Introduction
Gastric cancer (GC) is the fifth most frequently
diagnosed cancer and the third highest cause of cancer-related
deaths around the world (1).
Diagnosis often occurs in advanced or metastatic stages, and the
primary treatment strategy is chemotherapy based on the combination
of platinum drugs and 5-fluorouracil (5-FU) (2). Acting as a pyrimidine antagonist, 5-FU
inhibits DNA replication by competing with uracil for binding to
thymidylate synthase (TS) (3). The
clinical application of 5-FU is limited by the development of
chemoresistance after chemotherapy (4,5).
Molecular mechanisms involved in chemoresistance to 5-FU include
increased DNA damage repair, regulation of membrane drug
transporters, dysregulation of transcription factors (e.g.,
overexpression of EIF5A2, FOXM1 and GPC4, or downregulation of
RanBPM and TFAP2C) and tumor microenvironment (TME) factors such as
cancer-associated fibroblast (CAFs), endothelial-1 and aquaporin 1
(5). The TME comprises CAFs,
mesenchymal cells and immune components, such as tumor-associated
macrophages (TAM), which secrete cytokines and chemokines that
contribute to chemoresistance (6).
In this context, chemokine receptors, such as C-C motif chemokine
receptor 3 (CCR3), are possible therapeutic targets. CCR3 has G
protein associated transmembrane regions and is mainly expressed by
eosinophils and other immune cells (7). CCR3 can bind numerous ligands with
activator functions [chemokine (C-C motif) ligand (CCL) 3, 4, 5, 7,
13, 15, 23, 24, 26 and 28] and others as antagonists (CCL9, 10,
18), as well as CCL11 which has a dual function (7). Several of these ligands have been
reported to be associated with chemoresistance in cancer, including
CCL5, CCL11 and CCL15 (6), and our
previous study reported overexpression of CCL5 in
cisplatin-resistant gastric cancer cells (8). Furthermore, CCR3 expression has been
reported in certain cancers such as renal (9), colon (10) and, head and neck (11). In patients with GC, CCR3 expression
has been previously reported in peripheral blood CD4+ lymphocytes
(12). There are several CCR3
antagonists/inhibitors, including SB 297006 and SB 328437. SB
328437 is a potent and highly selective inhibitor of CCR3 and has
been reported to have attenuated spontaneous chronic colitis in
mice, reducing the number of eosinophils and regulatory molecules
in the colon (13). Furthermore, SB
328437 has been reported to have reversed resistance to pazopanib
and inhibited lung metastasis in a renal clear cell carcinoma model
(14). Computational biology has
contributed to the development of understanding of the interaction
between chemokines and their ligands. X-ray crystallography
techniques have revealed the three-dimensional configurations of
several chemokines, including CCR5, CCR2 and CCR3 (15). Although variations in the quaternary
structure have been observed, the monomeric unit, probably the key
functional form of CCR2 and CCR3 (15), remains conserved. Although the
effect of SB 328437 has been reported at the cellular level
(16), the specific interaction of
SB 328437 with CCR3 and its associated with resistance to 5-FU in
GC has not been elucidated. The present study evaluated the effect
of CCR3 receptor inhibition using the antagonist SB 328437 and
assessed the molecular dynamics of this interaction on resistance
to 5-FU in GC cells. In this study, a novel 5-FU-resistant gastric
cancer cell line was established and validated through viability
assays and TS gene-targeted RT-qPCR. The impact of CCR3 receptor
inhibition by SB 328437 and its molecular dynamism on 5-FU
resistance were investigated. Bioinformatics analysis utilized a
closely related CCR5 structure to predict CCR3 binding site,
refined within a stable membrane environment, and assessed ligand
binding stability and receptor conformational changes.
Materials and methods
Chemicals
The chemotherapeutic agent, 5-FU was purchased from
Selleck Chemicals and reconstituted at a concentration of 3.3 mM in
DMSO. The CCR3 antagonist, SB 328437, was purchased from
MedChemExpress and reconstituted at a concentration of 10 mM in
DMSO.
Cells and culture
The gastric adenocarcinoma AGS cell line was
purchased from the European Collection of Authenticated Cell
Cultures. The mycoplasma-free status of the parental cell line was
confirmed using the LookOut Mycoplasma PCR Detection Kit
(Sigma-Aldrich; Merck KGaA). Cells were grown in RPMI-1640 medium
supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher
Scientific, Inc.) and 1% (v/v) penicillin and streptomycin (Thermo
Fisher Scientific, Inc.) and maintained at 37°C in a 95% humidified
atmosphere and 5% CO2 conditions. Cells were subcultured
at 70–80% confluence and harvested after treatment with 0.25%
trypsin and 0.02% EDTA (Corning, Inc.).
Development of 5-FU resistant AGS
cells
AGS parental cells were used to establish
5-FU-resistant AGS cells (AGS R-5FU) by stepwise increases in 5-FU
drug doses according to the method previously reported by Coley
(17). Briefly, the starting
treatment dose was set at 20% of the half maximal effective
concentration (EC50) of the parental cells (17). Drug doses were gradually increased
until an arbitrarily defined resistance index (RI) ≥2 was reached.
RI values were calculated by dividing the EC50 values of
resistant cells by the EC50 values of parental cells.
Once the cells acquired 5-FU resistance, they were grown in a
drug-free medium for two weeks, frozen in liquid nitrogen and then
awakened in a medium containing 5-FU to confirm the level of
chemoresistance.
RNA extraction and quantitative
analysis
The mRNA expression levels of TS, a molecular marker
involved in 5-FU resistance (4) and
CCR3, were quantified using reverse transcription-quantitative
(RT-q) PCR. Total RNA was extracted from ~2.0×106 cells
using TRIzol (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. RNA concentration was determined using
the Infinite® NanoQuant spectrophotometer (Tecan Group,
Ltd.), and integrity was evaluated by measuring RNA 260/280
absorbance ratio and gel electrophoresis. The RNA was then treated
with DNase I (Promega Corporation) at 37°C for 30 min and
pre-incubated with 500 µg/ml random primers (Promega Corporation),
followed by denaturation at 70°C for 10 min. The first-strand
complimentary cDNA was prepared from 1 µg of RNA in a total
reaction volume of 20 µl using 25 mM dNTPs (Promega Corporation),
RNasin ribonuclease inhibitor (Promega Corporation), 200 U/µl M-MLV
reverse transcriptase (Promega Corporation) and M-MLV reverse
transcriptase 5X buffer (Promega Corporation) at 37°C for 45 min.
Subsequently, cDNA was amplified by qPCR using Brilliant II
Ultra-Fast SYBR® Green qPCR Master Mix according to the
manufacturer's protocol, using the Stratagene Mx-3000p real-time
PCR system (Agilent Technologies, Inc.). The RT-qPCR included an
initial denaturation step at 95°C for 10 min to ensure DNA
denaturation. Amplification comprised 40 cycles, consisting of
denaturation at 95°C for 15 sec, annealing at 60°C for 30 sec and
extension at 72°C for 30 sec. Subsequently, a melting curve was
performed to assess amplification specificity. The initial
temperature was 95°C for 15 sec, followed by a stage at 55°C for 1
min, with a gradual temperature increase of 0.15°C per sec until
95°C was reached. Relative mRNA expression levels were determined
using the 2−ΔΔCq method (18), using ACTB as the reference gene.
Primer sequences used in the present study are presented in
Table SI.
Cell viability assays
The viability of cells treated with 5-FU and/or SB
328437 was performed using a standard viability assay (MTT-formazan
assay). Briefly, 4×103 cells were seeded in 96-well
plates in 100 µl of culture medium and incubated at 37°C for 24 h
to allow cell attachment. Cells were exposed to the treatment
agents for 72 h at different pharmacological concentrations:
firstly, 0.01 to 1,000 µM 5-FU was used for the determination of
EC50 values and then, these values were used alone or in
combination with 50 µM SB 328437 to evaluate the effects of SB
328437 on resistance to 5-FU. Cells treated with DMSO (vehicle)
were used as controls. After 72 h of incubation, the culture medium
was removed and the cells were washed with 100 µl Dulbecco's
PBS/Modified (Thermo Fisher Scientific, Inc.) and were subsequently
treated with MTT at 0.5 mg/ml, followed incubation at 37°C for 2 h.
Absorbance was measured at 570 nm wavelength using an Infinite
NanoQuant spectrophotometer (Tecan Group, Inc.).
Protein preparation
The CCR3 crystal structure contains seven
transmembrane α-helices and an eighth α-helix in the intracellular
domain. The initial structure of CCR3 was obtained from the Protein
Data Bank (PDB) (19) database (PDB
ID, 7X9Y). The membrane environment, composed of
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, was generated
using the CHARMM-GUI server v3.8 (2–4). The
CHARMM force field parameters for SB328437 were acquired from the
‘Ligand Reader and Model’ tool on the CHARMM-GUI web server
(20–22). The system underwent restraint in the
xyz plane with a harmonic potential gradient, starting from 4,000
kJ/mol/nm2 in the initial NVT equilibration step and
reducing to 50 kJ/mol/nm2 in the final NPT equilibration
step. Subsequently, production runs were conducted without any
restraint for a duration of 100 ns.
Docking methodology
Autodock 4.2 (23)
was used to model docking in silico, employing the
Lamarckian genetic algorithm. The docking site was chosen based on
the relative position of well-studied CCR receptor structures,
following the CCR5-Maraviroc crystallographic structure (PDB:
4MBS). This docking region, situated closer to the extracellular
loops (ECL1, ECL2 and ECL3), was selected, as it has been reported
that binding sites for other molecules are typically located in
this region (24). The docking
active site was treated as a rigid molecule, while ligands were
considered flexible, with all non-ring torsions deemed active.
Cluster analysis was conducted after the docking experiment to
identify the global minimum conformation of SB 328437 in the
potential CCR3 binding site. The ligand poses with all-atoms root
mean square deviation <0.1 nm, were clustered, ranked by their
lowest docking energy and the representative binding mode was
selected. The 10 poses with lowest energy were chosen for further
investigation.
Simulation parameters
Molecular dynamics (MD) simulations were executed
using GROMACS software v.2019 (25)
under periodic boundary conditions. The minimization step comprised
50,000 steps with an emtol of 100 kJ/mol/nm. Equilibration steps
were divided into NVT and NPT equilibration, with temperature
coupling to T=310 K using the Berendsen thermostat. Pressure
coupling utilized the Berendsen barostat with a semi-isotropic
coupling scheme. Non-bonded interactions were computed as
Lennard-Jones potentials, and electrostatics were calculated as
Coulomb interactions. For the production run, electrostatic and
non-bonded interactions were computed using the equilibration
method, but temperature and pressure coupling schemes were changed
to Nose-Hoover and Parrinello-Rahman, respectively. LINCS
constraints for H-bonds were applied at each step, and a time step
of 2 fsec was used.
Principal component analysis (PCA)
calculation
PCA was performed using GROMACS as previously
described (26). Briefly, GROMACS
utilities gmx_covar and gmx_anaeig were used for PCA of CCR3-Alone
and CCR3-SB328437 complexes based on 100 ns MD simulations. The
first two eigenvectors with the largest eigenvalues were used to
create a 2D projection of independent trajectories to better
understand the conformational behavior pattern.
Image rendering
The Visual Molecular Dynamics (VMD) software package
(23) was used to extract and
calculate the data used for image rendering and the Protein Imager
webserver was used to render the data (27).
Statistical analysis
All experiments were performed in biological and
technical triplicates for each condition. Data were analyzed using
GraphPad Prism 10.0.3 (Dotmatics). EC50 values were
calculated from dose-response curves using non-linear regression.
RT-qPCR data were analyzed using the Mann-Whitney U test and the
Kruskal-Wallis test with Dunn's post-hoc test. Cell viability
assays were analyzed using the Kruskal-Wallis test with Dunn's
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference. Mean values are presented with a 95%
confidence interval.
Results
Establishment of 5-FU-resistant AGS
cells
Dose-response curves of parental and chemo-resistant
AGS cells are presented (Fig. 1A).
The dose-response curves allowed EC50 values to be
obtained for AGS WT and AGS R-5FU cell lines. The EC50
value for AGS R-5FU was 24.8 µM ±0.75, which represented a 2.6-fold
resistance index (RI) compared with the AGS WT (EC50=9.7
µM ±0.23). In addition, a significant increase in the relative
expression of TS (P<0.05) was observed in AGS R-5FU cells
compared with AGS WT (Fig. 1B).
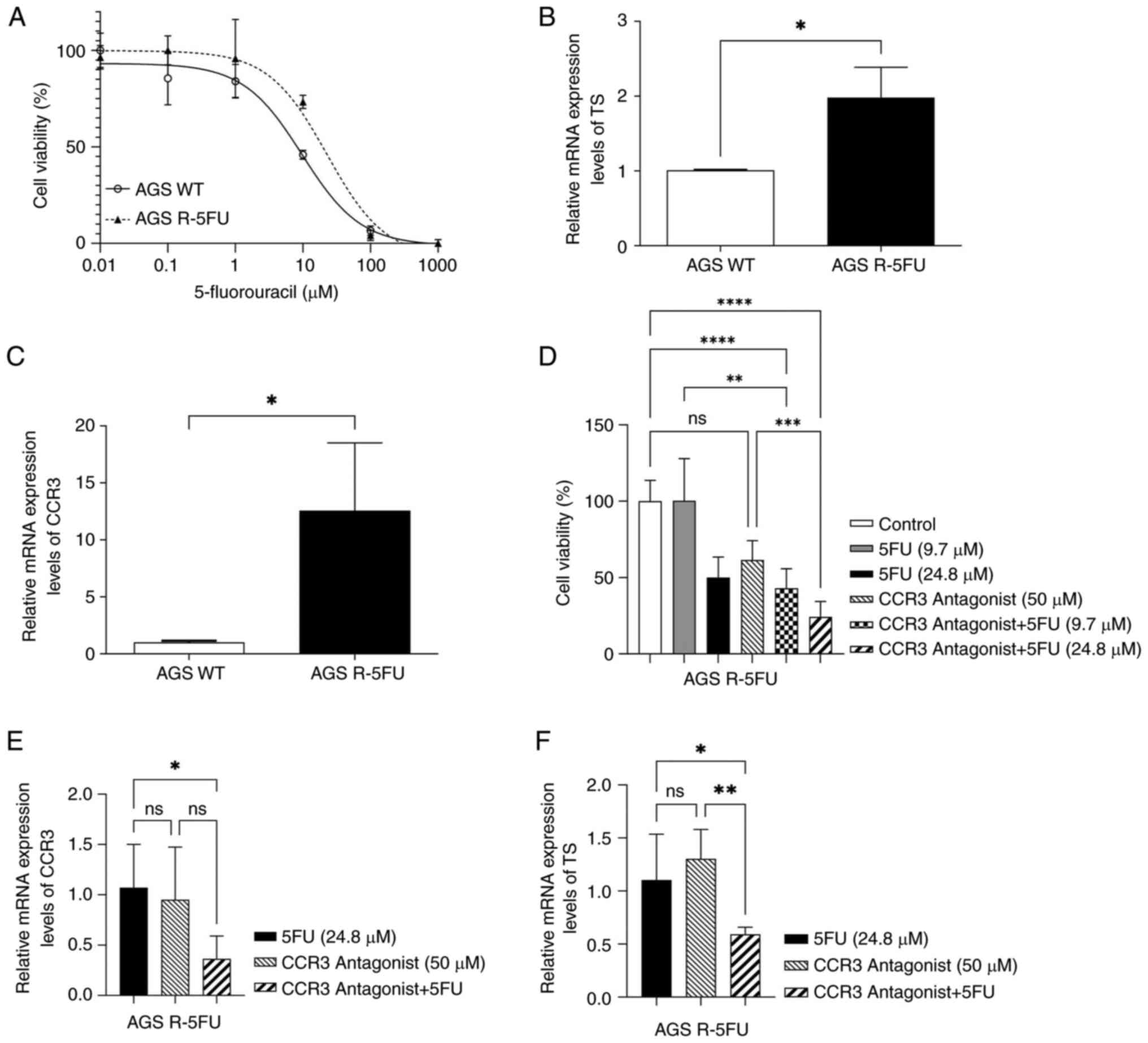 | Figure 1.In vitro assays. (A)
Dose-response curves for AGS R-5FU and AGS WT. mRNA expression
levels of (B) TS and (C) CCR3 in AGS R-5FU and AGS WT. (D) Cell
viability of AGS R-5FU cells treated with 5-FU and/or CCR3
antagonist, SB 328437, for 72 h. mRNA expression levels of (E) CCR3
and (F) TS after treatment with SB 328437 and/or 5-FU. Data were
expressed as mean ± standard deviation of three biological
replicates. *P<0.05, **P<0.01, ***P<0.001 and
****P<0.0001. AGS WT, wild type AGS cells; 5FU, 5-fluorouracil;
AGS R-5FU, 5-FU resistant AGS cells; TS, thymidylate synthase;
CCR3, C-C motif chemokine receptor 3; ns, not significant. |
CCR3 is overexpressed in
5-FU-resistant AGS cells
RT-qPCR was used to evaluate the transcriptional
expression of CCR3 in AGS R-5FU and AGS WT cells (Fig. 1C). This demonstrated that CCR3 mRNA
expression levels were significantly higher in AGS R-5FU cells
compared with AGS WT cells (P<0.05).
CCR3 antagonist in combination with
5-FU decreases cell viability and the relative expression of CCR3
and TS in 5-FU-resistant AGS cells
MTT assays were performed in AGS R-5FU cells treated
with the CCR3 antagonist, SB 328437, and/or 5-FU to evaluate their
effect on cell viability (Fig. 1D).
No significant differences in cell viability were demonstrated
between cells treated with SB 328437 and control cells. However, a
substantial decrease in cell viability was observed when SB 328437
was combined with 5-FU; cells treated with SB 328437 combined with
9.7 µM 5-FU demonstrated a significant reduction in cell viability
compared with control cells (P<0.0001) and cells treated with
9.7 µM of 5-FU only (P<0.01). Similarly, when SB 328437 was
combined with 24.8 µM 5-FU, cell viability was significantly
reduced when compared with control cells (P<0.0001), and cells
treated with SB 328437 only (P<0.001).
The transcriptional expression of CCR3 and TS in AGS
R-5FU cells treated for 72 h with SB 328437 and/or 5-FU. There were
no significant differences in the relative expression of CCR3
between cells treated with SB 328437 and those treated with 24.8 µM
5-FU. However, when SB 328437 was combined with 5-FU, a significant
decrease in the mRNA expression level of CCR3 was demonstrated
compared with the 5-FU group (P<0.05; Fig. 1E). Similarly, a significant
reduction in the relative expression of TS was observed in cells
treated with the SB 328437/5-FU combination compared to those
exposed to 5-FU (P<0.05) or SB 328437 (P<0.01) alone
(Fig. 1F).
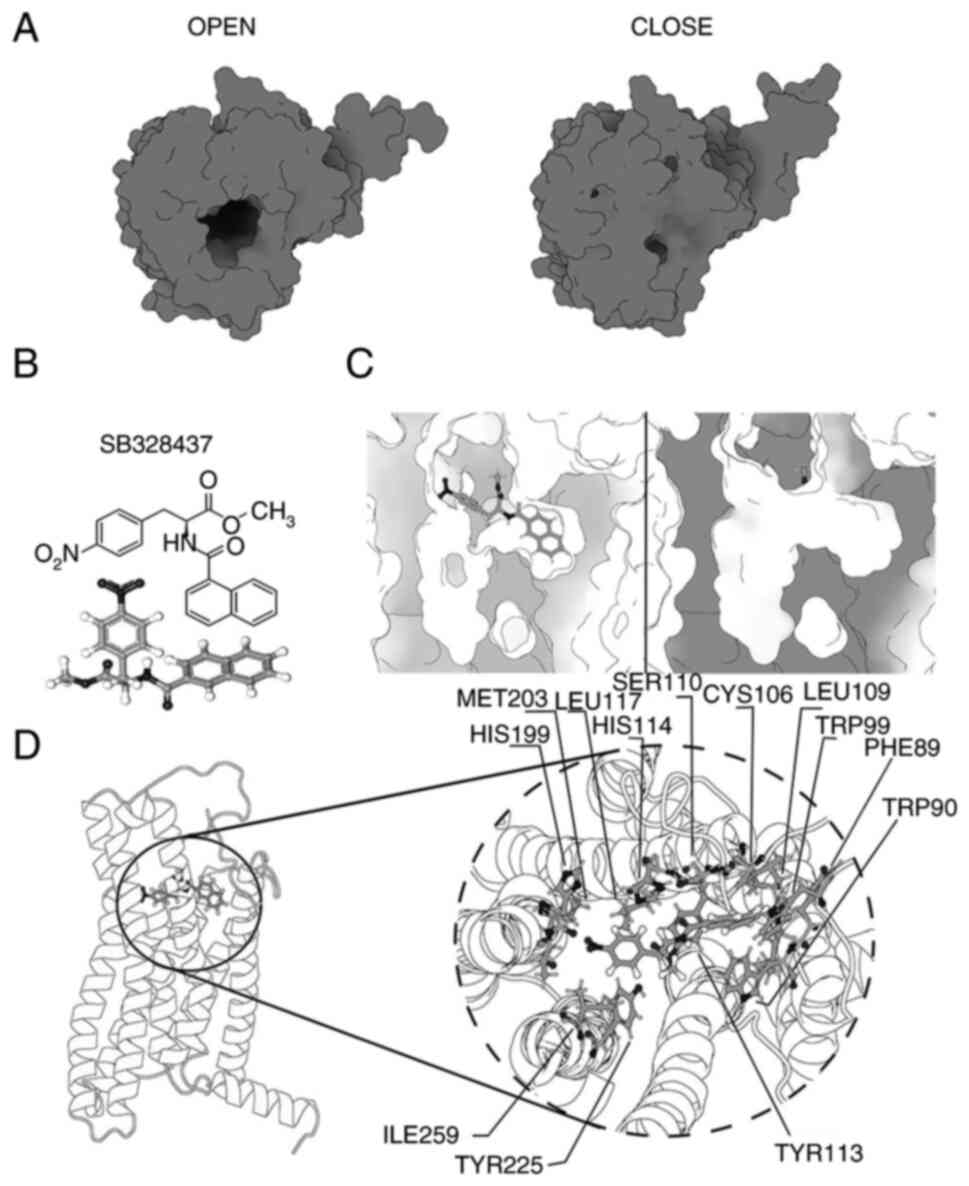
Changes in the structure of wild-type
CCR in the open/close states
To propose transition states, it is necessary to
identify cyclical changes in the system. These opening and closing
states can be observed as transitional microstates between the
opening of the binding pocket and its closure. The conformational
differences between the opening and closing of the binding pocket
were presented (Fig. 2A).
The receptor transition cycle, between opening and
closing, appears to be related to the proximity of the amino acids
which belong to the N-terminal region to the ECL2 loop, with a role
also indicated for ECL3 and TM5. For the structure of CCR3 to
change to its completely closed state, amino acids N271 and Y193
must be oriented toward each other which results in their
interaction; this interaction generates a displacement of the
extracellular ECL3 region towards the binding pocket of CCR3
functioning as a gate, which closes the possibility of interaction
with any ligand. However, for the ‘closed’ transition microstate to
be maintained over time, the Y193-N271 interaction must be
supported by another region of CCR3, the N-terminal region. For a
‘closed’ microstate to be fully achieved it is necessary for the
N-terminal region of CCR3, characterized by its alanine residue, to
be oriented toward the extracellular ECL2 loop. This orientation
establishes an interaction between A31 and E181, blocking the
capacity of the N-terminal region as ‘ligand recognition’, which is
known to be the region responsible for the identification of
possible ligands.
SB 328437 binds to the allosteric site
of CCR3 and blocks the entrance of endogenous ligands
To understand the interaction between CCR3 and SB
328437, a docking, guided specifically for the allosteric region of
CCR3, was performed to visualize a normal ligand relocation at the
specific site (Fig. 2B). During
each of the molecular dynamics performed the SB 328437 ligand was
positioned within the allosteric site of CCR3. CCR3 could be
characterized by 2 binding pockets, site A where the recognition
site for CCL3 or any CCR3 ligand is located and site B (where SB
328437 is bound), which would correspond to the allosteric site
(Fig. 2C). Given the conformational
shift of ECL2 that follows its contact and anchoring to the CCR3
allosteric site-which is strikingly similar to the previously
proposed change in the closed microstate-this binding has notable
implications for the potential impact of SB 328437 on CCR3.
The major conformational change visualized in CCR3
following the binding of SB 328437 is found in the ECL2 region,
which is repositioned, closing the passage to the N-terminal region
and the binding pocket, blocking the recognition of any ligand.
This conformational change occurs after ~50 nsec and is
characterized by the interaction between the ligand and, the ECL2
and N-terminal regions. This interaction occurs through a punctual
interaction of cysteine 183, which belongs to the ECL2 region, and
leucine 32, which belongs to the N-terminal region. Both residues
are oriented towards the ligand, initiating the conformational
change, blocking ligand recognition, and closing the receptor
opening.
SB 328437 possesses 2 aromatic rings and as such, it
needs a highly hydrophobic binding site to interact with CCR3. This
is demonstrated in the internal region of CCR3 which can be
characterized by the hydrophobic residues A31, L32, L87, C106,
L109, S110, C183, I259, S262 and S263, which can be seen to be
distributed around the binding site and so can be expected to
support the interaction of the binding site with the ligand.
Another feature of SB 328437 is the large number of atoms that can
act as proton donors or acceptors, these attract residues such as
T86, F89, W90, W99, Y113 and T200; interactions which hold the
ligand in place and ultimately affect the structure of CCR3. The
formation of an environment that makes possible the interaction,
understood as recognition and stabilization of the ligand inside
the binding pockets, is possible by the composition of highly
hydrophobic residues that generate a favorable environment,
supported lately by the interaction between the ligand and the
charged residues. Even if these residues promote a favorable
environment for ligand stabilization, specific residues generate
strong interactions with the ligand and keep the structure
stabilized, which can be seen in Fig.
2D.
Discussion
Although 5-FU is the chemotherapy treatment of first
choice for advanced GC, its effectiveness is limited by
chemoresistance (28). In the
present study, a new 5-FU-resistant GC cell line (AGS R-5FU) was
developed, the RI of which, as well as the overexpression of the TS
gene, made it possible to confirm the acquisition of resistance.
This study was initiated with two 5FU-resistant gastric cancer cell
lines, AGS R-5FU and MKN-28 R-5FU. However, as it was not possible
to confirm the overexpression of TS in MKN-28 R-5FU, its use was
discontinued. Consequently, the utilization of only a single
resistant cell line represents a limitation of the present
study.
As 5-FU exerts its anticancer effects through the
inhibition of TS and causing misincorporation of bases into DNA and
RNA, overexpression of TS is considered the main molecular
mechanism of resistance to 5-FU (4). CCR3, a chemokine receptor expressed by
eosinophils and other immune cells, has been identified in
peripheral blood CD4+ lymphocytes of patients with GC.
This clinical study reveals a positive regulatory relationship
between the expression of CCR5 and CCR3, indicating a complex
interaction between these molecules in the immune response linked
to cancer progression (12).
Furthermore, some of the ligands of CCR3, such as CCL5, have been
reported to be associated with chemoresistance (6,8).
However, the inhibition of CCR3 by the antagonist SB 328437 has
been reported to reverse resistance to pazopanib (14). The inhibition of CCR3 in
5-FU-resistant GC has not been previously assessed. Chemokines are
membrane proteins expressed in low quantities and as such, their
detection by techniques such as western blotting is limited.
Therefore, the present study focused on the transcriptional
expression of CCR3. CCR3 was overexpressed in AGS R-5FU, and its
inhibition using CCR3 antagonist, SB 328437, combined with 5-FU,
triggered sensitization to 5-FU, decreasing cell viability. Jöhrer
et al (9) proposed that CCR3
expression might facilitate proliferative responses of tumor cells
and ligands of CCR3 in renal cell carcinoma. As CCR3 actives
signaling pathways, such as MAPK and JAK/STAT, and affects
migration, cell growth/differentiation and apoptosis (29), the inhibition of CCR3 could explain
the decrease in cell viability. It was also observed that SB 328437
alone did not significantly decrease the transcriptional expression
of CCR3 or the cell viability of AGS R-5FU cells. However, when SB
328437 was combined with 5-FU, the mRNA expression levels of CCR3
and cell viability both significantly decreased.
To the best of our knowledge, no previous studies
have assessed the association of the combination of SB 328437 and
5-FU in drug chemoresistance. The present study evaluated whether
this combination also influenced the transcriptional expression of
TS. The results indicated that SB 328437 combined with 5-FU
significantly decreased the expression of TS. As TS overexpression
is a hallmark of chemoresistance to 5-FU, its inhibition served to
confirm the reduction in levels of chemoresistance. The potential
importance of these sensitization results necessitated the
description of the mechanism of interaction between SB 328437 and
CCR3, and so a computational biology analysis was performed.
Computational biology techniques support the prediction and
proposal of mechanisms of action for structures that have not yet
been resolved, such as the interactions of chemokines with
different ligands, as is the case of CCR3. Similar studies have
been previously reported for CCR5, one of the most studied
chemokines, which regulates the trafficking and functions of immune
cells (30). In-silico
analysis has shown that the upstream region of CCR5 and the
extracellular loop ECL2 were identified as critical in the
interaction of Maraviroc with relevant chemokine ligands (31). In the present study, a structural
analysis was performed of the behavior of CCR3 with SB 328437, a
ligand already known to be a selective receptor antagonist, but
which lacked a description of its mechanism of interaction. The
behavior of the CCR3-SB 328437 complex indicated a clear tendency
toward conformational change that caused receptor blockade and
promoted the displacement of the N-terminal region, relevant for
ligand recognition, disabling the ligand recognition function of
CCR3. In research related to CCRs, the functioning of CCR3
receptors has been described following the classical behavior of
2-site models. These refer to the relationship between an
allosteric site and an orthosteric site in the recognition and
modulation of conformational changes for the activation or
inhibition of a structure (15).
CCR3 is not exempt from this mechanism; based on the
in-silico modelling of the interaction of CCR3 with SB
328437, the receptor changed conformation between open and closed,
rendering the N-terminal region unable to exert its ligand
recognition effect. The relevance of the N-terminal region has been
described for most CCRs, identifying it as one of the most relevant
and functionally conserved regions for this type of receptor
(32). In addition to the relevance
of the N-terminal region, similar results have been reported in
relation to the relevance of ECL2 and ECL3 in ligand recognition by
CCR3 (33). Both regions were
modified by the effect of SB 328437 in the present study. Given the
ability of CCR3 to be modulated through different recognition
sites, the demonstrated of an allosteric activation is to be
expected. Such allosteric modulation has been described by other
researchers, who propose that in addition to allosteric modulation
at the binding site, cholesterol has the effect of being an
affinity modulator to different ligands (34,35).
Thus, describing the possible allosteric binding site of SB 328437
allows us to progressively approach a comprehensive understanding
of the mechanism of CCR3 activation. The results of the
in-silico modeling interaction of CCR3 and SB 328437 support
those of another investigation of CCR3 antagonists at the
allosteric site (36), highlighting
hydrophobic residues, which provide the correct environment for SB
328437 to interact with CCR3 and remain stable.
The present study demonstrated the impact of CCR3 on
chemoresistance to 5-FU in GC, emphasizing the potential
therapeutic effect of SB 328437. The structural changes observed
suggest a novel approach to sensitizing cancer cells by targeting
CCR3, presenting opportunities for further exploration in cancer
therapy. Future studies to clarify the synergistic interaction
between SB 328437 and 5-FU should be conducted using both in
vitro and in vivo assays. This interaction should also
be confirmed with the use of other CCR3 inhibitors, which would
provide a more comprehensive perspective on the role of this
possible therapeutic alternative in reducing drug resistance in
GC.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mr. Javier Retamal
(Laboratory of Integrative Biology, Universidad de La Frontera) for
their advice on the experimental design..
Funding
This work was supported by the National Research and Development
Agency through the National Doctoral Scholarship (grant no.
21222011), FONDECYT Postdoctorado (grant no. 3210629), FONDECYT
Regular (grant no. 1210440) and FONDEF Idea (grant no. ID21I10027)
grants. This work was also supported by the Millennium Institute on
Immunology and Immunotherapy IMII (grant nos. ICN09_016/ICN
2021_045; former P09/016-F) and CORFO BMRC, Biomedical research
consortium-Chile.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
AG and BML confirm the authenticity of all the raw
data. AG, MER, CR, PB and BML designed the study. AG and BM-L
conducted experiments. AG, MER, CR, PB and BML analyzed the data,
designed the figures and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ychou M, Boige V, Pignon JP, Conroy T,
Bouché O, Lebreton G, Ducourtieux M, Bedenne L, Fabre JM,
Saint-Aubert B, et al: Perioperative chemotherapy compared with
surgery alone for resectable gastroesophageal adenocarcinoma: An
FNCLCC and FFCD multicenter phase III trial. J Clin Oncol.
29:1715–1721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang N, Yin Y, Xu SJ and Chen WS:
5-Fluorouracil: Mechanisms of resistance and reversal strategies.
Molecules. 13:1551–1569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sethy C and Kundu CN: 5-Fluorouracil
(5-FU) resistance and the new strategy to enhance the sensitivity
against cancer: Implication of DNA repair inhibition. Biomed
Pharmacother. 137:1112852021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reyes ME, de La Fuente M, Hermoso M, Ili
CG and Brebi P: Role of CC chemokines subfamily in the platinum
drugs resistance promotion in cancer. Front Immunol. 11:9012020.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zajkowska M and Mroczko B: Eotaxins and
their receptor in colorectal cancer-a literature review. Cancers
(Basel). 12:13832020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mora-Lagos B, Cartas-Espinel I, Riquelme
I, Parker AC, Piccolo SR, Viscarra T, Reyes ME, Zanella L,
Buchegger K, Ili C and Brebi P: Functional and transcriptomic
characterization of cisplatin-resistant AGS and MKN-28 gastric
cancer cell lines. PLoS One. 15:e02283312020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jöhrer K, Zelle-Rieser C, Perathoner A,
Moser P, Hager M, Ramoner R, Gander H, Höltl L, Bartsch G, Greil R
and Thurnher M: Up-regulation of functional chemokine receptor CCR3
in human renal cell carcinoma. Clin Cancer Res. 11:2459–2465. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee YS, Kim SY, Song SJ, Hong HK, Lee Y,
Oh BY, Lee WY and Cho YB: Crosstalk between CCL7 and CCR3 promotes
metastasis of colon cancer cells via ERK-JNK signaling pathways.
Oncotarget. 7:36842–36853. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang WY, Lin YS, Lin YC, Nieh S, Chang
YM, Lee TY, Chen SF and Yang KD: Cancer-associated fibroblasts
promote tumor aggressiveness in head and neck cancer through
chemokine ligand 11 and C-C motif chemokine receptor 3 signaling
circuit. Cancers (Basel). 14:31412022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Andalib A, Doulabi H, Hasheminia S, Maracy
M and Rezaei A: CCR3, CCR4, CCR5, and CXCR3 expression in
peripheral blood CD4+ lymphocytes in gastric cancer patients. Adv
Biomed Res. 2:312013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Filippone RT, Dargahi N, Eri R, Uranga JA,
Bornstein JC, Apostolopoulos V and Nurgali K: Potent CCR3 receptor
antagonist, SB328437, suppresses colonic eosinophil chemotaxis and
inflammation in the winnie murine model of spontaneous chronic
colitis. Int J Mol Sci. 23:77802022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang C, Wang Y, Hong T, Cheng B, Gan S,
Chen L, Zhang J, Zuo L, Li J and Cui X: Blocking the autocrine
regulatory loop of Gankyrin/STAT3/CCL24/CCR3 impairs the
progression and pazopanib resistance of clear cell renal cell
carcinoma. Cell Death Dis. 11:1172020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shao Z, Tan Y, Shen Q, Hou L, Yao B, Qin
J, Xu P, Mao C, Chen LN, Zhang H, et al: Molecular insights into
ligand recognition and activation of chemokine receptors CCR2 and
CCR3. Cell Discov. 8:442022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
White JR, Lee JM, Dede K, Imburgia CS,
Jurewicz AJ, Chan G, Fornwald JA, Dhanak D, Christmann LT, Darcy
MG, et al: Identification of potent, selective non-peptide CC
chemokine receptor-3 antagonist that inhibits eotaxin-, eotaxin-2-,
and monocyte chemotactic protein-4-induced eosinophil migration. J
Biol Chem. 275:36626–36631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coley HM: Development of drug-resistant
models. Methods Mol Med. 88:267–274. 2004.PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Berman HM, Westbrook J, Feng Z, Gilliland
G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE: The protein
data bank. Nucleic Acids Res. 28:235–242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jo S, Kim T, Iyer VG and Im W: CHARMM-GUI:
A web-based graphical user interface for CHARMM. J Comput Chem.
29:1859–1865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brooks BR, Brooks CL III, Mackerell AD Jr,
Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C,
Boresch S, et al: CHARMM: The biomolecular simulation program. J
Comput Chem. 30:1545–1614. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu EL, Cheng X, Jo S, Rui H, Song KC,
Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et
al: CHARMM-GUI membrane builder toward realistic biological
membrane simulations. J Comput Chem. 35:1997–2004. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morris GM, Huey R, Lindstrom W, Sanner MF,
Belew RK, Goodsell DS and Olson AJ: AutoDock4 and AutoDockTools4:
Automated docking with selective receptor flexibility. J Comput
Chem. 30:2785–2791. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tan Q, Zhu Y, Li J, Chen Z, Han GW,
Kufareva I, Li T, Ma L, Fenalti G, Li J, et al: Structure of the
CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex.
Science. 341:1387–1390. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Berendsen HJC, van der Spoel D and van
Drunen R: GROMACS: A message-passing parallel molecular dynamics
implementation. Comput Phys Commun. 91:43–56. 1995. View Article : Google Scholar
|
|
26
|
Amadei A, Linssen AB and Berendsen HJ:
Essential dynamics of proteins. Proteins. 17:412–425. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tomasello G, Armenia I and Molla G: The
protein imager: A full-featured online molecular viewer interface
with server-side HQ-rendering capabilities. Bioinformatics.
36:2909–2911. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu ZY, Tang JN, Xie HX, Du YA, Huang L, Yu
PF and Cheng XD: 5-Fluorouracil chemotherapy of gastric cancer
generates residual cells with properties of cancer stem cells. Int
J Biol Sci. 11:284–294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hembruff SL and Cheng N: Chemokine
signaling in cancer: Implications on the tumor microenvironment and
therapeutic targeting. Cancer Ther. 7:254–267. 2009.PubMed/NCBI
|
|
30
|
Oppermann M: Chemokine receptor CCR5:
Insights into structure, function, and regulation. Cell Signal.
16:1201–1210. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Salmas RE, Yurtsever M and Durdagi S:
Investigation of inhibition mechanism of chemokine receptor CCR5 by
micro-second molecular dynamics simulations. Sci Rep. 5:131802015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wasilko DJ, Johnson ZL, Ammirati M, Che Y,
Griffor MC, Han S and Wu H: Structural basis for chemokine receptor
CCR6 activation by the endogenous protein ligand CCL20. Nat Commun.
11:30312020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Duchesnes CE, Murphy PM, Williams TJ and
Pease JE: Alanine scanning mutagenesis of the chemokine receptor
CCR3 reveals distinct extracellular residues involved in
recognition of the eotaxin family of chemokines. Mol Immunol.
43:1221–2131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Aalst EJ, McDonald CJ and Wylie BJ:
Cholesterol biases the conformational landscape of the chemokine
receptor CCR3: A MAS SSNMR-filtered molecular dynamics study. J
Chem Inf Model. 63:3068–3085. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Malgija MB, Rajendran HAD, Darvin SS,
Nachimuthu S and Priyakumari J: In silico exploration of HIV entry
co-receptor antagonists: A combination of molecular modeling,
docking and molecular dynamics simulations. Acta Sci Pharm Sci.
3:60–67. 2019.
|
|
36
|
Manna M, Niemelä M, Tynkkynen J,
Javanainen M, Kulig W, Müller DJ, Rog T and Vattulainen I:
Mechanism of allosteric regulation of β2-adrenergic
receptor by cholesterol. Elife. 5:e184322016. View Article : Google Scholar : PubMed/NCBI
|