Introduction
Gastric cancer is the second most common cause of
cancer-related death in the world, and accounts for 989,600 new
cases and 738,000 deaths annually (1,2).
Although the incidence of gastric cancer has been substantially
declining for several decades, the disease is associated with a
very poor prognosis, and the 5-year survival rate is ~20% (3). Surgery and chemotherapy are the
primary treatments. However, surgery and chemotherapy have limited
value in advanced disease. In a number of cases, the disease is
usually detected after invasion of the muscularis propria, and
tumor metastasis serves as an impediment to successful treatment
(4). Furthermore, there is an
absence of molecular markers for targeted therapy (1). Thus, new perspectives in
epidemiological and experimental research are important to
establish novel strategies for primary prevention.
N-acetylglucosaminyltransferase V (GnT-V) is a key
enzyme that catalyzes the formation of 1,6 N-acetylglucosamine
(GlcNAc) through the action of adding antennae branching structures
on a common core structure of Man3GlcNAc2 in the medial-Golgi
apparatus (5). Abnormalities in the
expression of GnT-V are thought to be associated with tumor
metastasis and invasion in various types of cancer. However, the
role of GnT-V in different cancers remains controversial. In the
case of colon cancer and hepatocarcinoma, for instance, high GnT-V
expression is associated with a poor prognosis (6–8). In
contrast, low GnT-V levels are linked to a poor prognosis in lung,
bladder carcinoma and neuroblastoma patients (9–11). The
role of GnT-V in gastric cancer and its invasive behavior has
scarcely been studied. Tian et al(12) reported that high GnT-V expression
was observed in 46% (23/50) of gastric cancer tissues and was
significantly correlated with lymph node metastases, peritoneal
dissemination and liver metastases, respectively. Our group
previously demonstrated that high GnT-V expression was associated
with a poor prognosis in patients with gastric cancer, and GnT-V
was expressed at a higher level in gastric cancer BGC823 cells than
in GES-1 cells (gastric mucosal cell line) (unpublished data).
Thus, it is reasonable to assume that inhibition of gastric cancer
metastasis/invasion may be associated with the efficacy of
repression of GnT-V.
Cancer cell metastasis/invasion is a complex process
whereby tumor cells acquire the ability to dissociate from the
primary lesion through activation of the epithelial growth factor
receptor (EGFR) signaling pathway, through alterations in the
related phenotype and the degradation of the basement membrane by
matrix metalloproteinases (MMPs) consequently promoting invasion by
alterating cell motility and growth. Aberrations in EGFR lead to
the abating of downstream signaling molecules such as focal
adhesion kinase (FAK), the phosphatidylinositol-3 kinase (PI3K),
Ras-mitogen-activated protein kinase (Ras-MAPK) generating an
anti-metastatic/invasive effect (13–18).
In addition, GnT-V has been found to contribute to heparin-binding
EGF-like growth factor (HB-EGF)-mediated epidermal
hyperproliferation by inhibiting endocytosis of EGFRs bearing β1–6
GlcNAc on their N-glycans (19). In
addition, downregulation of β1–6 GlcNAc branching in mammary tumor
cells by overexpression of GnT-III
(N-acetylglucosaminyltransferase-III), which antagonizes GnT-V
activity through conformational changes in N-glycans, was found to
decrease EGFR signaling (20–22).
These reports highlight the importance of GnT-V mediated
glycosylation of EGFR for tumor cell function. Previous reports
have shown that aberrant EGFR signaling partly mediated the
epithelial-mesenchymal transition (EMT) phenotype in squamous cell
carcinoma and normal cells (23,24).
The EMT phenotype is characterized by loss of epithelial markers
(E-cadherin), increased expression of mesenchymal factors
(vimentin), increased migratory capacity, and resistance to
apoptosis, and appears to play an important role in tumor cell
metastasis/invasion (25).
Furthermore, EGFR expression has also been shown to correlate with
MMP expression in breast cancer cell lines and non-small cell lung
cancer (15,26). MMPs are a family of zinc-containing
endopeptidases, among which, MMP-2 and MMP-9 are highly expressed
in aggressive tumors (27–30). However, the precise mechanism of
GnT-V regarding the association of EGFR signaling, EMT and MMPs in
gastric cancer still remains largely unknown. Thus, given that
elevated expression of GnT-V in gastric cancer and aberrant
glycosylation by GnT-V are reported to modulate EGFR signaling
(19), we hypothesized that
downregulation of GnT-V inhibits gastric cancer metastasis/invasion
through EGFR-initiated EMT phenotype and MMP-2/9 expression.
Investigation of the interaction between GnT-V and EGFR expression
as well as the EMT phenotype and MMPs in gastric cancer may provide
insight into the underlying biological mechanism, and may offer a
plausible explanation for cell migration and invasion.
In the present study, oligo-siRNA-induced RNA
interference was employed to downregulate GnT-V mRNA expression in
BGC823 cells (gastric cancer cell line), and the biological
behavior was consequently observed. The expression levels of EGFRs,
E-cadherin/vimentin and MMP-2/MMP-9 were evaluated to further
determine the underlying mechanisms.
Materials and methods
Grouping
Cells in this study were divided into four groups as
listed in Table I.
 | Table IFour groups of cells in this
study. |
Table I
Four groups of cells in this
study.
| Name | Treatment |
|---|
| BGC823 | Cells cultured
under normal condition without any treatment |
| BGC823/NC | Cells transfected
with oligo-negative control (NC) siRNA and Lipofectamine 2000
reagent |
| BGC823/lipo | Cells treated with
Lipofectamine 2000 reagent only |
| BGC823/GnT-V | Cells transfected
with oligo-GnT-V siRNA and Lipofectamine 2000 reagent |
Cell culture and transfection
BGC823 cells (gastric cancer cell line) were
generously provided by the Cell Division of the Center Laboratory
in Tongji Hospital of Tongji University, Shanghai, China. Cells
were cultured in 90% RPMI-1640 (Gibco) supplemented with 100 U/ml
penicillin and streptomycin antibiotics (Gibco) and 10% fetal
bovine serum (Gibco) at 37°C with 5% CO2.
The synthesized oligo-GnT-V siRNA and oligo-NC were
purchased from Shanghai GenePharma Co., Ltd., Shanghai, China. The
sequences are listed in Table II.
Oligo-GnT-V siRNA and oligo-NC were transfected into BGC823 cells
by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Oligofectamine reagent for the cells (6×104/3 ml)
consisted of 20 μM oligo-pairs (22.5 μl), Lipofectamine 2000 (30
μl) and RPMI-1640 (3 ml). Lipofectamine 2000 reagent was also added
into the culture medium as a quality control. Transfected cells
were harvested at a set time and were named BGC823/GnT-V, BGC823/NC
and BGC823/ lipo cells, respectively.
| Table IISynthesized sequences. |
Table II
Synthesized sequences.
| Name | Sequences |
|---|
| Oligo-GnT-V siRNA
F |
5′-ggcggaaauucguacagautt-3′ |
| Oligo-GnT-V siRNA
R |
5′-aucuguacgaauuuccgcctt-3′ |
| Oligo-NC F |
5′-uucuccgaacgugucacgutt-3′ |
| Oligo-NC R |
5′-acgugacacguucggagaatt-3′ |
Levels of GnT-V mRNA were detected by quantitative
real-time reverse transcription-PCR analysis (qRT-PCR). Reverse
transcription reactions used the PrimeScript RT Master Mix (Takara
Biotechnology Co., Ltd., Dalian, Japan) and proceeded for 15 min at
37°C followed by 5 sec at 85°C for complementary DNA (cDNA)
synthesis. Real-time reactions were performed using the SYBR
PrimeScript™ RT-PCR kit (Takara Biotechnology) under the following
conditions: 30 sec at 95°C for 1 cycle, 5 sec at 95°C, 20 sec at
60°C for 40 cycles, 95°C for 0 sec, 65°C for 15 sec, and 95°C for 0
sec for melting curve analysis. The PCR primers (Sangon Biotech
Co., Ltd., Shanghai, China) are listed in Table III. The relative mRNA expression
level of GnT-V in each sample was calculated using the comparative
expression level 2−ΔΔCT method. The expression of GnT-V
protein was detected by western blot assay. Cells (107)
were harvested and lysed with ice-cold lysis buffer (RIPA and a
mixture of protease inhibitors; Beyotime Institute of
Biotechnology, Haimen, China). Protein concentration of the
supernatant was determined by the BCA protein assay procedure.
Equal amounts of proteins were separated by 10% SDS-PAGE,
respectively. Proteins were then transferred to polyvinylidene
difluoride membrane using a semi-dry transfer apparatus. The
membranes were blocked in Tris-buffered saline (TBS) with 5%
non-fat milk for 1 h at room temperature, followed by incubation
with the appropriate primary antibodies (1:500-diluted antibody of
GnT-V, Abcam) at 4°C overnight. After washing in TBS-Tween 20
buffer, membranes were incubated for 2 h with the appropriate
peroxidase-conjugated secondary antibodies, then the protein bands
on the membranes were visualized using an ECL kit (Beyotime
Institute of Biotechnology). The film was scanned and processed
with the Odyssey Infrared Imaging system. Protein bands were
quantified by Quantity One. The densitometric value of each protein
band was normalized to GAPDH.
| Table IIISequence of primers. |
Table III
Sequence of primers.
| Primers | Sequences |
|---|
| GnT-V F |
5′-gaaaatggaatctgaaccctca-3′ |
| GnT-V R |
5′-actttgccatacacaagggact-3′ |
| GAPDH F |
5′-attgccctcaacgaccactt-3′ |
| GAPDH R |
5′-aggtccaccaccctgttgct-3′ |
| EGFR F |
5′-cgcaaagtgtgtaacggaatag-3′ |
| EGFR R |
5′-ccagaggaggagtatgtgtgaa-3′ |
| ErbB2 F |
5′-ttggtcactctgctgctgtaag-3′ |
| ErbB2 R |
5′-cttcattttggtagagccgaac-3′ |
| ErbB3 F |
5′-tgtaaggctgctgggactatg-3′ |
| ErbB3 R |
5′-gaacctgactgggtgacttga-3′ |
| ErbB4 F |
5′-ggggaataacattgcttcagag-3′ |
| ErbB4 R |
5′-ttaggaaggacaaggagaccaa-3′ |
| E-cadherin F |
5′-tgcccagaaaatgaaaaagg-3′ |
| E-cadherin R |
5′-gtgtatggcaatgcgttc-3′ |
| Vimentin F |
5′-gggacctctacgaggaggag-3′ |
| Vimentin R |
5′-cgcattgtcaacatcctgtc-3′ |
| MMP-2 F |
5′-ccactgccttcgatacac-3′ |
| MMP-2 R |
5′-gagccactctctggaatcttaaa-3′ |
| MMP-9 F |
5′-gttcccggagtgagttga-3′ |
| MMP-9 R |
5′-tttacatggcactgccaaagc-3′ |
Lectin blot assay
Cells were harvested and lysed. Proteins extracted
from the cells were electrophoresed on 10% SDS-PAGE, and
protein-blotted PDVF membranes were prepared in exactly the same
way as described for western blot analysis. After blocking with
blocking solution [PBS containing 0.5% (w/v) Tween 20], the
membranes were incubated with 1 μg/ml L-PHA (Sigma) for 2 h.
Reactive bands were detected with a diluted BCIP/NBT in
ExtrAvidin-AP buffer. The colorimetric reaction is normally
completed within 10–20 min. The blotted proteins are colored in
blue.
Cell proliferation assay (CCK-8)
Cells were seeded in 96-well plates at
2×103 cells/well. At the indicated times (0, 24, 48, 72
and 96 h), 10 μl of Cell Counting Kit-8 (CCK-8; Beyotime Institute
of Biotechnology) solution and 100 μl RPMI-1640 plus 10% FBS were
added to each well. The cells were incubated for 60 min, and the
absorbance at 450 nm was measured to calculate cell growth rates.
Growth rate = (absorbance at 450 nm at × h - absorbance at 450 nm
at 0 h)/(absorbance at 450 nm at 0 h).
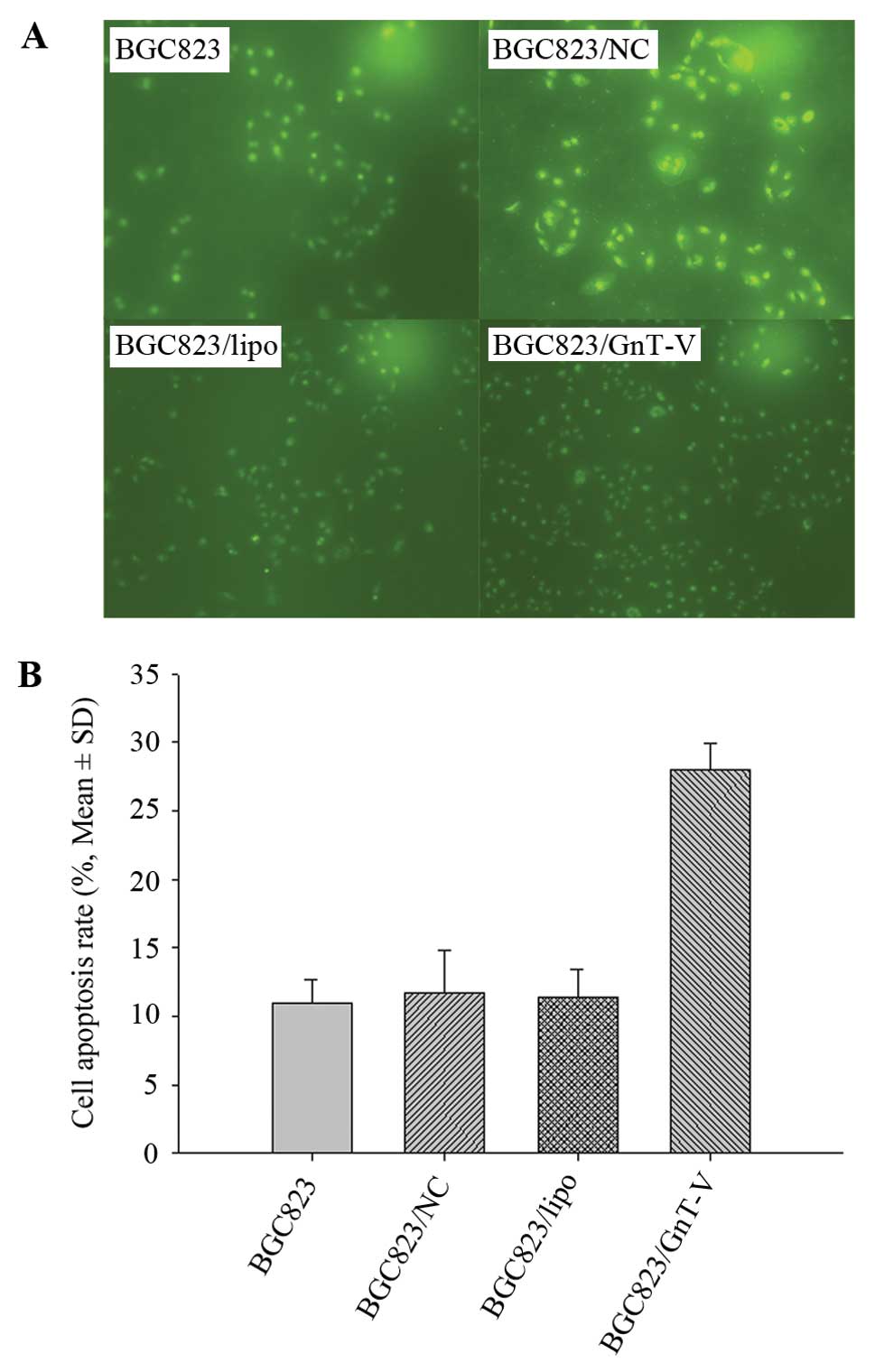
TUNEL assay for apoptosis detection
All-trans retinoic acid (ATRA) (80 mM) was
used to induce apoptosis in cells. Cells were treated with 80 mM
ATRA for 24 h then adherent cells were fixed with 4%
paraformaldehyde. Subsequently, the detection of apoptosis was
preceded by TdT-mediated dUTP nick end labeling (TUNEL) assay
according to the manufacturer’s instructions (in situ cell
death detection, Fluorescein kit, Roche). Apoptotic cells colored
green were observed under a fluorescence microscope. The numbers of
green fluorescence cells were counted in five different fields,
selected randomly.
Cell scratch-wound assay
Cells were seeded on 24-well plates and grown to a
monolayer. Wound areas were scraped using 20-μl plastic tips. At
the indicated times (0 and 24 h), the wound areas were photographed
and the wound healing rate was calculated. Healing rate = (width of
wound at 24 h - width of the wound at 0 h)/width of wound at 0
h.
Cell migration assay
To evaluate cell migration capability, Transwell
plates (Corning Incorporated) with a 6.5-mm diameter filter and an
8.0-μm pore size were used. Transwell chambers were inserted into a
24-well plate. Cells (1×105) were plated in the upper
compartment in 200 μl of serum-free medium/chamber, and 500 μl of
complete medium was added to the lower wells. The chambers were
incubated for 24 h at 37°C in 5% CO2 to allow cells to
migrate from the upper chamber to the lower well. Cells migrating
through the pores and adherent on the undersurface of the membrane
were stained with Giemsa reagent. The number of cells was counted
in five different fields. These fields were selected randomly.
Cell invasion assay
The cell invasion assay was performed using 24-well
Transwell units with 8-μm pore size polycarbonate inserts
(Matrigel™ Invasion Chamber, BD Biosciences). Cells that were
suspended in RPMI-1640 plus 10% FBS were added to each upper
compartment of the Transwell units. After being cultured for 24 h,
cells migrating through the Matrigel-coated polycarbonate membrane
were fixed using 4% paraformaldehyde, and then stained with Giemsa
reagent. The numbers of invasive cells were counted in five
different fields. These fields were selected randomly.
RNA isolation and quantitative real-time
polymerase chain reaction
Total RNA was isolated from cells using the TRIzol
reagent (Invitrogen). The product was reverse-transcribed into
first-strand complementary DNA (cDNA). Thereafter, the expression
levels of EGFR, ErbB2, ErbB3 and ErbB4, E-cadherin, vimentin,
MMP-2/MMP-9, were measured using the SYBR PrimeScript™ RT-PCR Mix
(Takara) according to the manufacturer’s protocol. GAPDH was used
to normalize the mRNA. Sequence-specific primers were designed as
shown in Table III. Real-time PCR
(40 cycles of denaturation at 92°C for 15 sec and annealing at 60°C
for 60 sec) was run on a LightCycler application system version
1.5.
Western blot assay
Cells were washed with phosphate-buffered saline
(PBS) twice and lysed with ice-cold lysis buffer (RIPA) (Beyotime
Institute of Biotechnology) and a mixture of protease inhibitors.
The cell lysates were centrifuged at 14,000 × g for 15 min at 4°C.
The supernatant was collected, and the protein concentration was
determined by BCA kit (Beyotime Institute of Biotechnology). Equal
amounts of proteins were separated by 10 or 8% SDS-PAGE,
respectively. Proteins were then transferred to PVDF membranes. The
membranes were blocked in Tris-buffered saline (TBS) with 5%
non-fat milk for 1 h at room temperature, followed by incubation
with the appropriate primary antibodies (a 1:500 dilution of the
antibody for MMP-2; a 1:5,000 dilution of the antibody for MMP-9; a
1:1,000 dilution of the antibody for E-cadherin; a 1:200 dilution
of the antibody for vimentin; a 1:500 dilution of the antibodies of
EGFR, ErbB2, ErbB3 and ErbB4) at 4°C overnight. All antibodies were
purchased from Abcam Co., Ltd., Cambridge, MA, USA. After washing
in TBS-Tween 20 buffer, membranes were incubated for 2 h with the
appropriate peroxidase-conjugated secondary antibodies. After
washing in TBS-Tween 20 buffer, the protein bands on the membranes
were visualized using an ECL kit. The film was scanned and
processed with Odyssey Infrared Imaging system. Protein bands were
quantified by Quantity One. The densitometric value of each protein
band was normalized to GAPDH.
Statistical analysis
Data are presented using means ± SD. Statistical
comparisons of groups were performed using one-way analysis of
variance (ANOVA), and statistical significance was defined as
P<0.05. All the analyses were performed using SPSS 13.0
software.
Results
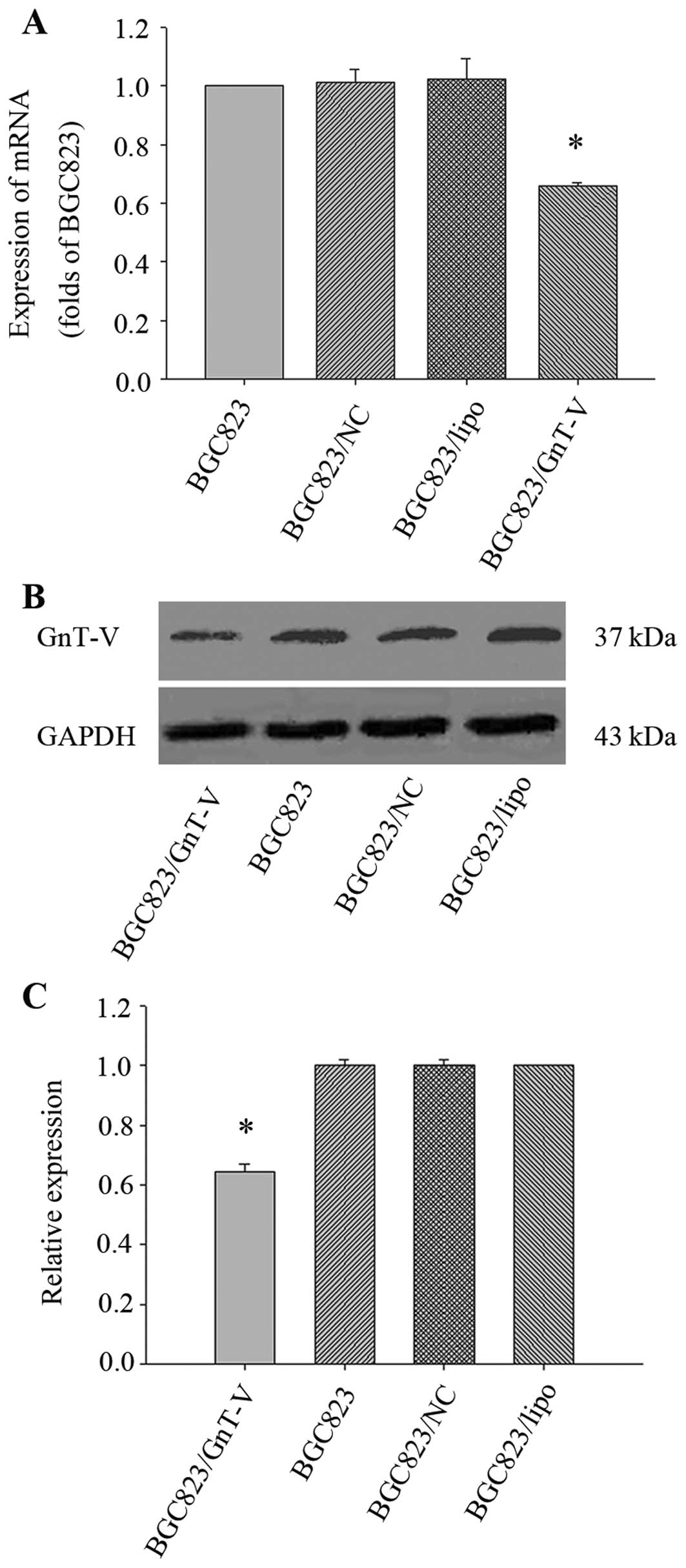
Downregulation of the GnT-V gene in
BGC823 cells
We first explored the effect of oligo-GnT-V siRNA on
GnT-V expression in BGC823 cells. After being transfected by the
constructed oligo-GnT-V siRNA, BGC823 cells were harvested at 24
and 48 h for future use. The result showed that the expression
levels of GnT-V mRNA and protein in BGC823/GnT-V cells were
decreased by 33.87±0.01 and 35.69±2.67% when compared to the BGC823
cells (P<0.05) (Fig. 1). Thus,
the BGC823/GnT-V cells were used in all subsequent experiments.
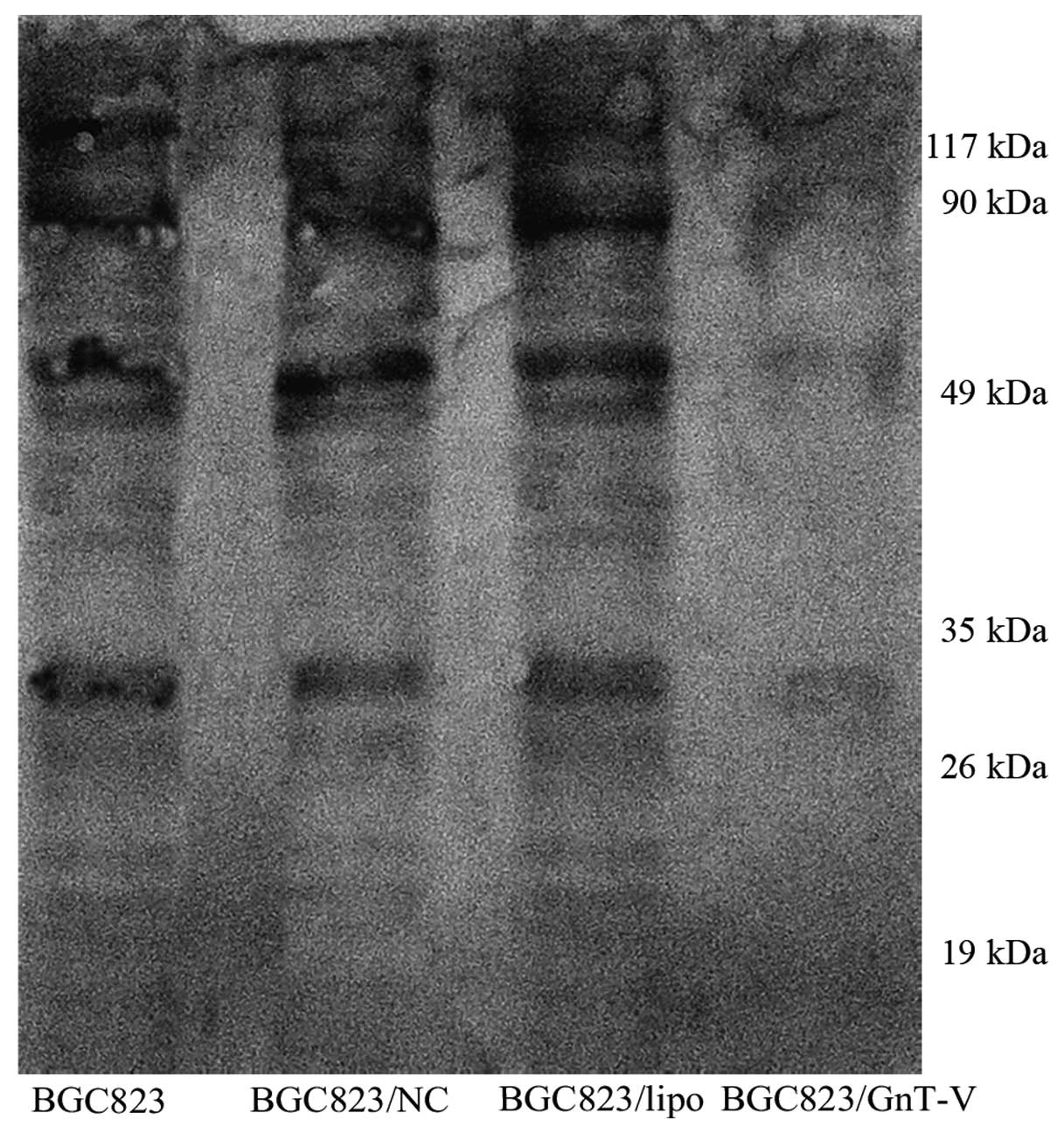
β1–6 branched N-oligosaccharides are
decreased in the BGC823 GnT-V cells
β1–6 branching of asparagine-linked
oligosaccharides, which are the products of GnT-V, have been shown
to correlate with tumor grade and metastasis. We verified here that
GnT-V suppression contributed to the reduction in β1–6 branched
N-oligosaccharides in the BGC823/GnT-V cells (Fig. 2). We performed lectin blot analysis
on total cellular proteins using L-PHA, which preferentially binds
to GlcNAc residues on β1–6 branches of tri- or tetra-antennary
sugar chains. This analysis showed that GnT-V catalyzed specific
glycosylation to target glycoproteins, of which major molecular
sizes were ~10–120 kDa. The result suggests that target substrates
of GnT-V were repressed efficiently.
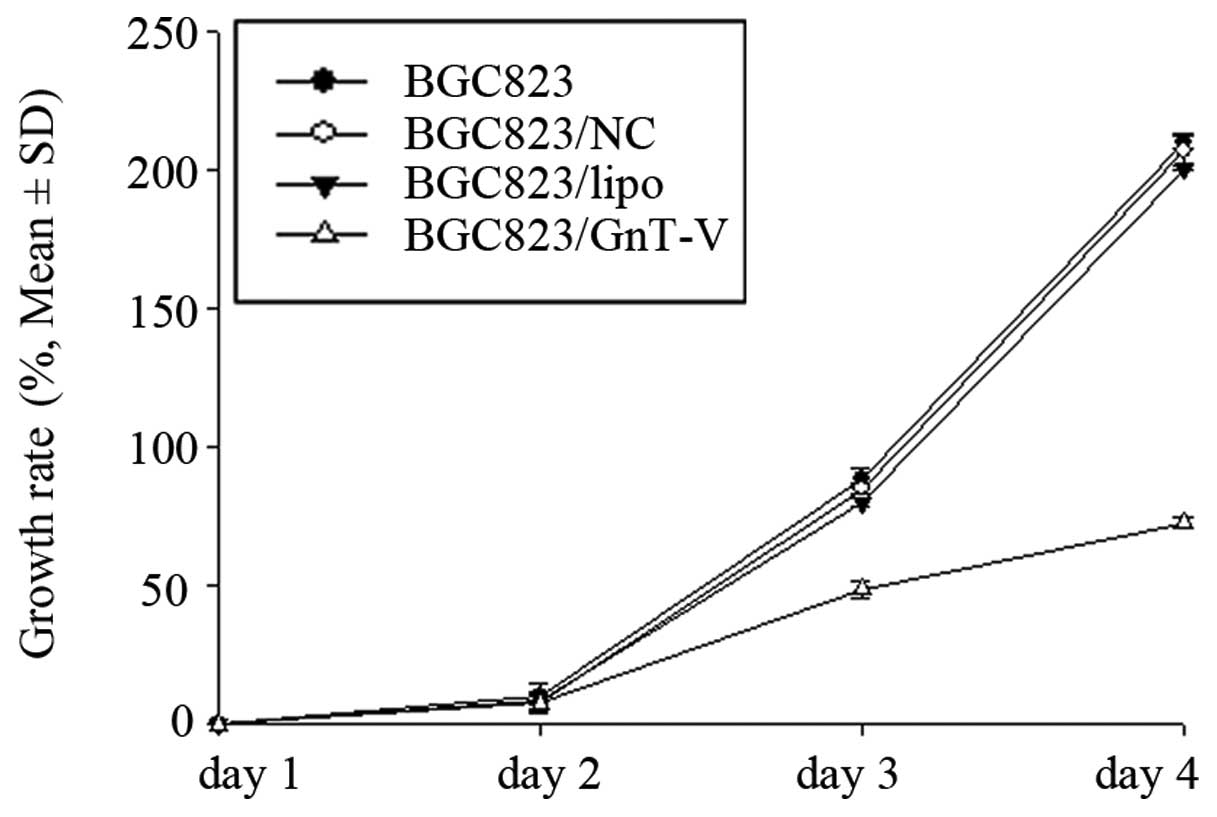
Proliferation of BGC823 cells is
inhibited by downregulation of GnT-V
A previous study showed that downregulation of GnT-V
induced significant growth suppression in a nasopharyngeal
carcinoma cell line (31). We aimed
to ascertain whether downregulation of GnT-V in gastric cancer
cells contributes to inhibition of cell proliferation. We addressed
this question using the CCK-8 assay to characterize the growth
ability and TUNEL assay to evaluate the apoptosis rates induced by
ATRA in BGC823 cells. As shown in Fig.
3, the growth rates of the BGC823/GnT-V cells were lower than
those of BGC823, BGC823/NC and BGC823/lipo cells over a 4-day
period. TUNEL assay revealed that the apoptosis rates of the
BGC823/GnT-V, BGC823, BGC823/NC and BGC823/lipo cells following
treatment with 80-mM ATRA for 24 h were 27.97±1.96, 11.00±1.67,
11.24±3.19 and 11.39±2.08%, respectively (P<0.05) (Fig. 4), which suggests that BGC823/GnT-V
cells are more susceptible to apoptosis induced by ATRA. These
results showed that downregulation of GnT-V inhibited the
proliferation of BGC823 cells.
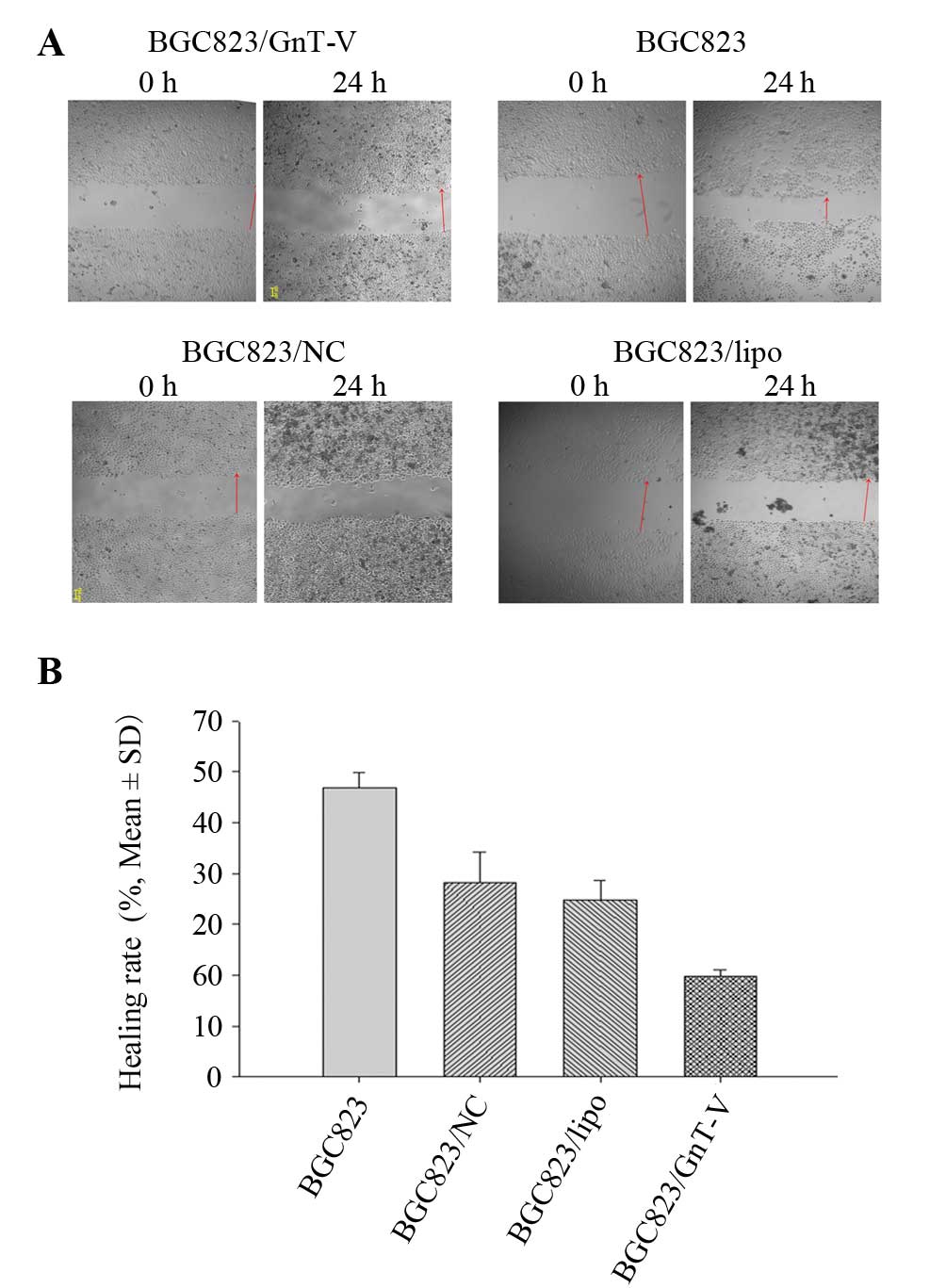
Migration and invasion of BGC823 cells
are inhibited by downregulation of GnT-V
The scratch-wound and Transwell migration assays
were applied to detect cell migration ability. Wound healing rates
of the BGC823, BGC823/NC and BGC823/lipo cells were higher than
that of the BGC823/GnT-V cells at 24 h (Fig. 5A and B). The result of the Transwell
migration assay was consistent with that of the scratch-wound
assay. More BGC823, BGC823/NC and BGC823/lipo cells migrated
through the membrane of the Transwell unit than BGC823/GnT-V cells
(P<0.05) (Fig. 5C and D).
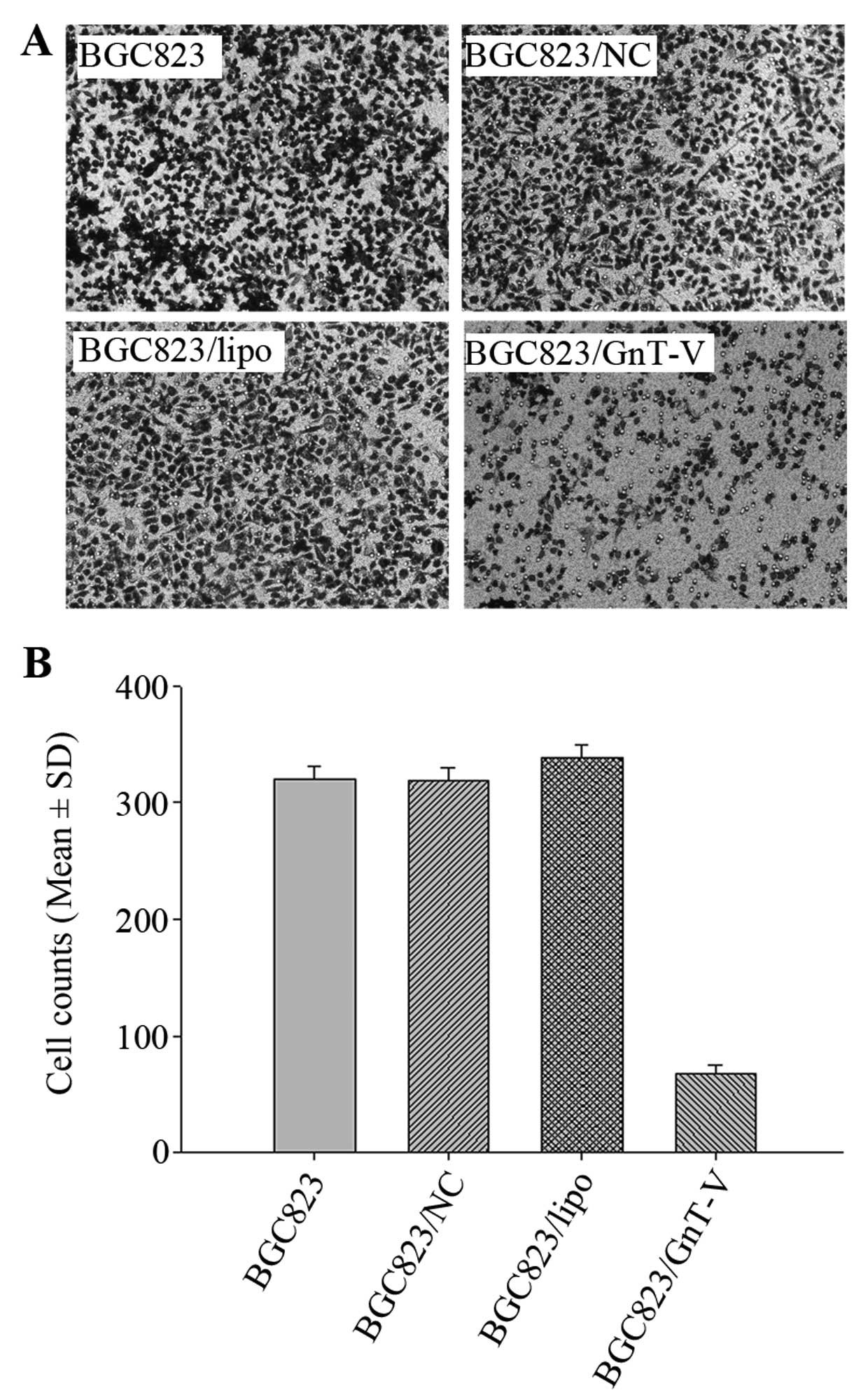
The invasive ability of the cells was determined by
a Matrigel-coated Transwell assay. Fig.
6 shows that less BGC823/GnT-V cells penetrated the
Matrigel-coated membrane when compared with the number of invading
BGC823, BGC823/NC and BGC823/lipo cells (P<0.05).
Expression of invasion-related factors in
the BGC823/GnT-V cells
To elucidate the mechanism by which GnT-V
downregulation affects EGFR-mediated aberration of the EMT
phenotype and MMP-9/MMP-2 expression, the expression levels of
EGFRs, E-cadherin/vimentin and MMP-2/MMP-9 were investigated. A
different expression pattern for EGFR/ErbB2 and ErbB3/ErbB4 was
found in the cells. EGFR and ErbB2 were decreased in the
BGC823/GnT-V cells both at the mRNA and protein levels. In
contrast, ErbB3 in the BGC823/GnT-V cells showed no difference when
compared with the other cells both at the mRNA and protein levels.
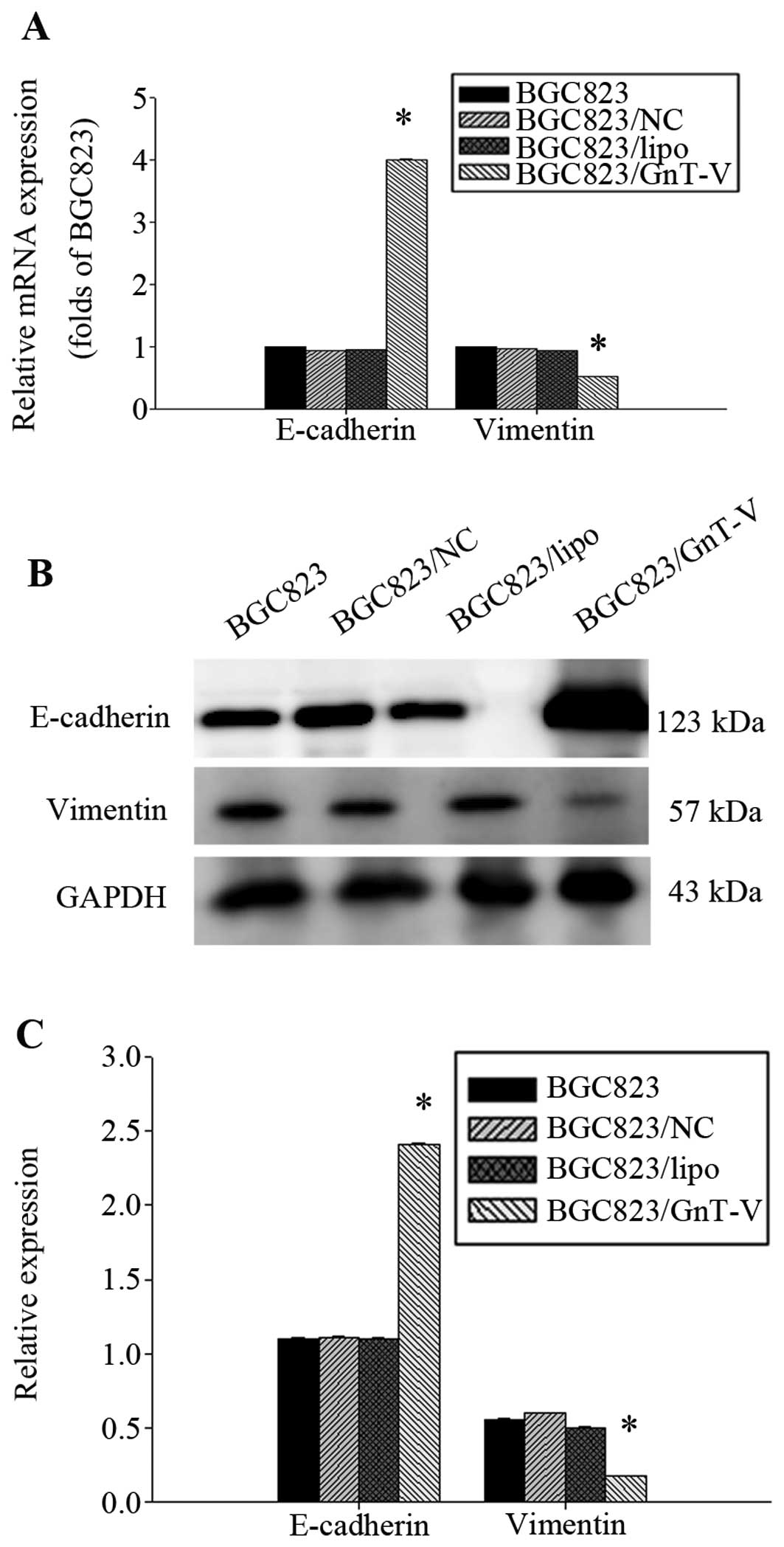
Yet, ErbB4 protein in the BGC823/GnT-V cells was deceased (Fig. 7). E-cadherin and vimentin are
important regulatory markers for EMT. We found that the E-cadherin
expression level was significantly higher in the BGC823/GnT-V
cells, and vimentin expression was lower in the BGC823/GnT-V cells
(Fig. 8). MMP-9 expression was
decreased in the BGC823 GnT-V cells both at the protein and mRNA
levels (Fig. 9). No MMP-2
expression was detected (data not shown). Taken together, our
results suggest that downregulation of GnT-V disturbed the levels
of EGFR, E-cadherin/vimentin and MMP-9 expression, thereby
subsequently inhibiting the process of metastasis and invasion.
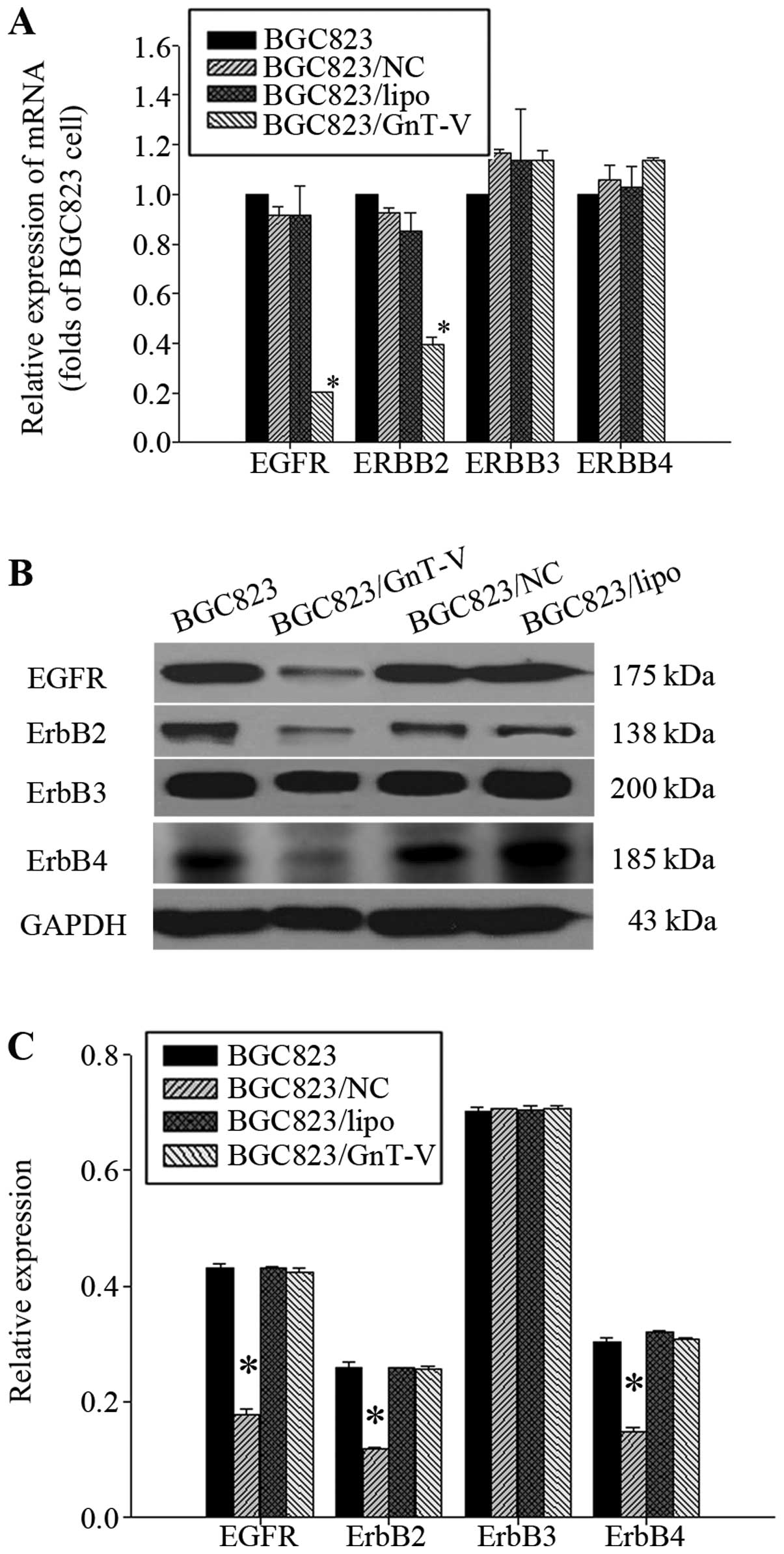 | Figure 7Expression of EGFRs. (A) Effect of the
downregulation of GnT-V on the mRNA expression of EGFRs was
determined by qRT-PCR. The mRNA expression of EGFR and ErbB2 in
BGC823/GnT-V cells was decreased respectively, by 81.07±0.06 and
60.50±1.19% when compared to the BGC823 cells (P<0.05). The mRNA
expression of ErbB3 and ErbB4 in BGC823/GnT-V, BGC823, BGC823/NC
and BGC823/lipo cells had no significant differences (P>0.05).
(B) EGFR, ErbB2, ErbB3 and ErbB4 protein expression in cells was
determined by western blot assay. (C) Protein bands were quantified
by Quantity One. The densitometric value of each protein band was
normalized to GAPDH. The result are displayed on a bar diagram. The
EGFR, ErbB2 and ErbB4 protein levels in BGC823/GnT-V cells when
compared with the BGC823 cells were decreased by 55.21±0.01,
50.54±0.00 and 47.02±0.19%, respectively (P<0.05). The
expression of ErbB3 protein in BGC823/GnT-V, BGC823, BGC823/NC and
BGC823/lipo cells had no significant differences (P>0.05).
*Statistical significant. |
Discussion
N-acetylglucosaminyltransferase V (GnT-V) catalyzes
β1–6 branching of N-acetylglucosamine on asparagine-linked
oligosaccharides of cell proteins (21,32,33).
The roles of GnT-V in the metastasis/invasion of various types of
tumors are conflicting. Upregulated GnT-V activity has been
reported in human colon cancer, hepatocarcinoma and breast cancer
tissues (6–8,34).
However, increased GnT-V activity has been found to be associated
with favorable stages as well as favorable prognosis in non-small
cell lung cancers, bladder carcinomas and neuroblastoma (9–11). In
the present study, we investigated the role of GnT-V in gastric
cancer during the process of metastasis and invasion. The
BGC823/GnT-V, BGC823, BGC823/NC and BGC823/lipo cells were cultured
for assaying. It was found that downregulation of GnT-V expression
was accompanied by a reduction in β1–6 N-acetylglucosamine
branches, inhibition of cell growth and enhanced cell apoptosis
induced by ATRA, resulting in inhibition of BGC823/GnT-V cell
proliferation in vitro. Furthermore, the metastatic and
invasive potential of BGC823/GnT-V cells was less than that of the
other cell groups. These results demonstrated that targeted
suppression of GnT-V reduced proliferation and invasion ability of
BGC823 cells in vitro.
It is well known that EGFR-mediated signaling plays
a crucial role in the control of cell metastasis and invasion in
epithelial and cancer cells. To examine whether EGFRs are also
required in the process of gastric cancer metastasis and invasion,
EGFR, ErbB1, ErbB2 and ErbB3 levels were evaluated in BGC823 cells.
The results indicated that suppression of GnT-V may decrease EGFR
and ErbB2 gene transcript activity by reducing glycosylation of the
transcription factors involved. Further investigation may be useful
to answer this problem in the future. The decreased expression of
ErbB4 at the protein level was induced by reduction of β1–6 GlcNAc
branching accompanied by GnT-V suppression as hypothesized before.
As EGFR signaling is essential for tumor cell proliferation and
migration/invasion, we assumed that downregulation of GnT-V
inhibits proliferation and migration/invasion of gastric cancer
cells by modulated EGFR signaling.
On the other hand, modification of E-cadherin
N-glycans by GnT-V plays a role in tumor metastasis, as GnT-V has
been reported to delocalize E-cadherin to the cytoplasm by
post-transcriptional modification of E-cadherin (35,36).
However, little is known about the post-transcriptional
modifications of E-cadherin and its role in E-cadherin-mediated
tumor progression in gastric cancer cells. A substantial body of
evidence has appeared to support the view that the E-cadherin
function may mainly be affected by mechanisms through
N-glycosylation at the post-translational level in some carcinomas
(37–39). However, our data demonstrated that
downregulation of GnT-V affected the transcripts of E-cadherin and
vimentin, and further influenced the protein expression of
E-cadherin and vimentin.
The MMP family is considered to be one of the most
important factors in tumor invasion. Particularly, the high
expression of MMP-9 is considered as a crucial factor for cell
migration/invasion of endothelial cells into the adjacent stroma
(30). It is essential to elucidate
the molecular mechanisms underlying the N-glycan regulation of the
invasive function of MMP-9 in gastric cancer biology. The present
study showed that MMP-9 expression in gastric cancer was
decreased.
In conclusion, downregulation of the GnT-V gene
inhibits invasion of gastric cancer BGC823 cells in vitro.
The underlying mechanisms may be linked to EGFR signaling-initiated
EMT phenotype and MMP-9 expression. These findings suggest that
GnT-V may be a potential target for predicting the invasive
potential of gastric cancer.
Acknowledgements
The present study was supported by a grant from the
National Science Foundation of China (no. 81170333) and the
Shanghai Committee of Science and Technology Key Projects for Basic
Research (no. 12JC1408402).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Nagini S: Carcinoma of the stomach: a
review of epidemiology, pathogenesis, molecular genetics and
chemoprevention. World J Gastrointest Oncol. 4:156–169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Danaei G, Vander Hoorn S, Lopez AD, Murray
CJ and Ezzati M; Comparative Risk Assessment collaborating group
(cancers). Causes of cancer in the world: comparative risk
assessment of nine behavioural and environmental risk factors.
Lancet. 366:1784–1793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Catalano V, Labianca R, Beretta GD, Gatta
G, de Braud F and Van Cutsem E: Gastric cancer. Crit Rev Oncol
Hematol. 71:127–164. 2009. View Article : Google Scholar
|
|
5
|
Przybyło M, Pocheć E, Link-Lenczowski P
and Lityńska A: Beta 1–6 branching of cell surface glycoproteins
may contribute to uveal melanoma progression by up-regulating cell
motility. Mol Vis. 14:625–636. 2008.
|
|
6
|
Murata K, Miyoshi E, Kameyama M, et al:
Expression of N-acetylglucosaminyltransferase V in colorectal
cancer correlates with metastasis and poor prognosis. Clin Cancer
Res. 6:1772–1777. 2000.PubMed/NCBI
|
|
7
|
Guo P, Chen HJ, Wang QY and Chen HL:
Downregulation of N-acetylglucosaminyltransferase V facilitates
all-trans retinoic acid to induce apoptosis of human
hepatocarcinoma cells. Mol Cell Biochem. 284:103–110.
2005.PubMed/NCBI
|
|
8
|
Xu YY, Lu Y, Fan KY and Shen ZH: Apoptosis
induced by all-trans retinoic acid in
N-acetylglucosaminyltransferase V repressed human hepatocarcinoma
cells is mediated through endoplasmic reticulum stress. J Cell
Biochem. 100:773–782. 2007.
|
|
9
|
Dosaka-Akita H, Miyoshi E, Suzuki O, Itoh
T, Katoh H and Taniguchi N: Expression of
N-acetylglucosaminyltransferase V is associated with prognosis and
histology in non-small cell lung cancers. Clin Cancer Res.
10:1773–1779. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishimura H, Takahashi T, Nakagawa H, et
al: N-acetylglucsaminyltransferase V and beta1–6 branching N-linked
oligosaccharides are associated with good prognosis of patients
with bladder cancer. Clin Cancer Res. 12:2506–2511. 2006.
|
|
11
|
Inamori K, Gu J, Ohira M, et al: High
expression of N-acetylglucosaminyltransferase V in favorable
neuroblastomas: involvement of its effect on apoptosis. FEBS Lett.
580:627–632. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian H, Miyoshi E, Kawaguchi N, et al: The
implication of N-acetylglucosaminyltransferase V expression in
gastric cancer. Pathobiology. 75:288–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsieh CY, Tsai PC, Tseng CH, Chen YL,
Chang LS and Lin SR: Inhibition of EGF/EGFR activation with
naphtho(1,2-b)furan-4,5-dione blocks migration and invasion of
MDA-MB-231 cells. Toxicol In Vitro. 27:1–10. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baselga J and Arteaga CL: Critical update
and emerging trends in epidermal growth factor receptor targeting
in cancer. J Clin Oncol. 23:2445–2459. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rothhut B, Ghoneim C, Antonicelli F and
Soula-Rothhut M: Epidermal growth factor stimulates matrix
metalloproteinase-9 expression and invasion in human follicular
thyroid carcinoma cells through focal adhesion kinase. Biochimie.
89:613–624. 2007. View Article : Google Scholar
|
|
16
|
Mascia F, Cataisson C, Lee TC, et al: EGFR
regulates the expression of keratinocyte-derived
granulocyte/macrophage colony-stimulating factor in vitro and in
vivo. J Invest Dermatol. 130:682–693. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastore S, Mascia F, Mariani V and
Girolomoni G: The epidermal growth factor receptor system in skin
repair and inflammation. J Invest Dermatol. 128:1365–1374. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pastore S and Mascia F: Novel acquisitions
on the immunoprotective roles of the EGF receptor in the skin.
Expert Rev Dermatol. 3:525–527. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kimura A, Terao M, Kato A, et al:
Upregulation of N-acetylglucosaminyltransferase-V by
heparin-binding EGF-like growth factor induces keratinocyte
proliferation and epidermal hyperplasia. Exp Dermatol. 21:515–519.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao Y, Sato Y, Isaji T, et al: Branched
N-glycans regulate the biological functions of integrins and
cadherins. FEBS J. 275:1939–1948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gu J, Zhao Y, Isaji T, et al:
Beta1,4-N-acetylglucosaminyltransferase III down-regulates neurite
outgrowth induced by costimulation of epidermal growth factor and
integrins through the Ras/ERK signaling pathway in PC12 cells.
Glycobiology. 14:177–186. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao Y, Nakagawa T, Itoh S, et al:
N-acetylglucosaminyl transferase III antagonizes the effect of
N-acetylglucosaminyl transferase V on α3β1 integrin-mediated cell
migration. J Biol Chem. 281:32122–32130. 2006.
|
|
23
|
Chavez MG, Buhr CA, Petrie WK,
Wandinger-Ness A, Kusewitt DF and Hudson LG: Differential
downregulation of E-cadherin and desmoglein by epidermal growth
factor. Dermatol Res Pract. 2012:3095872012.PubMed/NCBI
|
|
24
|
Terao M, Ishikawa A, Nakahara S, et al:
Enhanced epithelial-mesenchymal transition-like phenotype in
N-acetylglucosaminyltransferase V transgenic mouse skin promotes
wound healing. J Biol Chem. 286:28303–28311. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dilly M, Hambruch N, Haeger JD and Pfarrer
C: Epidermal growth factor (EGF) induces motility and upregulates
MMP-9 and TIMP-1 in bovine trophoblast cells. Mol Reprod Dev.
77:622–629. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Delassus GS, Cho H, Park J and Eliceiri
GL: New pathway links from cancer-progression determinants to gene
expression of matrix metalloproteinases in breast cancer cells. J
Cell Physiol. 217:739–744. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fink K and Boratyński J: The role of
metalloproteinases in modification of extracellular matrix in
invasive tumor growth, metastasis and angiogenesis. Postepy Hig Med
Dosw (Online). 66:609–628. 2012.(In Polish).
|
|
29
|
Partridge JJ, Madsen MA, Ardi VC,
Papagiannakopoulos T, Kupriyanova TA, Quigley JP and Deryugina EI:
Functional analysis of matrix metalloproteinases and tissue
inhibitors of metalloproteinases differentially expressed by
variants of human HT-1080 fibrosarcoma exhibiting high and low
levels of intravasation and metastasis. J Biol Chem.
282:35964–35977. 2007. View Article : Google Scholar
|
|
30
|
Di Carlo A: Matrix metalloproteinase-2 and
-9 in the sera and in the urine of human oncocytoma and renal cell
carcinoma. Oncol Rep. 28:1051–1056. 2012.PubMed/NCBI
|
|
31
|
Zhuo E, He J, Wei T, et al:
Down-regulation of GnT-V enhances nasopharyngeal carcinoma cell
CNE-2 radiosensitivity in vitro and in vivo. Biochem Biophys Res
Commun. 424:554–562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Beheshti Zavareh R, Sukhai MA, Hurren R,
et al: Suppression of cancer progression by MGAT1 shRNA knockdown.
PLoS One. 7:e437212012.PubMed/NCBI
|
|
33
|
Xu Q, Isaji T, Lu Y, et al: Roles of
N-acetylglucosaminyltrans-ferase III in epithelial-to-mesenchymal
transition induced by transforming growth factor β1 (TGF-β1) in
epithelial cell lines. J Biol Chem. 287:16563–16574.
2012.PubMed/NCBI
|
|
34
|
Li D, Li Y, Wu X, et al: Knockdown of
Mgat5 inhibits breast cancer cell growth with activation of
CD4+ T cells and macrophages. J Immunol. 180:3158–3165.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pinho SS, Reis CA, Paredes J, et al: The
role of N-acetylglucosaminyltransferase III and V in the
post-transcriptional modifications of E-cadherin. Hum Mol Genet.
18:2599–2608. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pinho SS, Seruca R, Gärtner F, Yamaguchi
Y, Gu J, Taniguchi N and Reis CA: Modulation of E-cadherin function
and dysfunction by N-glycosylation. Cell Mol Life Sci.
68:1011–1020. 2011.PubMed/NCBI
|
|
37
|
Zhao H, Liang Y, Xu Z, et al:
N-glycosylation affects the adhesive function of E-Cadherin through
modifying the composition of adherens junctions (AJs) in human
breast carcinoma cell line MDA-MB-435. J Cell Biochem. 104:162–175.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu T, Zhang X, Shang M, et al:
Dysregulated expression of Slug, vimentin, and E-cadherin
correlates with poor clinical outcome in patients with basal-like
breast cancer. J Surg Oncol. 107:188–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Roberti MP, Arriaga JM, Bianchini M, et
al: Protein expression changes during human triple negative breast
cancer cell line progression to lymph node metastasis in a
xenografted model in nude mice. Cancer Biol Ther. 13:1123–1140.
2012. View Article : Google Scholar : PubMed/NCBI
|