Introduction
MicroRNAs (miRNAs or miRs), one of the most abundant
classes of post-transcriptional regulators, are attracting
increasing attention. MiRNAs inhibit translation initiation by
binding to 3′ untranslated regions (3′UTRs) (1). The upregulation of certain miRNAs in
carcinoma suggests that they can function as oncogenes, and
inversely, the downregulation of certain miRNAs suggests that they
can also function as tumor suppressors (2–4). Let-7
miRNAs are the earliest discovered miRNAs, which are critical for
cell generation and differentiation (5). Let-7 miRNAs have been reported to be
tumor suppressors, playing critical roles in tumorigenesis and the
metastatic progression of many types of cancer, such as breast,
lung, colorectal and ovarian cancer, as well as nervous system
neoplasms (6,7). Let-7 miRNAs have been demonstrated to
regulate multiple oncogenes, such as RAS, high-mobility group
AT-hook 2 (HMGA2), c-Myc and caspase-3, by targeting and degrading
their mRNAs (6,8,9).
Let-7a, a member of the let-7 family, has been selected as a
representative by a number of studies (10–13).
Estrogen has been implicated in breast diseases for
a number of years, particularly in estrogen receptor (ER)-positive
breast cancer. ER consists of two isoforms, ERα and ERβ. The
biological function of estrogen is mediated mainly by
nucleus-localized ERα, which is a critical component in breast
cancer initiation and progression. Along with the discovery of
anti-estrogen drugs and estrogen receptor antagonists, such as
tamoxifen and letrozole, endocrine therapy plays a vital role in
combating breast cancer. However, patients inevitably develop
resistance to these drugs. Finding a novel strategy to inhibit the
function of estrogen is crucial. Cancer stem cells (CSCs) are a
group of cells showing a high capacity for sphere formation, self
renewal, tumorigenicity and migration (14). Based on these characteristics, CSCs
are considered responsible for resistance to chemo- and
radiotherapy (14,15), eventually promoting the regeneration
and recurrence of cancer. Therefore, therapies targeting CSCs may
be promising strategies for the treatment of breast cancer.
However, it is remarkable that CSCs can remain dormant (in
‘hibernation’) for long periods of time, a quiescence associated
with slow cell cycle kinetics, which renders CSCs resistant to
anticancer therapies and enhances their tumor initiating potential
(16,17).
Recently, scientists discovered that let-7 degrades
ERα as a target, and then inhibits cell proliferation and promotes
cell apoptosis (18). This
discovery suggests that let-7 miRNAs may be candidates for drug
therapies against estrogen and cancer in the future. However, the
function of let-7 related to ERα in breast cancer stem cells
(BCSCs) has not yet been investigated. The isolation of CSCs has
provided us with the opportunities to explore their origin,
self-renewal, differentiation, invasiveness, metastatic ability,
resistance to chemotherapy and radiotherapy (19,20).
The side population (SP) method was created as a classic and
non-specific method for the separation of CSCs, which relies on
their characteristics (21), and is
particularly useful in a situation where molecular markers of these
stem cells are unclear (22,23).
Previous studies have confirmed that SP cells are an enriched
source of CSCs, and based on the lack of accumulation of the
Hoechst dye, SP cells have been proven to enrich primitive and
undifferentiated cells in a variety of tumors and cell lines
(24). In this study, we applied
the SP method to separate SP cells from breast cancer cell lines
for research purposes.
In order to verify our hypothesis that let-7 is a
regulator of the ERα signaling pathway in breast tumors and BCSCs,
we firstly examined clinical specimens to search for clues of the
correlation between let-7 and ERα, and then detected their
expression in BCSCs to identify the signaling pathway associated
with let-7. Finally, we transfected let-7a into SP cells to
investigate the changes in the expression of target genes and
biological characteristics.
Materials and methods
Expression of let-7 and ERα in clinical
specimens
To examine the expression of hsa-let-7 and ERα,
specimens from 43 patients with ER-positive breast cancer were
collected. These patients underwent surgery at the First Affiliated
Hospital of Xi’an Jiaotong University (Xi’an, China) from February
2009 to February 2011. Standard modified radical mastectomy was
performed for each patient, with or without sentinel lymph node
dissection and complete axillary lymph node dissection. No
pre-operative chemo- or radiotherapy was performed on any of these
patients. Tumor tissue specimens were from the central part of the
tumor, except for the necrotic part; normal tissue specimens were
obtained from tissue located >5 cm away from the tumor margin.
Fresh specimens were stored in liquid nitrogen. All the patients
were diagnosed and confirmed by pathological examination. Their
clinical data are summarized in Table
I.
 | Table IClinical and pathological features of
the breast cancer patients. |
Table I
Clinical and pathological features of
the breast cancer patients.
| Parameter | No. of cases | Percentage |
|---|
| Age (years) |
| ≤50 | 19 | 44.2 |
| >50 | 24 | 55.8 |
| Menopause |
| Pre | 20 | 45.1 |
| Post | 23 | 55.9 |
| Tumor size
(cm) |
| ≤2 | 27 | 62.8 |
| >2 | 16 | 37.2 |
| Grade of
malignancy |
| G1 + G2 | 31 | 72.1 |
| G3 | 12 | 27.9 |
| Progesterone
status |
| Positive | 33 | 76.7 |
| Negative | 10 | 23.3 |
| HER2 status |
|
Overexpression | 8 | 18.6 |
| Normal
expression | 35 | 81.4 |
| Ki-67 |
| ≤25% | 32 | 74.4 |
| >25% | 11 | 25.6 |
The expression levels of let-7 and ERα were examined
by real-time quantitative reverse transcription-polymerase chain
reaction (qRT-PCR). The cDNA templates for qRT-PCR were synthesized
from extracted RNA samples. The primers of the let-7 miRNAs are
listed in Table II. Primers of ERα
were: 5′-ATG ACC ATG ACC CTC CAC ACC AAA GCA-3′ and 5′-TTC AGA CCG
TGG CAG GGA AAC CCT CT-3′. miR-U6 and β-actin were used as the
internal reference. Gene expression was detected using SYBR Premix
Ex TaqII (Takara, Inc., Dalian, China). Real-time PCR was carried
out in a 20.0 μl reaction volume, including 10.0 μl of SYBR Premix
Ex TaqII, 0.8 μl certain let-7 miRNA primer and 0.8 μl Uni-miR qPCR
Primer (Takara, Inc.). ERα was determined by adding 1.0 μl forward
primer and 1.0 μl reverse primer of ERα (AuGCT DNA; Syn
Biotechnology, Xi’an, China). All analyses were performed in
triplicate.
| Table IILet-7 miRNAs and U6 primers. |
Table II
Let-7 miRNAs and U6 primers.
| Gene | Primer | Annealing temp
(°C) |
|---|
| miR-let-7a |
TGAGGTAGTAGGTTGTATAGTT | 54.7 |
| miR-let-7b |
TGAGGTAGTAGGTTGTGGTT | 58.3 |
| miR-let-7c |
TGAGGTAGTAGGTTGTATGGTT | 56.3 |
| miR-let-7d |
TCTCCATCATCCAACGTATCAA | 57.1 |
| miR-let-7e |
ACTCCATCCTCCAACATATCAA | 57.3 |
| miR-let-7f |
GATATGTTAGATAACGGAAGGG | 56.8 |
| miR-let-7g |
ACTCCATCACCAAACATGTCAA | 59.0 |
| miR-let-7i |
GACGCGTTCGATGACGGAACGA | 58.5 |
| U6-F |
CTCGCTTCGGCAGCACA | 56.0 |
Cell culture
The human breast cancer cell lines, MCF-7 and T47-D,
were kept in the Central Laboratory of the Medical College of Xi’an
Jiaotong University. RPMI-1640 medium (Thermo Scientific Co., Ltd.,
Shanghai, China) was supplemented with 10% fetal bovine serum,
penicillin 100 U/ml and streptomycin 100 mg/ml (Gibco-Invitrogen,
Grand Island, NY, USA). MCF-7 and T47-D were incubated at 37°C in a
5% CO2 atmosphere. SP cells separated from MCF-7 and
T47-D cells were cultured in stem cell growth medium (SCGM), which
was DMEM/F12 (Thermo Scientific Co., Ltd.) medium supplemented with
1% non-essential amino acid, 5 mM HEPES, 50 μg/ml insulin, 20 ng/ml
human epidermal growth factor (hEGF), 10 ng/ml human basic
fibroblast growth factor (hbFGF) (3,19,25,26).
Isolation of breast CSCs (SP cells)
In this study, we used a FACSAria cell soter (BD
Biosciences, San Jose, CA, USA) in order to separate SP cells from
MCF-7 and T47-D cell lines. The SP protocol was essentially
performed as previously described (21). DNA-specific Hoechst 33342 dye and
verapamil hydrochloride were purchased from Sigma-Aldrich Co., Ltd.
(St. Louis, MO, USA) MCF-7 and T47-D cells were collected
(1×106 cells/ml), and incubated in RPMI-1640 medium as
described above. The positive group contained 5 μg/ml Hoechst
33342, and was incubated at 37°C for 90 min in SCGM. The control
group contained the ABCG inhibitor, verapamil (1 mM), with the
other conditions being the same. Four groups of two breast cancer
cell lines were analyzed by flow cytometry. After the SP and non-SP
(NSP) cells were successfully separated, we re-analyzed the cells
to evaluate sorting purity. We separated the SP cells in three
rounds. Total RNA and protein were extracted and identified and the
average expressions were calculated.
Analysis of let-7 and ERα
Total RNA was extracted using Takara RNAiso Plus
(Takara, Inc.), then cDNA was synthesized using 1.0 μl (~1.0 μg)
total RNA in a reaction volume of 20.0 μl by following the protocol
of Takara One Step PrimeScript miRNA cDNA Synthesis Kit (Takara,
Inc.). Following dilution for five times, 2.0 μl cDNA was absorbed
from 100.0 μl mix for real-time PCR in a 20.0 μl reaction volume,
including 10.0 μl of SYBR Premix Ex TaqII, 0.8 μl certain let-7
miRNA primer and 0.8 μl Uni-miR qPCR Primer (Takara, Inc.). The
primers of let-7 family members used in this reaction are listed in
Table II. The level of each miRNA
was calculated and presented following the 2−ΔΔCt method
[ΔΔCt = ΔCt (SP cells) - ΔCt (NSP cells)], using miR-U6 as the
internal reference for BCSCs.
To examine ERα expression in SP and NSP cells, total
RNA was extracted, and the protocols were the same as those
described above. When the mixture was ready for real-time PCR in a
20.0 μl reaction volume, 1.0 μl forward primer and 1.0 μl reverse
primer of ERα (AuGCT DNA; Syn Biotechnology) were added, not using
Uni-miR qPCR Primer. The primers for ERα were: 5′-ATG ACC ATG ACC
CTC CAC ACC AAA GCA-3′ and 5′-TTC AGA CCG TGG CAG GGA AAC CCT
CT-3′. The expression level of ERα was presented following the
2−ΔΔCt method, using β-actin as the internal reference,
of which the primers were 5′-GGT GGC TTT TAG GAT GGC AAG-3′ and
5′-ACT GGA ACG GTG AAG GTG ACA G-3′. All analyses were performed in
triplicate.
Analysis of protein expression
Cyclin D1 and pS2 are the direct downstream genes of
ERα, and play an important role in the cell cycle, cell
differentiation and proliferation. We investigated ERα, cyclin D1
and pS2 expression by western blot analysis in the SP cells.
Firstly, total protein was extracted from the isolated SP and NSP
cells by radioimmunoprecipitation assay (RIPA) lysis buffer.
Following centrifugation, the supernatants were collected. Total
protein concentrations were measured using the BCA assay kit.
Clarified protein lysates were electrophoretically resolved on a
denaturing SDS polyacrylamide gel (8–12%) and electrotransferred
onto nitrocellulose membranes.
The membranes were firstly blocked in 5% non-fat dry
milk in Tris-buffered saline (TBS) for 2 h, and then probed with
specific primary antibodies against ERα (ab10286), cyclin D1
(ab62151), pS2 (ab92377), purchased from Abcam (Hong Kong, China).
Immunopositive bands were detected using the chemiluminescence
detection system (Amersham Biosciences, Piscataway, NJ, USA) and
autoradiography. The western blot analysis results were scored as
positive if the band of interest was present at the expected
molecular weight. All analyses were performed in triplicate.
Transfection of let-7a into SP cells
The let-7a sequence was synthesized and cloned into
the carrier plasmid, pGenesil, by GenePharma Co., Ltd. (Shanghai,
China), which contained neo gene; pGenesil-control was also
constructed by GenePharma Co., Ltd. The resulting plasmids were
labeled pGenesil-let-7a and pGenesil-control, confirmed by
restriction enzyme digestion and agarose gel electrophoresis. In
order to determine the effects of let-7a on ERα, the
pGenesil-let-7a plasmid was transfected into the SP cells from the
MCF-7 and T47-D cell lines, using Lipofectamine 2000 according to
the manufacturer’s instructions. After 48 h, the cells were placed
in selection medium, and let-7a was detected. After the
transfection of let-7a into MCF-7 and T47-D SP cells, we examined
the mRNA and protein expression of ERα. We also examined the
protein expression levels of cyclin D1 and pS2 after transfection.
The protocols were as described above.
Flow cytometry
MCF-7 and T47-D SP cells which were transfected with
the let-7a and pGenesil-control plasmids were divided into the
following four groups: i) MCF-7 let-7a positive group, ii) MCF-7
control group, iii) T47-D let-7a positive group and iv) T47-D
control group.
For analysis of the apoptosis rates,
5×105 cells from each group were collected, and then
washed with PBS twice. Flow cytometric analysis was carried out
according to standard procedures. The cells were resuspended in 500
μl binding buffer, and then 5 μl Annexin V-FITC and 10 μl PI (both
from BD Biosciences) were added. The mixture was gently vortexed
and then incubated for 15 min at 18–28°C in the dark. The cells
were analyzed by flow cytometry within 1 h of incubation and all
analyses were performed in triplicate.
For cell cycle analysis, 5×105 cells were
collected, washed twice with PBS, and then fixed with ice-cold 70%
ethanol for 24 h at 4°C. The fixed cells were then stained with PI
for 30 min at 37°C; finally all samples were analyzed by flow
cytometry. Cell cycle analysis of DNA contents was performed using
MultiCycle software. All analyses were performed in triplicate.
Statistical analysis
All statistical analyses were performed using
SPSS13.0 software (IBM Corp., Armonk, NY, USA). The results are
presented as the means ± standard deviation (SD). The intragroup
differences of the miRNAs and protein expression were assessed by
the analysis of variance (ANOVA) test. The statistical differences
of let-7 and its downstream genes between the four groups were
determined using the Student’s t-test. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Expression of let-7 and ERα in clinical
specimens
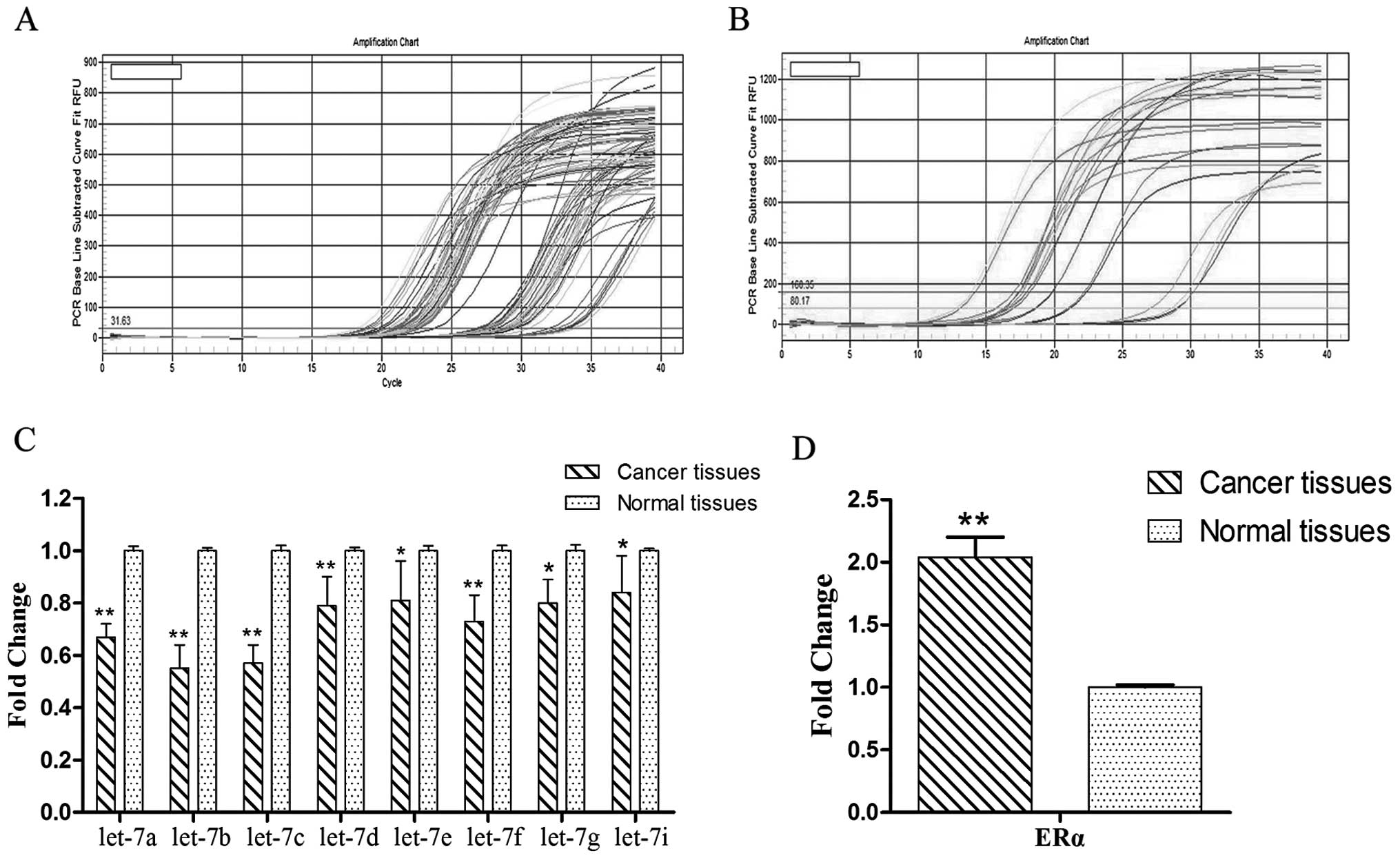
The results of quantitative real-time PCR showed
that let-7 miRNAs were downregulated in tumor tissues compared to
normal tissues. Among all let-7 miRNAs, the differences of let-7a,
let-7b and let-7c between the two groups were the most significant
(Fig. 1A and C); on the contrary,
ERα was significantly upregulated in the cancer tissues compared to
the normal tissues (Fig. 1B and D).
A significant inverse correlation was observed.
Isolation of SP cells
The SP cells from the MCF-7 and T47-D cells
accounted for 5.0 and 3.3% of the total population, respectively.
As expected, verapamil inhibited the proportion of SP cells. The
proportion of MCF-7 and T47-D SP cells dropped to 0.4 and 0.1%,
respectively when verapamil was added into the process of
separation in the control group. Representative data are shown in
Fig. 2; these data are in general
agreement with previous studies (26–30).
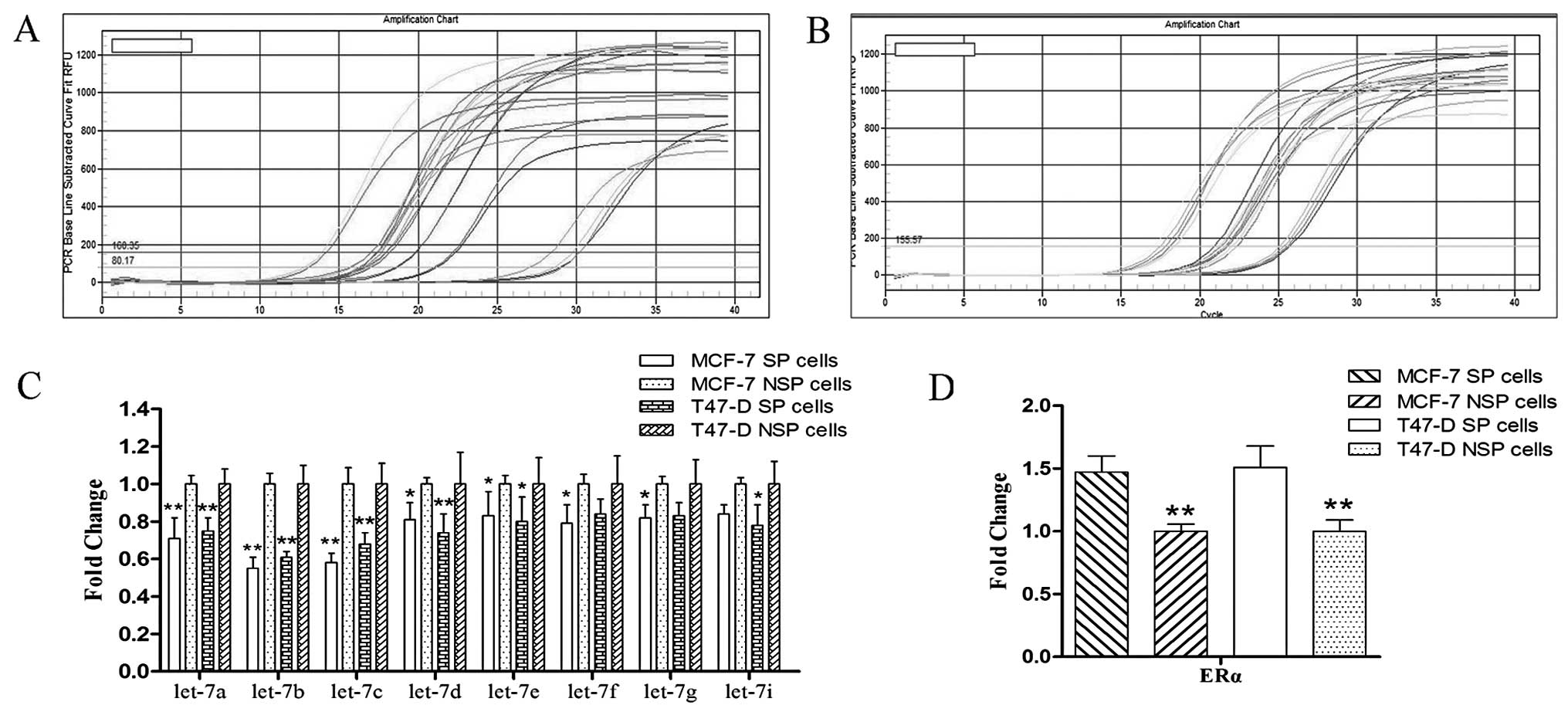
Expression of let-7 and its correlation
with ERα mRNA
The results of quantitative real-time PCR showed
that let-7 miRNAs were downregulated in the SP cells compared to
the NSP cells; among them, let-7a/b/c were significantly
downregulated (Fig. 3A and C); on
the contrary, ERα was upregulated in the SP cells compared to the
NSP cells (Fig. 3B and D). These
two factors had a significant inverse correlation.
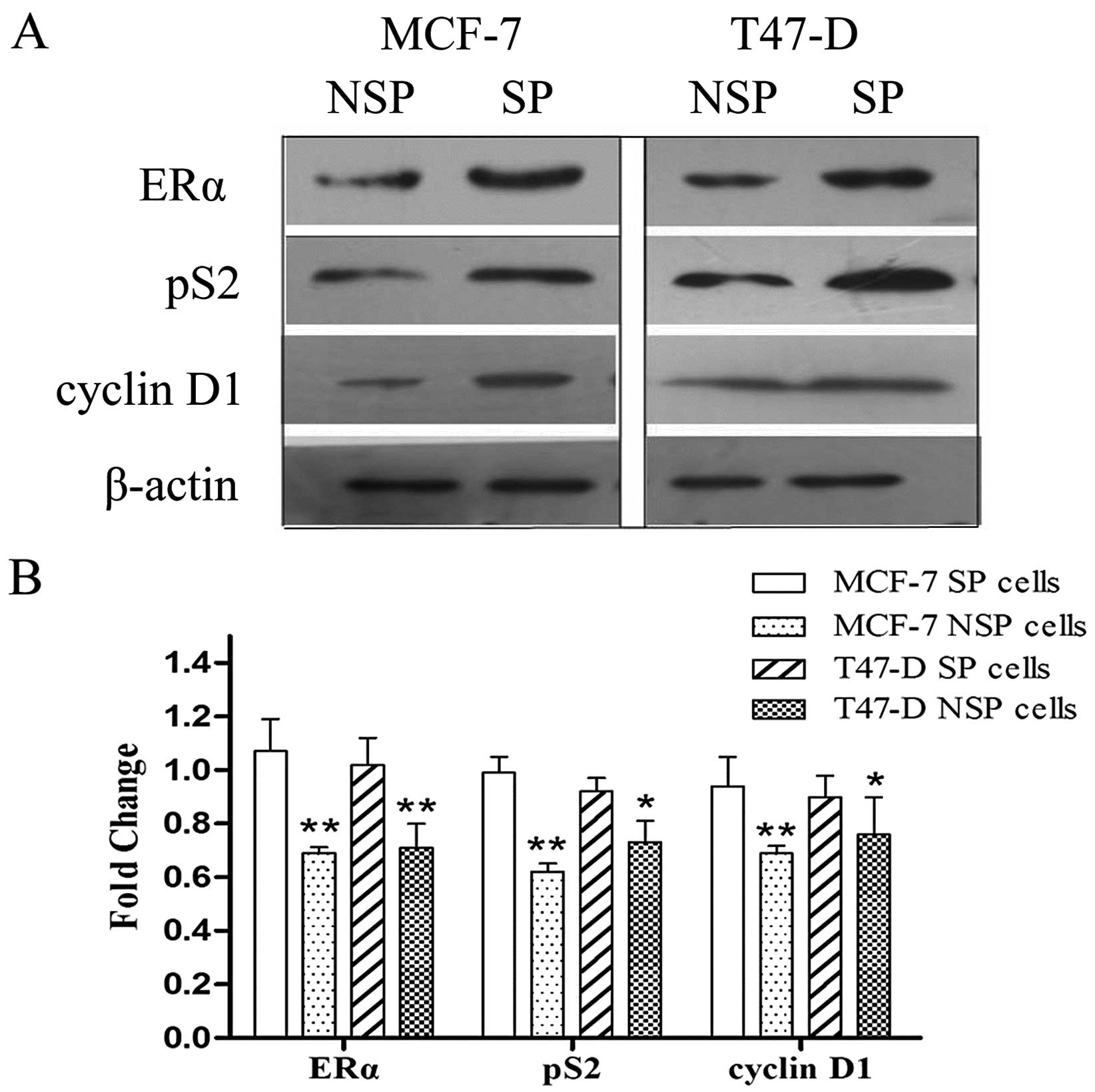
Expression of ERα downstream genes
We assessed the protein expression level of ERα,
cyclin D1 and pS2 in SP and NSP cells. The expression of ERα,
cyclin D1 and pS2 were significantly higher in the SP compared to
the NSP cells; importantly, we detected an inverse correlation
between cyclin D1, pS2 and let-7 (Fig.
4).
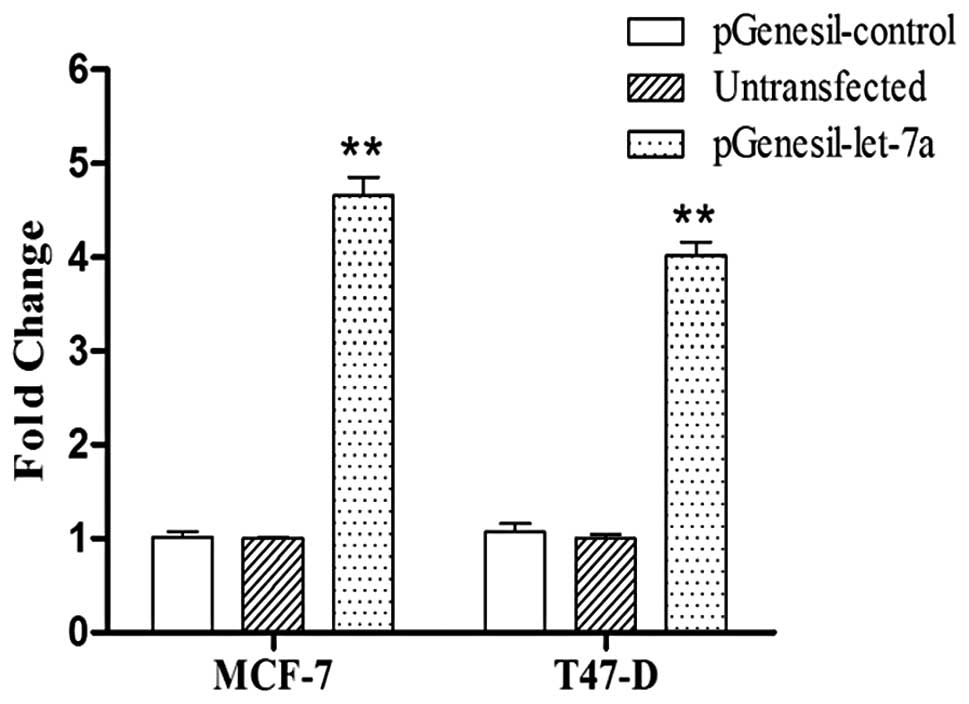
Let-7 targets ERα signaling pathway
PGenesil-let-7a and pGenesil-control plasmids were
constructed; the recombinant vector was verified by sequencing and
examined by qRT-PCR. Let-7a expression was significantly enhanced
in the SP cells transfected with pGenesil-let-7a compared to the SP
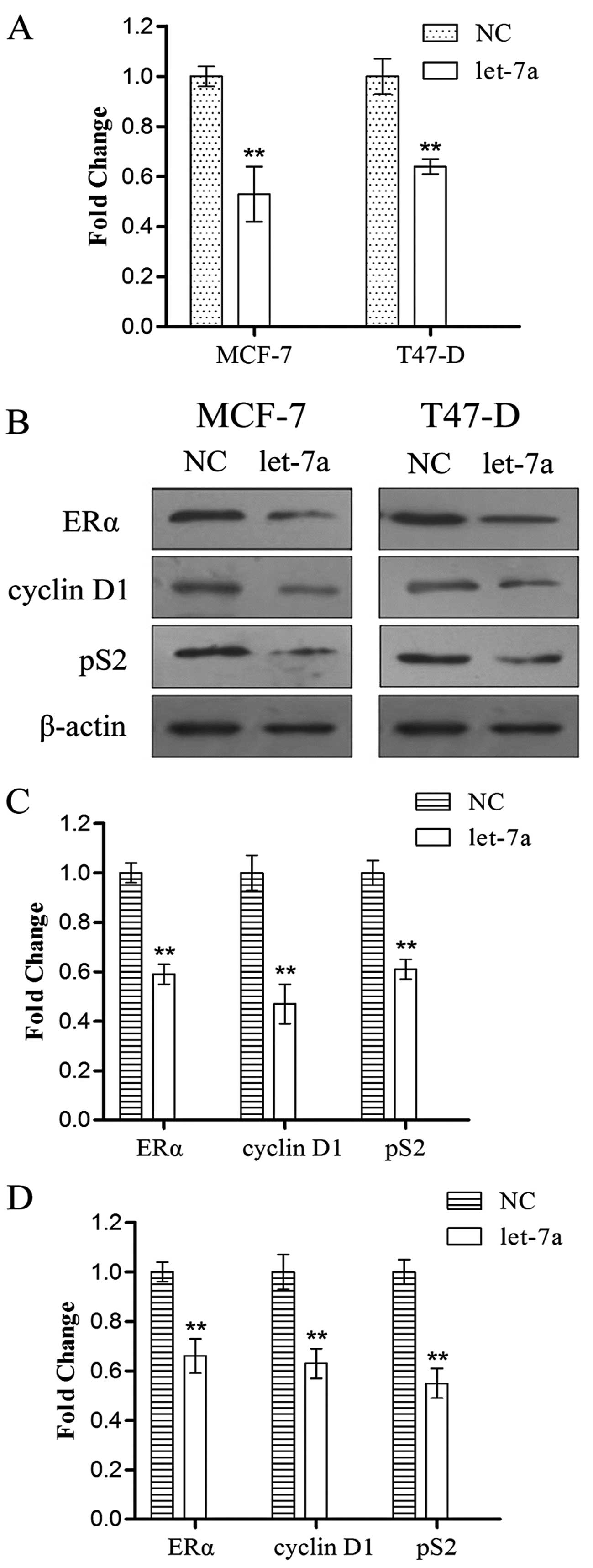
cells transfected with pGenesil-control after two days (Fig. 5). At the mRNA level, the results
showed that let-7a inhibited ERα mRNA expression after two days
(28–61% of negative control, P<0.01) (Fig. 6A), indicating that let-7a promotes
the degradation of ERα mRNA. At the protein level, let-7a
suppressed the expression of ERα (Fig.
6B). Furthermore, we examined the downstream genes of ERα
following transfection for two days; the results showed that let-7a
significantly inhibited the expression of pS2 and cyclin D1
(Fig. 6B). The gray value results
of western blot analysis are shown in Fig. 6C and D.
Cell cycle and apoptosis analysis of
let-7a SP cells
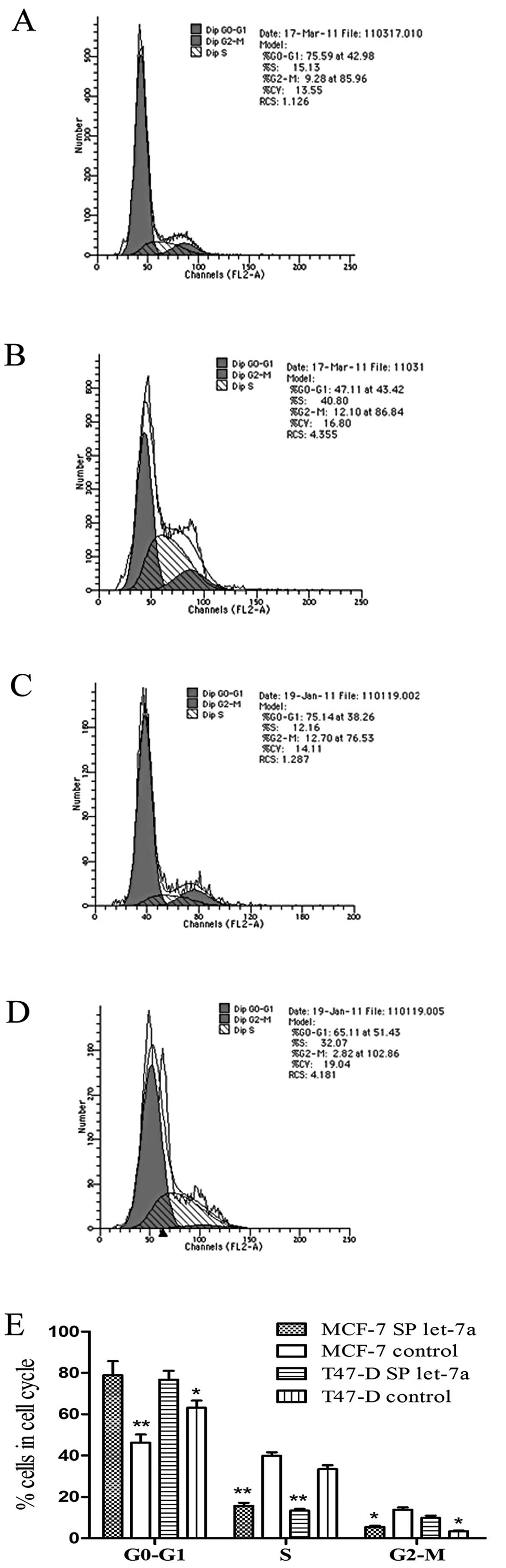
Cell cycle analysis showed that let-7a increased the
percentage of SP cells in the G0-G1 phase, and decreased the
percentage of SP cells in the S phase, compared with the control
group. The results of MCF-7 SP cells are shown in Fig. 7A and B, and the results of T47-D SP
cells are shown in Fig. 7C and D,
P<0.05. These results indicate that let-7a promotes the G0-G1
transition, and thereby inhibits SP cell proliferation. We then
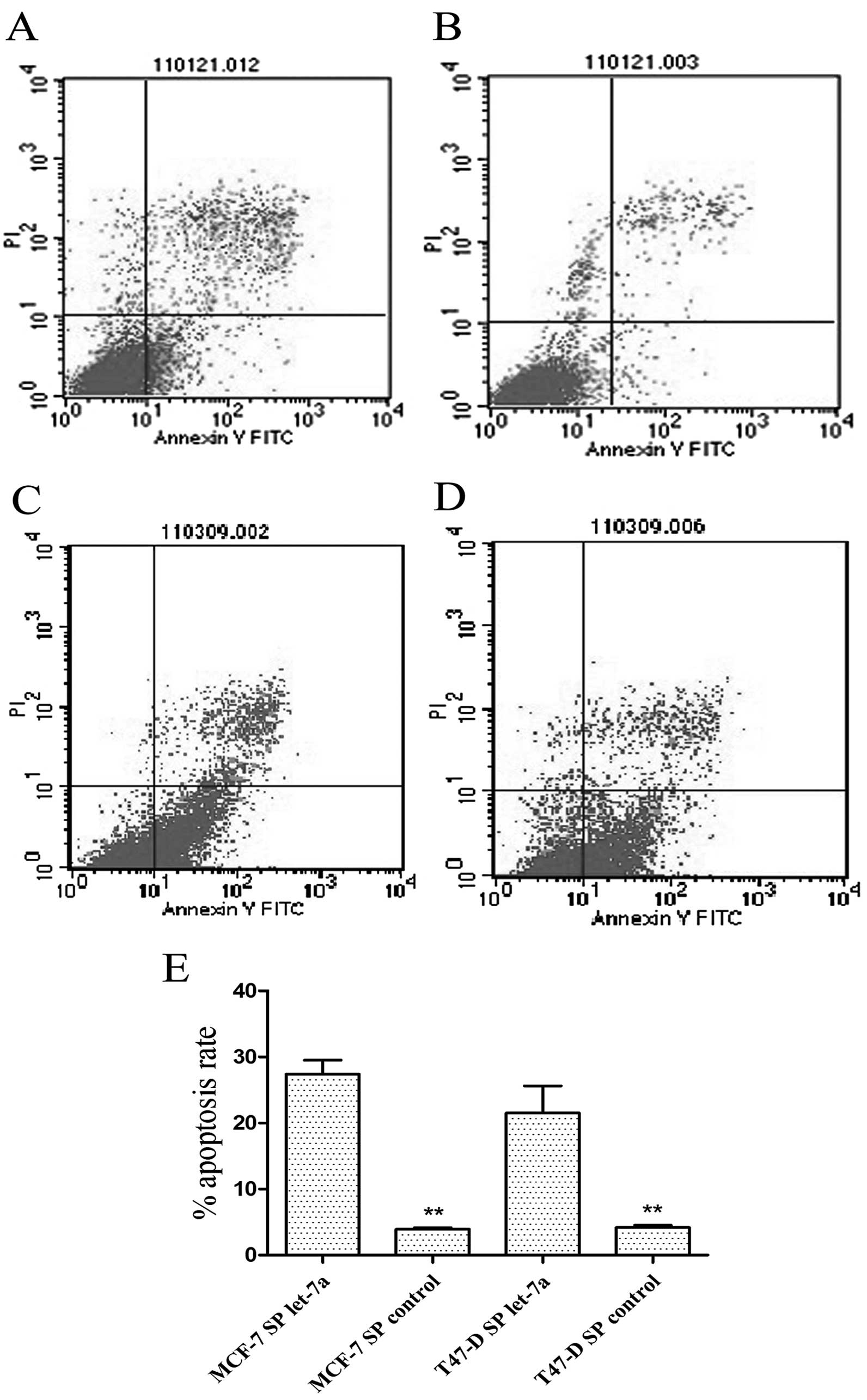
assessed SP cells from the two cell lines to examine the effect of
let-7a on CSC apoptosis. The results showed that let-7a increased
the apoptotic rate of MCF-7 and T47-D SP cells (Fig. 8).
Discussion
CSCs possess lots of fundamental characteristics
which are similar to normal stem cells (31,32),
and have an enhanced capacity for tumor generation and metastasis,
eventually leading to resistance to chemotherapy and tumor
recurrence (14). To investigate
CSCs, separation is the first and the most crucial procedure. The
separation of CSCs from tumor tissues or cell lines is usually
based on SP cells, CD44+/CD24−/low and
ALDH+ phenotypes (28,29,33–35).
The use of SP cells is widely accepted, and these cells are
identified as an enriched group of CSCs. Based on several
experimental and clinical analyses of CSCs in breast tumors, a
correlation between the proportion of CSCs and poor prognosis has
been established (36–39). Therefore, the search for effective
therapies and novel strategies which could specifically perish and
kill BCSCs is an urgent issue which may aid in the treatment of
breast cancer.
The close correlation between breast cancer and ER
has been emphasized for decades. The upregulation of ERα is
considered an important poor prognostic factor, which can induce
aggressive tumorigenesis through the activation of NF-κB, MAPK and
other signaling pathways in breast cancer cells. However, the exact
function of the ERα signaling pathway in BCSCs has not yet been
elucidated. Till now, little information is available regarding ERα
in BCSCs (30). It has been
confirmed that let-7 can silence multiple oncogenes, such as c-Myc,
RAS, HMGA2 and newly discovered ERα. The possible interference of
let-7 in the ERα transcription and translation process has been
reported in a variety of human malignancies (18,40,41);
however, the biological effects of let-7 on ERα have not been
elucidated in BCSCs. ERα can be directly regulated by let-7 as
there is an evident binding site (18,41).
Therefore, ERα may be triggered by the downregulation of let-7
miRNAs in BCSCs.
In this study, we demonstrate the possible
interactions between let-7 and the ERα signaling pathway; let-7
targets and degrades ERα, both in breast tumor tissues and BCSCs.
We began with the detection of clinical samples, and observed an
inverse correlation between let-7 miRNAs and ERα. We then detected
the expression levels of ERα downstream genes in SP cells,
including cyclin D1 and pS2 to verify our hypothesis. The
significant correlation between let-7a and ERα status in SP cells
suggests that let-7 is an important regulator of ERα expression and
can influence estrogen-dependent CSC proliferation. Finally, we
transfected let-7a into SP cells to examine changes in the ERα
signaling pathway. The results provide further evidence to support
our hypotheses that let-7 regulates the cell cycle and apoptosis of
ER-positive BCSCs by influencing ERα and the ERα signaling
pathway.
Taken together, our findings indicate that the low
expression of let-7 in BCSCs results in estrogen signaling pathway
activation, which has a significant effect on estrogen-dependent
cells. The low expression of let-7 in MCF-7 and T47-D SP cells did
not allow let-7 the opportunity to suppress ERα; therefore, ERα was
upregulated, accompanied by the high expression of direct
downstream genes, such as cyclin D1 and pS2, which play important
roles in maintaining the function and character of BCSCs in
ER-positive breast cancer. These data shed light onto novel and
critical mechanisms by which BCSCs divide and repopulate
themselves. As CSCs play a crucial role in cancer recurrence and
metastasis, our results may lead to the development of novel
strategies for the treatment of patients with ER-positive breast
cancer; let-7 miRNAs may be potential agents for anticancer
therapy.
Acknowledgements
This study was supported by the Natural Science
Foundation of China, approved ID: 81272418. The authors acknowledge
the assistants at the Department of Oncology and the staff of the
Key Laboratory of Environment and Genes Related to Disease,
Ministry of Education, Faculty of Public Health, College of
Medicine, Xi’an Jiaotong University for their technical
assistance.
References
|
1
|
Kinch LN and Grishin NV: The human Ago2 MC
region does not contain an eIF4E-like mRNA cap binding motif. Biol
Direct. 4:22009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Z, Li Y, Ahmad A, et al: Targeting
miRNAs involved in cancer stem cell and EMT regulation: An emerging
concept in overcoming drug resistance. Drug Resist Updat.
13:109–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zimmerman AL and Wu S: MicroRNAs, cancer
and cancer stem cells. Cancer Lett. 300:10–19. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Medina PP and Slack FJ: microRNAs and
cancer: an overview. Cell Cycle. 7:2485–2492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Büssing I, Slack FJ and Grosshans H: let-7
microRNAs in development, stem cells and cancer. Trends Mol Med.
14:400–409. 2008.
|
|
7
|
Roush S and Slack FJ: The let-7 family of
microRNAs. Trends Cell Biol. 18:505–516. 2008. View Article : Google Scholar
|
|
8
|
Johnson CD, Esquela-Kerscher A, Stefani G,
et al: The let-7 microRNA represses cell proliferation pathways in
human cells. Cancer Res. 67:7713–7722. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee YS and Dutta A: The tumor suppressor
microRNA let-7 represses the HMGA2 oncogene. Genes Dev.
21:1025–1030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Muller DW and Bosserhoff AK: Integrin beta
3 expression is regulated by let-7a miRNA in malignant melanoma.
Oncogene. 27:6698–6706. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He XY, Chen JX, Ou-Yang X, Zhang Z and
Peng HM: Construction of let-7a expression plasmid and its
inhibitory effect on k-Ras protein in A549 lung cancer cells. Nan
Fang Yi Ke Da Xue Xue Bao. 30:2427–2431. 2010.(In Chinese).
|
|
12
|
Sureban SM, May R, Ramalingam S, et al:
Selective blockade of DCAMKL-1 results in tumor growth arrest by a
Let-7a MicroRNA dependent mechanism. Gastroenterology. 137:649–659.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Oliveras-Ferraros C, Cufi S,
Vazquez-Martin A, et al: Micro(mi)RNA expression profile of breast
cancer epithelial cells treated with the anti-diabetic drug
metformin: induction of the tumor suppressor miRNA let-7a and
suppression of the TGFβ-induced oncomiR miRNA-181a. Cell Cycle.
10:1144–1151. 2011.PubMed/NCBI
|
|
14
|
Clevers H: The cancer stem cell: premises,
promises and challenges. Nat Med. 17:313–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jordan CT, Guzman ML and Noble M: Cancer
stem cells. N Engl J Med. 355:1253–1261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ito K, Bernardi R, Morotti A, et al: PML
targeting eradicates quiescent leukaemia-initiating cells. Nature.
453:1072–1078. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shachaf CM, Kopelman AM, Arvanitis C, et
al: MYC inactivation uncovers pluripotent differentiation and
tumour dormancy in hepatocellular cancer. Nature. 431:1112–1117.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao Y, Deng C, Wang J, et al: Let-7
family miRNAs regulate estrogen receptor alpha signaling in
estrogen receptor positive breast cancer. Breast Cancer Res Treat.
127:69–80. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Charafe-Jauffret E, Ginestier C, Iovino F,
et al: Breast cancer cell lines contain functional cancer stem
cells with metastatic capacity and a distinct molecular signature.
Cancer Res. 69:1302–1313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Iliopoulos D, Hirsch HA, Wang G and Struhl
K: Inducible formation of breast cancer stem cells and their
dynamic equilibrium with non-stem cancer cells via IL6 secretion.
Proc Natl Acad Sci USA. 108:1397–1402. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goodell MA, Brose K, Paradis G, Conner AS
and Mulligan RC: Isolation and functional properties of murine
hematopoietic stem cells that are replicating in vivo. J Exp Med.
183:1797–1806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Challen GA and Little MH: A Side order of
dtem vells: the SP phenotype. Stem Cells. 24:3–12. 2006. View Article : Google Scholar
|
|
23
|
Ho MM, Ng AV, Lam S and Hung JY: Side
population in human lung cancer cell lines and tumors is enriched
with stem-like cancer cells. Cancer Res. 67:4827–4833. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hadnagy A, Gaboury L, Beaulieu R and
Balicki D: SP analysis may be used to identify cancer stem cell
populations. Exp Cell Res. 312:3701–3710. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hirsch HA, Iliopoulos D, Tsichlis PN and
Struhl K: Metformin selectively targets cancer stem cells, and acts
together with chemotherapy to block tumor growth and prolong
remission. Cancer Res. 69:7507–7511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Geng S, Wang Q, Wang J, et al: Isolation
and identification of a distinct side population cancer cells in
the human epidermal squamous cancer cell line A431. Arch Dermatol
Res. 303:181–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lu X, Xu K, Lu H, et al: CD44(+)/CD24(−)
cells are transit progenitors and do not determine the molecular
subtypes and clinical parameters in breast carcinomas. Ultrastruct
Pathol. 35:72–78. 2011.
|
|
28
|
Wang J, Guo L-P, Chen L-Z, Zeng Y-X and Lu
SH: Identification of cancer stem cell-like side population cells
in human nasopharyngeal carcinoma cell line. Cancer Res.
67:3716–3724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakanishi T, Chumsri S, Khakpour N, et al:
Side-population cells in luminal-type breast cancer have
tumour-initiating cell properties, and are regulated by HER2
expression and signalling. Br J Cancer. 102:815–826. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi GM, Xu Y, Fan J, et al: Identification
of side population cells in human hepatocellular carcinoma cell
lines with stepwise metastatic potentials. J Cancer Res Clin Oncol.
134:1155–1163. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dick JE: Stem cell concepts renew cancer
research. Blood. 112:4793–4807. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bhattacharyya S and Khanduja KL: New hope
in the horizon: cancer stems cells. Acta Biochim Biophys Sin.
42:237–242. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Idowu MO, Kmieciak M, Dumur C, et al:
CD44(+)/CD24(−/low) cancer stem/progenitor cells are more abundant
in triple-negative invasive breast carcinoma phenotype and are
associated with poor outcome. Hum Pathol. 43:364–373. 2012.
|
|
34
|
Abraham BK, Fritz P, McClellan M,
Hauptvogel P, Athelogou M and Brauch H: Prevalence of
CD44+/CD24−/low cells in breast cancer may
not be associated with clinical outcome but may favor distant
metastasis. Clin Cancer Res. 11:1154–1159. 2005.PubMed/NCBI
|
|
35
|
Charafe-Jauffret E, Ginestier C, Iovino F,
et al: Aldehyde dehydrogenase 1-positive cancer stem cells mediate
metastasis and poor clinical outcome in inflammatory breast cancer.
Clin Cancer Res. 16:45–55. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiao X, Katiyar S, Willmarth NE, et al:
c-Jun induces mammary epithelial cellular invasion and breast
cancer stem cell expansion. J Biol Chem. 285:8218–8226. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chuthapisith S, Eremin J, El-Sheemey M and
Eremin O: Breast cancer chemoresistance: Emerging importance of
cancer stem cells. Surg Oncol. 19:27–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Silva IA, Bai S, McLean K, et al: Aldehyde
dehydrogenase in combination with CD133 defines angiogenic ovarian
cancer stem cells that portend poor patient survival. Cancer Res.
71:3991–4001. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iinuma H, Watanabe T, Mimori K, et al:
Clinical significance of circulating tumor cells, including cancer
stem-like cells, in peripheral blood for recurrence and prognosis
in patients with Dukes’ stage B and C colorectal cancer. J Clin
Oncol. 29:1547–1555. 2011.PubMed/NCBI
|
|
40
|
Giovannetti E, Erozenci A, Smit J, Danesi
R and Peters GJ: Molecular mechanisms underlying the role of
microRNAs (miRNAs) in anticancer drug resistance and implications
for clinical practice. Crit Rev Oncol Hematol. 81:103–122. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sayed D and Abdellatif M: MicroRNAs in
development and disease. Physiol Rev. 91:827–887. 2011. View Article : Google Scholar : PubMed/NCBI
|