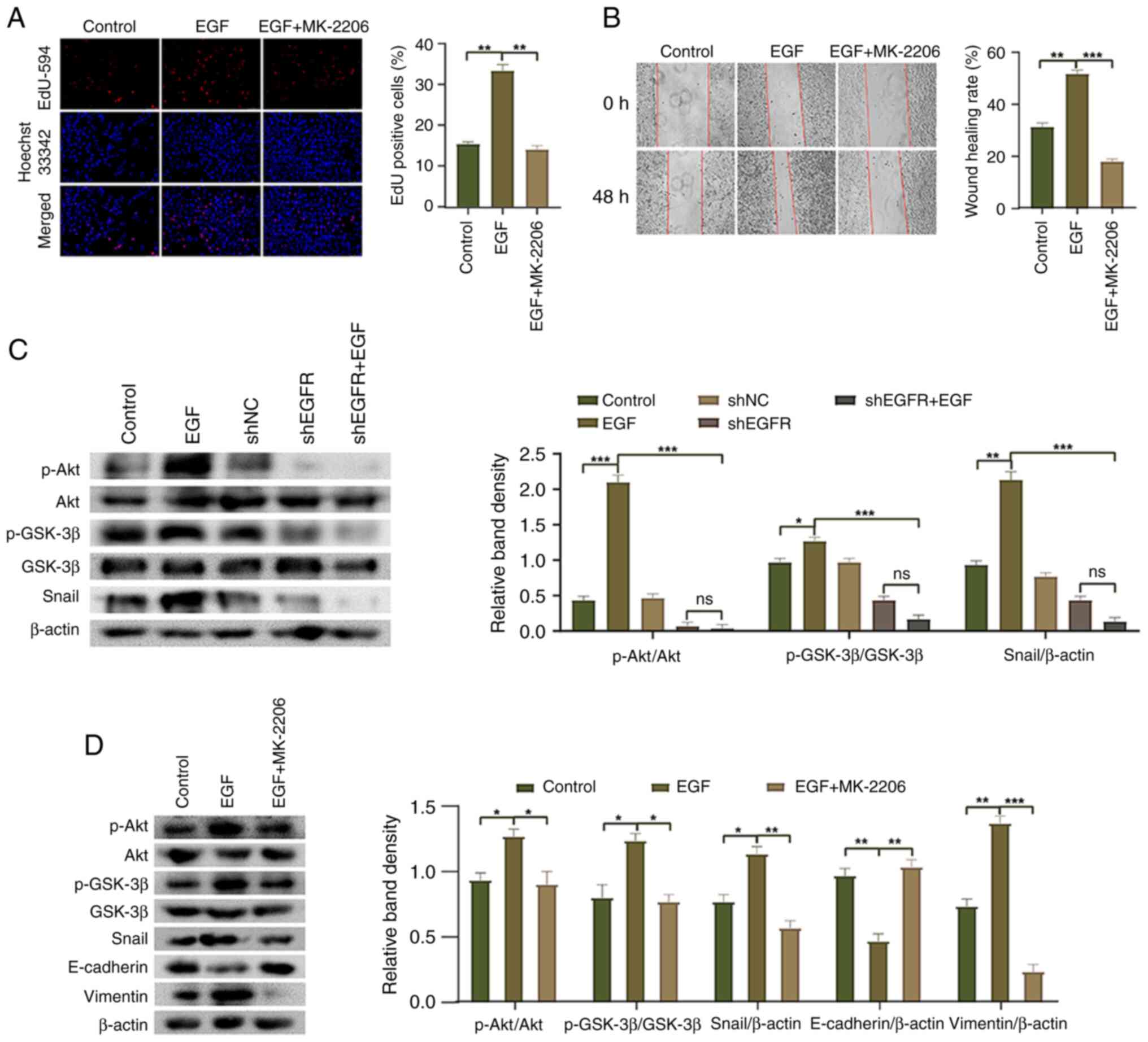 | Figure 5.Molecular mechanism underlying
EGFR-mediated epithelial-mesenchymal transition in HepG2 cells. (A)
EdU-594 assay was used to detect the proliferation of HepG2 cells
(magnification, ×200). (B) Wound healing assay was used to detect
the migration of HepG2 cells (magnification, ×100). (C) Western
blotting was used to detect the phosphorylation of Akt and GSK-3β,
and the expression of Snail in HepG2 cells. (D) Western blotting
was used to detect the phosphorylation of Akt and GSK-3β, and the
expression levels of Snail, E-cadherin and Vimentin in HepG2 cells
after MK-2206 treatment. *P<0.05, **P<0.01 and ***P<0.001.
EGFR, epidermal growth factor receptor; NC, negative control; ns,
not significant; p-, phosphorylated; sh, short hairpin. |
Discussion
The present study aimed to determine the molecular
mechanism by which EGFR mediates EMT to promote the progression of
liver cancer. The results revealed that EGFR mediated EMT through
the Akt/GSK-3β/Snail signaling pathway, thereby promoting cell
proliferation and migration. EGFR and p-EGFR were highly expressed
in HCC tissues, and knockdown of EGFR significantly inhibited the
proliferation and migration of HepG2 cells.
EMT is the process by which epithelial cells acquire
mesenchymal cell phenotypes, which is an important step in tumor
development and metastasis (31).
One of the markers of EMT is the loss of E-cadherin function
(32); notably, Snail can strongly
inhibit the expression of the E-cadherin protein (33). EMT is regulated by various signaling
pathways, including receptor tyrosine kinases, transforming growth
factor-β and STAT3 (34). In the
present study, treatment with EGF to activate EGFR led to a
decrease in E-cadherin expression and an increase in vimentin
expression in HepG2 cells. The opposite results were obtained in
response to EGFR knockdown, thus indicating that EGFR can promote
EMT. Investigations into the molecular mechanism underlying
EGFR-mediated EMT indicated that activation of EGFR promoted the
phosphorylation of Akt and GSK-3β, whereas knocking down EGFR
inhibited the phosphorylation of Akt and GSK-3β. Notably, the
present study revealed that activation of EGFR also promoted the
expression of the downstream cytoplasmic transcription factor
Snail. The effects of EGF treatment were reversed following EGFR
knockdown. Moreover, the Akt inhibitor (MK-2206) inhibited the
phosphorylation of Akt and GSK-3β, decreased Snail expression, and
partially reversed the cell EMT phenotype, thus reducing cell
migration and proliferation. These findings indicated that EGF/EGFR
may be involved in regulating the EMT of HCC through the
Akt/GSK-3β/Snail signaling pathway, promoting proliferation and
migration.
Although previous studies have reported the effect
of EGFR on EMT in HCC (35,36), to the best of our knowledge, the
mechanism by which EGFR mediates EMT through the Akt/GSK-3β/Snail
signaling pathway in HCC has not been reported. The present study
revealed that the HepG2 cell EMT phenotype could be reversed, and
the proliferation and migration of HepG2 cells could be reduced, by
inhibiting the Akt/GSK-3β/Snail signaling pathway. However, a
number of factors are involved in the progression of HCC;
therefore, further research is required to better define the
mechanisms underlying the occurrence of HCC and to identify
possible treatment methods.
In conclusion, the present study revealed that
EGF/EGFR can mediate EMT through the Akt/GSK-3β/Snail signaling
pathway, thus promoting the proliferation and migration of HepG2
cells. Inhibiting EGFR activation may partially reverse the EMT
phenotype, thus inhibiting the proliferation and migration of HepG2
cells. This knowledge may lead to innovative approaches for the
treatment of liver cancer.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Dr Huaiyong Gan
(Department of Pathology, The First Affiliated Hospital of Bengbu
Medical College) for his work in clinical tumor tissue sample
collection.
Funding
This work was supported by grants from the 2022 Graduate
Innovation Fund Project of Anhui University of Science &
Technology (grant no. 2022CX2144), the National Natural Science
Fund of China (grant nos. 82071862 and 81872017), and the Medical
Special Cultivation Project of Anhui University of Science &
Technology (grant no. YZ2023H1A005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JG, ZH, XS, QS, WR and XH designed and performed the
research. SZ and XT analyzed data and drafted the manuscript. JG
and XT confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Anhui University of Science & Technology (approval no.
2021005). The study obtained the written informed consent of all
participants.
Patient consent for publication
Patients provided written informed consent for
publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fujimoto A, Totoki Y, Abe T, Boroevich KA,
Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al:
Whole-genome sequencing of liver cancers identifies etiological
influences on mutation patterns and recurrent mutations in
chromatin regulators. Nat Genet. 44:760–764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dat VHX, Nhung BTH, Chau NNB, Cuong PH,
Hieu VD, Linh NTM and Quoc NB: Identification of potential microRNA
groups for the diagnosis of hepatocellular carcinoma (HCC) using
microarray datasets and bioinformatics tools. Heliyon.
8:e089872022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo Y, Ren Y, Dong X, Kan X and Zheng C:
An overview of hepatocellular carcinoma after insufficient
radiofrequency ablation. J Hepatocell Carcinoma. 9:343–355. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pfister D, Núñez NG, Pinyol R, Govaere O,
Pinter M, Szydlowska M, Gupta R, Qiu M, Deczkowska A, Weiner A, et
al: NASH limits anti-tumour surveillance in immunotherapy-treated
HCC. Nature. 592:450–456. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Steeg PS and Theodorescu D: Metastasis: A
therapeutic target for cancer. Nat Clin Pract Oncol. 5:206–219.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang Y, Liang J, Cao N, Gao J, Xie Y,
Zhou S and Tang X: ASIC1α up-regulates MMP-2/9 expression to
enhance mobility and proliferation of liver cancer cells via the
PI3K/AKT/mTOR pathway. BMC Cancer. 22:7782022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang C, Zhang H, Zhang L, Zhu AX, Bernards
R, Qin W and Wang C: Evolving therapeutic landscape of advanced
hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol.
20:203–222. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Llovet JM, Montal R, Sia D and Finn RS:
Molecular therapies and precision medicine for hepatocellular
carcinoma. Nat Rev Clin Oncol. 15:599–616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu
Z, Li Q, Luo M, Liang R and Ye J: Molecular targeted and immune
checkpoint therapy for advanced hepatocellular carcinoma. J Exp
Clin Cancer Res. 38:4472019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faivre S, Rimassa L and Finn RS: Molecular
therapies for HCC: Looking outside the box. J Hepatol. 72:342–352.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang A, Yang XR, Chung WY, Dennison AR
and Zhou J: Targeted therapy for hepatocellular carcinoma. Signal
Transduct Target Ther. 5:1462020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lanaya H, Natarajan A, Komposch K, Li L,
Amberg N, Chen L, Wculek SK, Hammer M, Zenz R, Peck-Radosavljevic
M, et al: EGFR has a tumour-promoting role in liver macrophages
during hepatocellular carcinoma formation. Nat Cell Biol.
16:972–977. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z: ErbB Receptors and Cancer. Methods
Mol Biol. 1652:3–35. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thompson SM, Jondal DE, Butters KA,
Knudsen BE, Anderson JL, Stokes MP, Jia X, Grande JP, Roberts LR,
Callstrom MR and Woodrum DA: Heat stress induced,
ligand-independent MET and EGFR signalling in hepatocellular
carcinoma. Int J Hyperthermia. 34:812–823. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reynolds AR, Tischer C, Verveer PJ, Rocks
O and Bastiaens PI: EGFR activation coupled to inhibition of
tyrosine phosphatases causes lateral signal propagation. Nat Cell
Biol. 5:447–453. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Niu J, Li W, Liang C, Wang X, Yao X, Yang
RH, Zhang ZS, Liu HF, Liu FY, Pei SH, et al: EGF promotes DKK1
transcription in hepatocellular carcinoma by enhancing the
phosphorylation and acetylation of histone H3. Sci Signal.
13:eabb57272020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma Y, Xu R, Liu X, Zhang Y, Song L, Cai S,
Zhou S, Xie Y, Li A, Cao W and Tang X: LY3214996 relieves acquired
resistance to sorafenib in hepatocellular carcinoma cells. Int J
Med Sci. 18:1456–1464. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krebs AM, Mitschke J, Lasierra Losada M,
Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D,
Reichardt W, Bronsert P, et al: The EMT-activator Zeb1 is a key
factor for cell plasticity and promotes metastasis in pancreatic
cancer. Nat Cell Biol. 19:518–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang WH, Lan HY, Huang CH, Tai SK, Tzeng
CH, Kao SY, Wu KJ, Hung MC and Yang MH: RAC1 activation mediates
Twist1-induced cancer cell migration. Nat Cell Biol. 14:366–374.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yeh HW, Hsu EC, Lee SS, Lang YD, Lin YC,
Chang CY, Lee SY, Gu DL, Shih JH, Ho CM, et al: PSPC1 mediates
TGF-β1 autocrine signalling and Smad2/3 target switching to promote
EMT, stemness and metastasis. Nat Cell Biol. 20:479–491. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin J, Song T, Li C and Mao W: GSK-3β in
DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy
of cancer. Biochim Biophys Acta Mol Cell Res. 1867:1186592020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pecoraro C, Faggion B, Balboni B, Carbone
D, Peters GJ, Diana P, Assaraf YG and Giovannetti E: GSK3β as a
novel promising target to overcome chemoresistance in pancreatic
cancer. Drug Resist Updat. 58:1007792021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lv Q, Wang J, Xu C, Huang X, Ruan Z and
Dai Y: Pirfenidone alleviates pulmonary fibrosis in vitro and in
vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3
signaling pathways. Mol Med. 26:492020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Amin MB, Edge S, Greene F, Byrd DR,
Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR,
Sullivan DC, et al: AJCC Cancer Staging Manual. 8th edition.
Springer International Publishing; Cham, Switzerland: pp.
2912017
|
|
29
|
Liu X, Adorno-Cruz V, Chang YF, Jia Y,
Kawaguchi M, Dashzeveg NK, Taftaf R, Ramos EK, Schuster EJ,
El-Shennawy L, et al: EGFR inhibition blocks cancer stem cell
clustering and lung metastasis of triple negative breast cancer.
Theranostics. 11:6632–6643. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Y, Cao N, Gao J, Liang J, Liang Y,
Xie Y, Zhou S and Tang X: ASIC1a stimulates the resistance of human
hepatocellular carcinoma by promoting EMT via the AKT/GSK3β/Snail
pathway driven by TGFβ/Smad signals. J Cell Mol Med. 26:2777–2792.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Babaei G, Aziz SG and Jaghi NZZ: EMT,
cancer stem cells and autophagy; The three main axes of metastasis.
Biomed Pharmacother. 133:1109092021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Montanari M, Rossetti S, Cavaliere C,
D'Aniello C, Malzone MG, Vanacore D, Di Franco R, La Mantia E,
Iovane G, Piscitelli R, et al: Epithelial-mesenchymal transition in
prostate cancer: An overview. Oncotarget. 8:35376–35389. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Giannelli G, Koudelkova P, Dituri F and
Mikulits W: Role of epithelial to mesenchymal transition in
hepatocellular carcinoma. J Hepatol. 65:798–808. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang B, Liu T, Wu JC, Luo SZ, Chen R, Lu
LG and Xu MY: STAT3 aggravates TGF-β1-induced hepatic
epithelial-to-mesenchymal transition and migration. Biomed
Pharmacother. 98:214–221. 2018. View Article : Google Scholar : PubMed/NCBI
|