Introduction
The epidermal growth factor receptor (EGFR) is a
member of the EGFR receptor tyrosine kinase (RTK) family, together
with ErB2/Neu/HER2, Erb3/HER3 and ErbB4/HER4 (1). Activating ligands of these receptors
include epidermal growth factor (EGF), transforming growth factor α
(TGF-α), epiregulin, neuregulins and amphiregulin. Binding of the
ligands to the extracellular domain of EGFR RTKs causes the
formation of homodimers or heterodimers, cross-activation of
tyrosine kinase domains and autophosphorylation. These events
result in the stimulation of intracellular signaling cascades,
including the phosphatidylinositol-3 kinase (PI3K)/Akt and
mitogen-activated protein kinase (MAPK) cascades. Although EGFR
signaling is essential for normal tissue development and
homeostasis, the abnormal activity of EGFR RTK members plays a key
role in tumor initiation and progression (2).
Glioblastoma multiforme (GBM) is the most frequent
and malignant neoplasm of the human nervous system. Despite current
therapies, the prognosis for patients with GBMs remains poor, with
a median survival time that ranges from 8.8 months (patients <50
years of age) to 1.8 months (patients >80 years of age)
(3,4). EGFR gene amplification and EGFR
overexpression are common features of GBM; however, they are rare
in low-grade gliomas, which suggests a causal role of aberrant EGFR
signaling in the pathogenesis of GBM (5). The amplification of EGFR is
present in 35–70% of cases, typically in primary de novo GBM
(4,6,7).
In our previous study, we reported the unusual loss
of EGFR gene copy in GBM obtained from a patient with an
unusually favorable course of the disease who experienced a
survival of 5.5 years between diagnosis and death (8). Here, we present further detailed
analysis of the HGG-02 cell line derived from this tumor when
compared with the GM7 reference cell line that contains a standard
EGFR gene copy number (9).
Materials and methods
Cell lines and cell culture
The HGG-02 (8) and
GM7 (9) established glioblastoma
cell lines were used in the present study. Cell cultures were
maintained in DMEM supplemented with 20% fetal calf serum, 2 mM
glutamine and antibiotics (100 IU/ml of penicillin and 100 μg/ml of
streptomycin) (PAA Laboratories, Linz, Austria) under standard
conditions at 37°C in an atmosphere of 95% air and 5%
CO2. The cells were subcultivated once a week. Where
indicated, the cells were starved in serum-free media.
RT-PCR
For RT-PCR, total RNA was extracted with the
GenElute™ Mammalian Total RNA Miniprep kit (Sigma-Aldrich, St.
Louis, MO, USA). For all samples, equal amounts of RNA (25 ng of
RNA/1 μl of total reaction content) were reverse transcribed into
cDNA using M-MLV (Top-Bio, Prague, Czech Republic) and oligo-dT
(Qiagen Inc., Valencia, CA, USA) priming. Primers for EGFR, ErbB2,
ErbB3, ErbB4, TGF-α, EGF, epiregulin (EREG), neuroglycan C (NGC),
NRG-1, NRG-2, PDGFRα, PDGFRβ, PDGFA, PDGFB, HSP90AB1 and GAPDH were
used as described in Table I.
 | Table IPrimer sequences used for RT-PCR. |
Table I
Primer sequences used for RT-PCR.
| Gene symbol | Gene full name | Forward primer | Reverse primer | Size (bp) |
|---|
| EGFR | Epidermal growth
factor receptor |
5′-AGAAAGGCAGCCACCAAATTAGCC-3′ |
5′-TTCCTGGCTAGTCGGTGTAAACGT-3′ | 304 |
| ErbB2 | V-erb-b2
erythroblastic leukemia viral oncogene homolog 2 |
5′-CCTGAATATGTGAACCAGC-3′ |
5′-ACCTCTTGATGCCAGCAGAA-3′ | 440 |
| ErbB3 | V-erb-b2
erythroblastic leukemia viral oncogene homolog 3 |
5′-GGGTATGAAGAGATGAGAGC-3′ |
5′-TCAAGAGCCTGAGAGGAA-3′ | 496 |
| ErbB4 | V-erb-b2
erythroblastic leukemia viral oncogene homolog 4 |
5′-TACCGAGATGGAGGTTTTGC-3′ |
5′-GTTGGCAAAGGTGTTGAGGT-3′ | 447 |
| TGF-α | Transforming growth
factor α |
5′-CCTGCTGCCCGCCCGCCCGT-3′ |
5′-GCTGGCAGCCACCACGGCCA-3′ | 305 |
| EGF | Epidermal growth
factor |
5′-TGCCAACTGGGGGTGCACAG-3′ |
5′-CTGCCCGTGGCCAGCGTGGC-3′ | 339 |
| EREG | Epiregulin |
5′-TCCAGTGTCAGAGGGACACA-3′ |
5′-GGTTGGTGGACGGTTAAAAA-3′ | 488 |
| NGC | Neuroglycan C |
5′-CCCCACCACATCCTTTTATG-3′ |
5′-GGGGAGCACTAGGATCATCA-3′ | 680 |
| NRG-1 | Neuregulin 1 |
5′-GACCTCTACTTCTCGTGACA-3′ |
5′-TCCAATCTGTTAGCAATGTG-3′ | 256 |
| NRG-2 | Neuregulin 2 |
5′-CGTTGGTAAAGGTGCTGGAC-3′ |
5′-ACGCAATAGGACTTGGCTGT-3′ | 586 |
| PDGFRα | Platelet-derived
growth factor receptor α |
5′-TCCCGAGACTCCTGTAACCT-3′ |
5′-TTCACTTCTCCAGGGTAAGTC-3′ | 300 |
| PDGFRβ | Platelet-derived
growth factor receptor β |
5′-AGGTGTCATCCATCAACGTCT-3′ |
5′-CTCTCACTTAGCTCCAGCAC-3′ | 646 |
| PDGFA | Platelet-derived
growth factor α polypeptide |
5′-TGCCCATTCGGAGGAAGAGA-3′ |
5′-TTGGCCACCTTGACGCTGC-3′ | 223 |
| PDGFB | Platelet-derived
growth factor β polypeptide |
5′-TTGGACCTGAACATGACCCG-3′ |
5′-ACGTTGCGGTTGTTGCAGCA-3′ | 239 |
| HSP90AB1 | Heat shock protein
90 kDa α (cytosolic), class B member 1 |
5′-CGCATGAAGGAGACACAGAA-3′ |
5′-TCCCATCAAATTCCTTGAGC-3′ | 169 |
| GAPDH |
Glyceraldehyde-3-phosphate
dehydrogenase |
5′-AGCCACATCGCTCAGACACC-3′ |
5′-GTACTCAGCGCCAGCATCG-3′ | 302 |
Phospho-RTK array analysis
The Human Phospho-RTK Array kit (R&D Systems,
Minneapolis, MN, USA) was used to determine the relative levels of
tyrosine phosphorylation of 42 distinct RTKs according to the
manufacturer’s protocol. The arrays were incubated with 450 μg of
protein lysate. The levels of phosphorylation were quantified using
ImageJ software and normalized to phosphotyrosine positive-control
spots.
Phospho-MAPK array analysis
For the detection of the phosphorylation status of
MAPKs and other serine/threonine kinases, the Human Phospho-MAPK
Array kit (R&D Systems) was used according to the
manufacturer’s protocol, and 250 μg of protein lysate was used for
each array. The arrays were analyzed using ImageJ software, and the
levels of phosphorylation were normalized to positive control
spots.
Expression profiling
Total RNA was extracted with the GenElute™ Mammalian
Total RNA Miniprep kit, and the samples were assessed
spectrophotometrically. Purified biotin-labeled cRNA was generated
using the Oligo GEArray® Reagent kit and was hybridized
to the OHS-802 Oligo GEArray® Human Cancer Microarray,
which profiles 440 genes related to cancer, in accordance with the
manufacturer’s instructions (SABiosciences, Frederick, MD, USA).
The GEArray Expression Analysis Suite software (SABiosciences) was
used for data analyses.
Results
EGFR and EGFRvIII mRNA status
As our previous findings suggested that the loss of
the EGFR gene copy may have contributed to the unusually
prolonged survival in a GBM patient (8), we continued a study of this case with
a more detailed analysis of EGFR expression and dysregulation of
cell signaling pathways in HGG-02 cells derived from the
corresponding primary tumor tissue. Using RT-PCR, we assessed the
EGFR status at the mRNA level in the HGG-02 and GM7 cell lines.
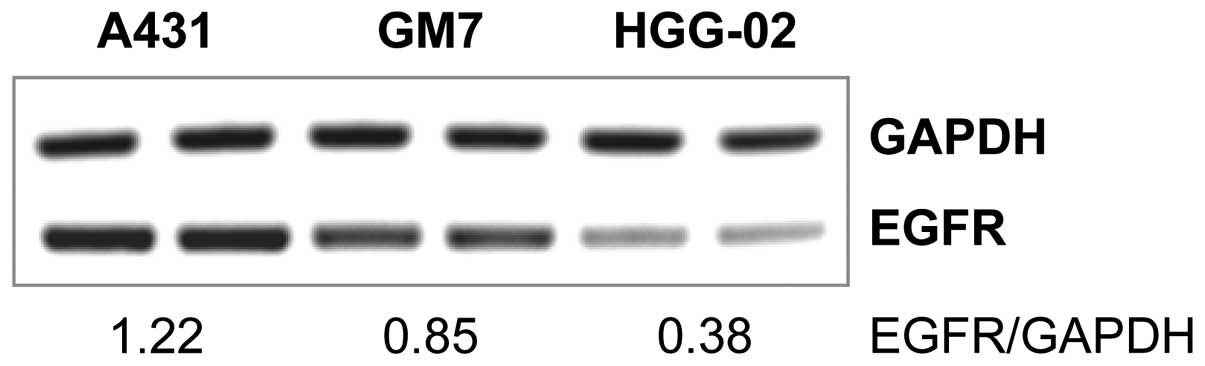
HGG-02 cells showed significantly (2.24-fold) lower expression of
EGFR when compared to the GM7 cells derived from a glioblastoma
with a standard EGFR gene copy number. Relative expression
levels of EGFR in HGG-02 and GM7 were 0.38 and 0.85, respectively
(Fig. 1). Furthermore, we examined
the presence of the most common mutant form of EGFR, i.e.,
EGFRvIII. HGG-02 and GM7 cells did not express the EGFRvIII variant
(data not shown). These results support the hypothesis that the
decreased EGFR mRNA expression in HGG-02 cells was caused by the
lower EGFR gene dosage.
RTK signaling
As EGFR-downstream pathways may be activated and
modulated by other RTKs aside from EGFR family receptors, we
performed human phospho-RTK arrays to explore the phosphorylation
status of 42 RTKs in the HGG-02 cells (Fig. 2A). Consistent with our previous
data, we detected lower levels of phosphorylated EGFR in the HGG-02
cell line, which was in contrast with the GM7 cells (Fig. 2B). However, HGG-02 cells showed
higher levels of ErbB2 and ErbB3 activation. ErbB4, another
receptor of the EGFR family, did not show a significant difference
in the phosphorylation state between these two cell lines.
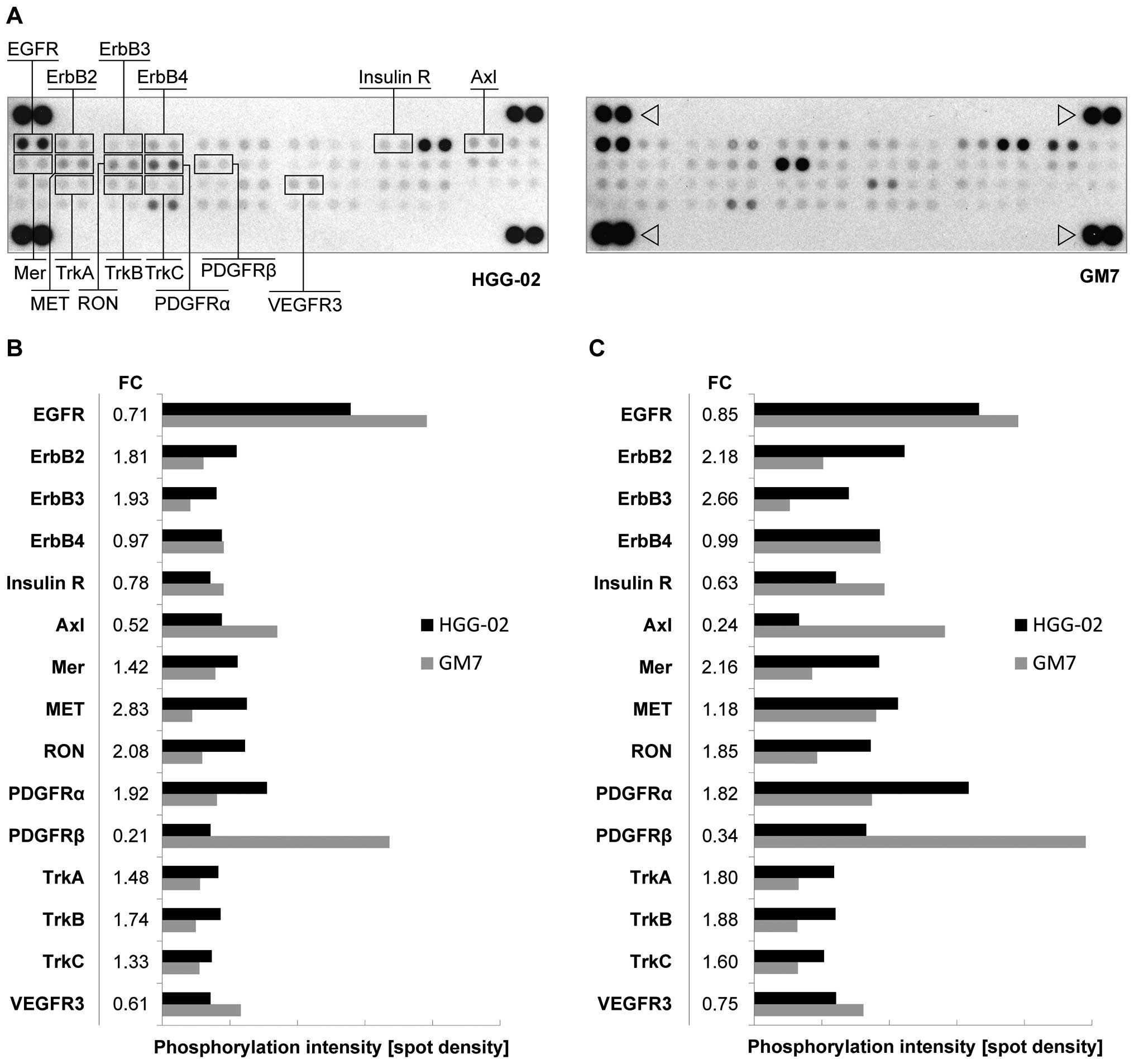 | Figure 2Phospho-RTK array in the HGG-02 cells
demonstrates the downregulation of EGFR in comparison with the GM7
reference cell line. (A) Simultaneous detection of the
phosphorylation status of 42 RTKs in the HGG-02 and GM7 cell lines
using a human phospho-RTK array. Relevant receptors are noted.
Phosphotyrosine-positive control spots are marked with an
arrowhead. (B) In comparison with the GM7 cell line, HGG-02 cells
exhibited downregulation of the EGFR, PDGFRβ, Axl, VEGFR3 and
insulin receptors. PDGFRα, Mer, MET, RON, TrkA, TrkB and TrkC were
upregulated. The level of phosphorylation was quantified using
ImageJ software and normalized to phosphotyrosine-positive control
spots. (C) The phosphorylation status after a 24-h serum-free
cultivation. Note the overall higher phosphorylation levels and
changes in ErbB2, ErbB3, Axl and Mer phosphorylation between the
cell lines in comparison with the serum-containing conditions. FC,
phosphorylation fold-change values of the HGG-02 cell line when
compared to the GM7 reference cell line. RTK, receptor tyrosine
kinase; EGFR, epidermal growth factor receptor. |
In addition, we identified the downregulation of the
phosphorylation of the PDGFRβ, Axl, VEGFR3 and insulin receptors as
well as the upregulation of the phosphorylation of PDGFRα, MET,
RON, Mer, TrkA, TrkB and TrkC in the HGG-02 cells (Fig. 2B).
To eliminate the influence of serum (which contains
growth factors, hormones and other components) on the
phosphorylation status of RTKs, we performed phospho-RTK arrays
simultaneously using the cell lines after a 24-h serum-free
cultivation. Notably, the serum-free conditions did not markedly
change the phosphorylation status in the HGG-02 and GM7 cells;
however, the overall level of phosphorylation was higher when
compared to the serum-containing conditions (Fig. 2C). In the serum-free conditions,
increased upregulation of Mer, ErbB2 and ErbB3 as well as
downregulation of Axl were detected.
Signaling of MAPK and other
serine/threonine kinases
To examine RTK downstream pathways activated in the
HGG-02 cells, we employed phospho-MAPK arrays. We also performed
the phospho-MAPK arrays on cells cultivated under serum-containing
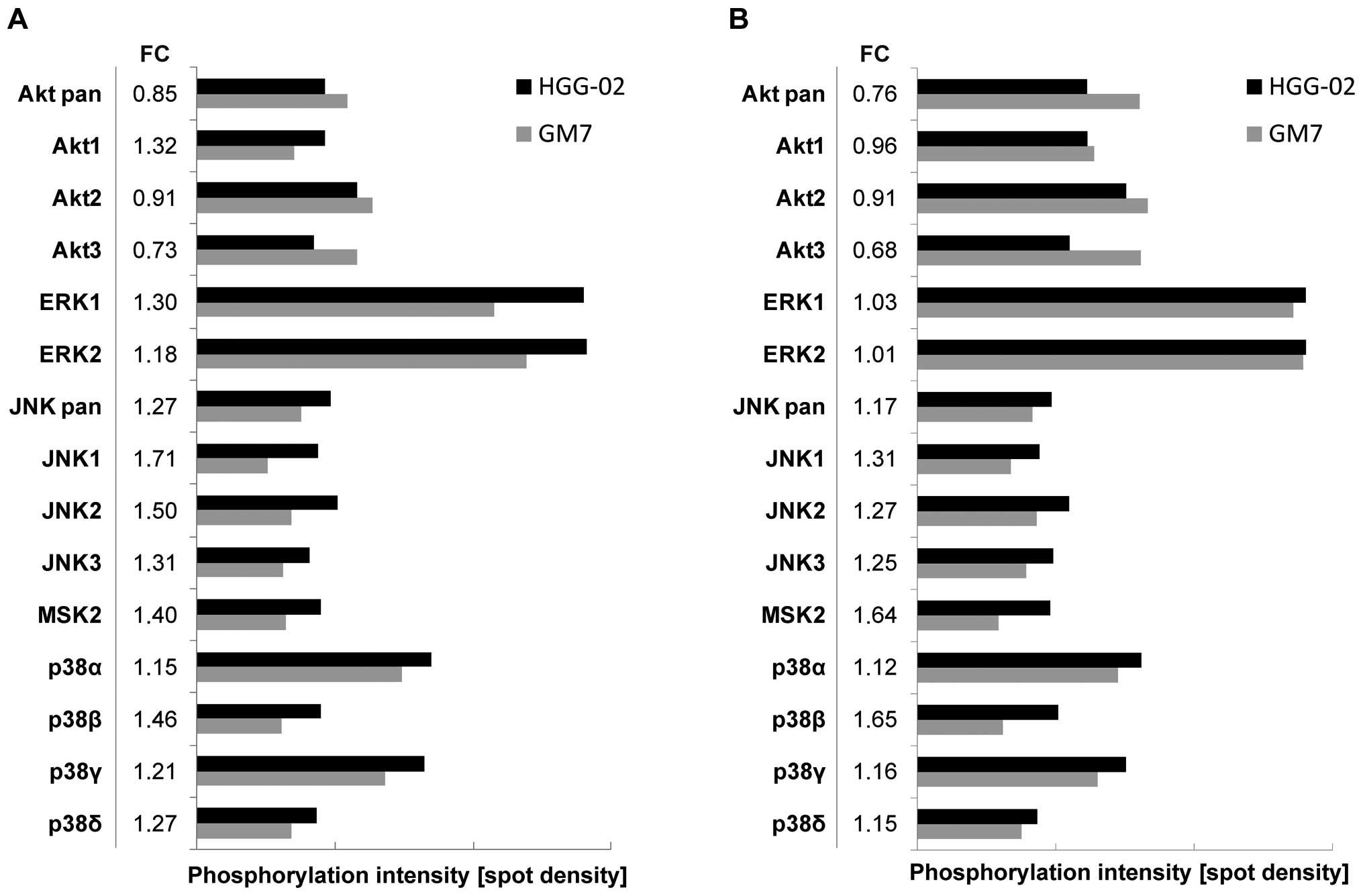
and serum-free conditions, as described above. An Akt pan-specific
antibody revealed an overall diminished level of signaling through
the Akt pathway in the HGG-02 cells when compared to the GM7 cells
in the serum-containing and serum-free conditions (Fig. 3). However, differences in the
phosphorylation levels of Akt1, Akt2 and Akt3 kinases were
observed. Akt1 was more phosphorylated in the HGG-02 cells in the
serum-containing but not in the serum-free conditions, whereas the
levels of Akt2 and Akt3 phosphorylation were decreased regardless
of the conditions. In addition, we detected an upregulation of
signaling through the p38 pathway, JNK pathway and MSK2 in the
HGG-02 cells. ERK1 and ERK2 signaling was also slightly elevated in
the HGG-02 cells but only under the serum-containing conditions
(Fig. 3A).
Expression of the EGFR and PDGFR families
and their ligands
The results of the phospho-RTK arrays obtained under
serum-free conditions indicated the presence of constitutively
active receptor tyrosine-kinase pathways in the HGG-02 and GM7 cell
lines. To investigate this possibility, we performed RT-PCR of the
receptors in the EGFR and PDGFR families and their respective
ligands after cultivation of the cells under serum-containing and
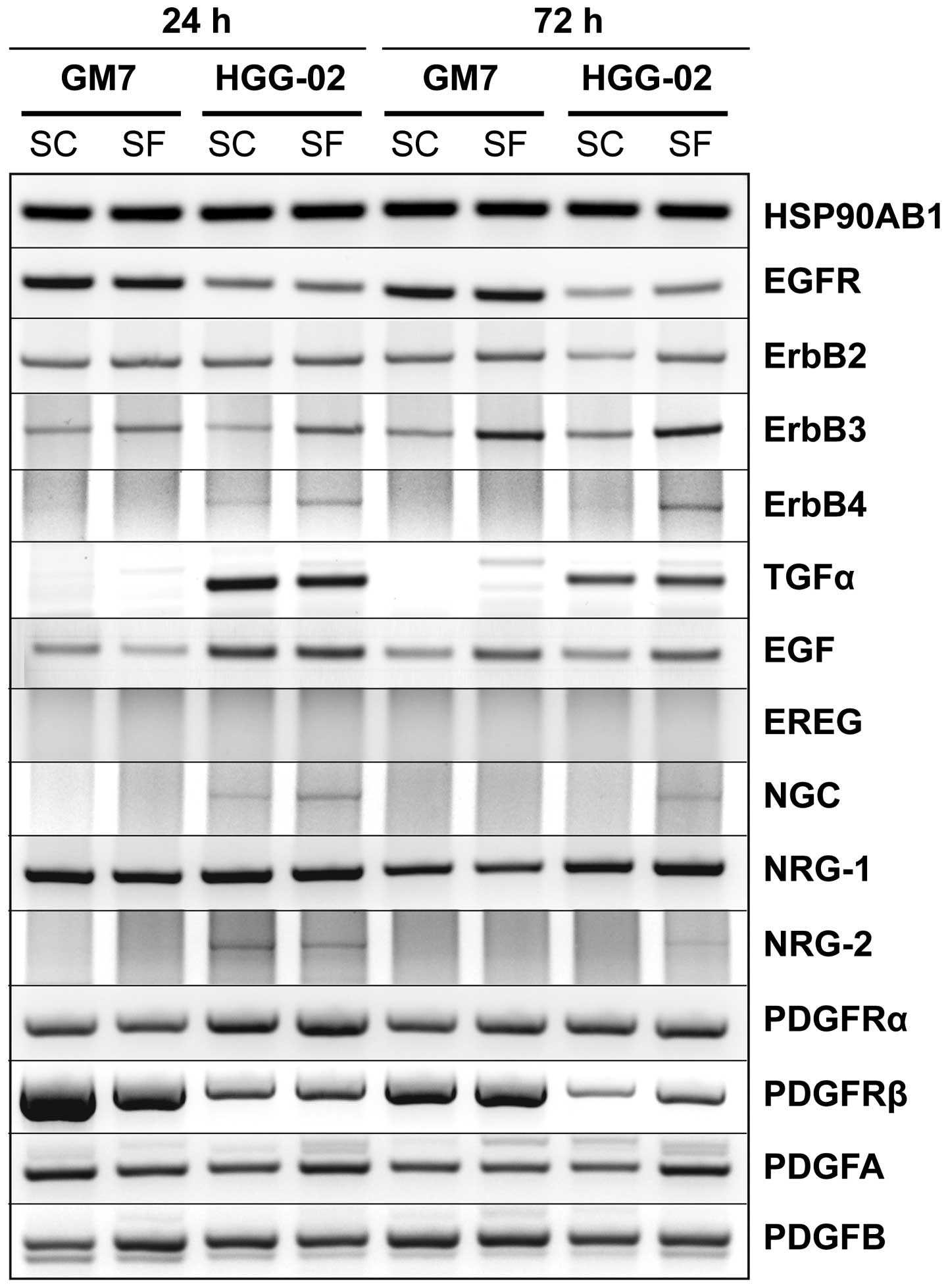
serum-free conditions (Fig. 4).
As expected, expression of EGFR was decreased in the
HGG-02 cells and did not significantly vary with time or under
serum-free conditions. Although there was no difference in the
expression of ErbB2, ErbB3 expression was higher under serum-free
conditions and increased with time in both cell lines. ErbB4
expression was detected in the HGG-02 cells only and was stronger
in the serum-free conditions. These patterns of expression were
observable for its ligand NRG-2 and for the ErbB3-specific ligand
NGC. In contrast to the low levels of EGFR mRNA, strong expression
of the EGFR-specific ligand TGF-α was observed in the HGG-02 cells
but not in the GM7 cells. EGF, the second EGFR-specific ligand, was
also upregulated in the HGG-02 cells. Nevertheless, after 72 h of
cultivation, the expression of EGF did not differ between cell
lines and was higher in the serum-free conditions. Finally, the
expression of NRG-1 increased in the HGG-02 cells after 72 h of
cultivation, and EREG was not expressed in any of the samples.
We observed an apparent downregulation of PDGFRβ in
the HGG-02 cells. In contrast, the expression of PDGFRα was
slightly increased when compared with that in the GM7 cells.
However, there were no obvious trends in the expression of the
ligands for these receptors, including PDGFA and PDGFB.
Cancer-related gene expression
Gene expression profiling affirmed a decrease in
EGFR expression at the mRNA level in the HGG-02 cells. Out of the
440 investigated genes, the HGG-02 expression profile revealed the
upregulation of 108 genes (FC of 2–38.8) and downregulation of 10
genes (FC of 0.2–0.44) in comparison with the GM7 cells. Functional
clustering showed that most of the upregulated genes are involved
in the cell cycle, signaling pathways induced by growth factors,
focal adhesions and apoptosis (Table
II). The downregulated genes encode proteins responsible for
organizing the extracellular matrix (ECM) or interacting with the
ECM.
| Table IIFunctional clustering of the
molecular or biochemical pathways associated with genes that were
differentially expressed in the HGG-02 cell line when compared to
the GM7 cell line. |
Table II
Functional clustering of the
molecular or biochemical pathways associated with genes that were
differentially expressed in the HGG-02 cell line when compared to
the GM7 cell line.
| Molecular or
biochemical pathway | No. of deregulated
genes | Percentage of
deregulated genes | P-value |
|---|
| Upregulated gene expression
(>2-fold change vs. GM7) |
| Pathways in
cancer | 20 | 19.05 | <0.001 |
| Cell cycle | 12 | 11.43 | <0.001 |
| MAPK signaling
pathway | 12 | 11.43 | <0.001 |
| Glioma | 8 | 7.62 | <0.001 |
| Focal
adhesions | 8 | 7.62 | 0.017 |
| Neurotrophin
signaling pathway | 7 | 6.67 | 0.006 |
| p53 signaling
pathway | 6 | 5.71 | 0.002 |
| Apoptosis | 6 | 5.71 | 0.006 |
| T cell receptor
signaling pathway | 6 | 5.71 | 0.014 |
| B cell receptor
signaling pathway | 5 | 4.76 | 0.017 |
| Toll-like receptor
signaling pathway | 5 | 4.76 | 0.045 |
| Downregulated gene expression
(<0.5-fold change vs. GM7) |
| ECM-receptor
interactions | 3 | 30.00 | 0.004 |
| Focal
adhesions | 3 | 30.00 | 0.021 |
The 17 genes that were upregulated in the HGG-02
cells more than 5-fold were CDKN2A, TFAP2C, IGFBP3, CDKN2B, FRZB,
SRPX and Siva1. The downregulated genes included COL6A3, COL1A1,
DCN, FN1, FBN1 and GDF15.
Discussion
Despite many advances in the treatment of GBM, the
outcomes of patients with this tumor remain poor. Previously, we
reported the case of a patient with GBM with a mild clinical course
and a loss of EGFR gene copy (8). As both of these aspects are uncommon
in GBMs, we performed a detailed analysis of the HGG-02 cell line
that was derived from this patient’s tumor to understand the cancer
regulatory pathways that may have contributed to the favorable
clinical course in this case. As control cells, the reference GM7
cell line that was derived from a highly aggressive tumor type and
showed a standard EGFR gene copy number (9) was used.
Using RT-PCR, a phospho-RTK array and expression
profiling, we confirmed the downregulation of EGFR expression and
signaling in HGG-02 cells. However, this downregulated signaling
did not affect the downstream MAPKs, ERK1 or ERK2 and was possibly
compensated for by the activation of the ErbB2 and ErbB3 receptors.
These results are in agreement with another study that recently
reported the compensatory activation of ErbB2 and ErbB3 receptors
in GBM deprived of EGFR signaling, which suggests an intrinsic
mechanism of GBM resistance to EGFR-targeted therapy (10). RT-PCR analysis revealed weak but
detectable ErbB4 expression in the HGG-02 cells, while ErbB4
expression was not detected in the GM7 reference cell line. In
contrast, the ErbB4 phosphorylation status was the same in these
two cell lines, and we assume that this discrepancy may point to
the previously questioned correlation of RNA and protein levels
(11). However, the RT-PCR results
of EGFR family-related ligands showed a strong elevation in TGF-α,
which was accompanied by an increased expression of EGF in the
HGG-02 cells. Previous studies have suggested that EGFR and its
ligands EGF and TGF-α play an important role in the
autocrine/paracrine loop and support cell proliferation in human
gliomas (12,13). Nevertheless, considering that the
HGG-02 cells acquired the loss of EGFR gene copy and the
phospho-RTK array revealed a downregulation in EGFR signaling, we
assume that the HGG-02 cells and the respective tumor do not
represent EGFR-driven GBM.
Apart from the EGFR family, we identified changes in
the phosphorylation of 11 other RTKs, including Axl. The
phosphorylation of Axl was significantly reduced in comparison with
the GM7 cells. This reduction was even more obvious under
serum-free conditions, which suggests the constitutive activation
of Axl in the GM7 reference cell line. Previous studies have found
that Axl is constitutively phosphorylated in many glioma cell lines
and that the downstream MAPK and PI3K pathways are activated
(14). Moreover, Axl overexpression
has been reported as a frequent event in human gliomas and is
correlated with a poor prognosis in these patients (15). Therefore, the downregulation of Axl
phosphorylation in HGG-02 cells may indicate a less aggressive
phenotype of the respective tumor. Nevertheless, in the HGG-02
cells, we detected an upregulation of signaling through Mer,
another member of the TAM family of RTKs (16). Recently, 2 independent studies
demonstrated that Mer promotes invasion, protects cells from
apoptosis in GBM and has a similar function to Axl (17,18).
However, the reduction in Axl phosphorylation in the HGG-02 cells
was ~2-fold greater than Mer upregulation. Overall, these results
suggest that Axl and Mer play an important role in the
aggressiveness of GBM and emphasize the need for further research
of the TAM receptor tyrosine kinases in GBM.
Recently, it has been observed that MET, an HGF
receptor, is activated during EGFR-targeted therapy resistance, and
various studies have suggested that silencing EGFR is compensated
by MET signaling in these cases (19). In accordance with these
observations, we detected an upregulation of signaling through MET
in the HGG-02 cells, which may partially compensate for the
downregulation of EGFR signaling caused by the loss of EGFR
gene copy. MET overexpression has been associated with a shorter
survival time and poor response (20), and it has been suggested that MET
activation is required for the acquisition of the glioblastoma
cancer stem cell phenotype (21).
Furthermore, upregulated phosphorylation of RON, the MSP receptor,
may indicate another compensatory mechanism in HGG-02 cells.
Eckerich et al reported that although RON signaling showed
no mitogenic effect on GBM cells, it can induce glioma cell
migration (22). However, these
findings are in contrast with the unusually non-aggressive
phenotype of the HGG-02 tumor, and we hypothesize that the
activation of MET and RON signaling is not strong enough to
overcome the loss of EGFR gene copy number in this case.
Nevertheless, HGG-02 cells exhibited increased
phosphorylation of Trk receptors. It is well established that Trk
receptors support the survival and differentiation of the nervous
system; however, there is growing evidence that the Trk family can
also induce or enhance cell death in certain tumor types, primarily
in pediatric tumors of neural origin (23). It has been shown that expression of
TrkA and TrkC correlates with a favorable outcome in neuroblastoma
and medulloblastoma patients. In GBM, immunoreactivity for TrkA was
shown to be inversely associated with the grade of malignancy
(24). Moreover, TrkA
overexpression was found to induce autophagic cell death that was
mediated through the JNK pathway (25,26).
Therefore, we hypothesized that elevated signaling through Trk
receptors and JNK MAPKs in HGG-02 cells contributed to the
non-aggressive phenotype of the primary tumor.
Notably, we also detected an apparent difference in
PDGF signaling in both of the cell lines studied. Whereas HGG-02
cells preferentially signaled through PDGFRα, PDGFRβ was strongly
phosphorylated in the GM7 reference cell line. Moreover, these
results, which were obtained using the phospho-RTK array,
corresponded with the results obtained by RT-PCR. Using RT-PCR, we
detected PDGFRβ overexpression in the GM7 cells, whereas PDGFRα was
overexpressed in the HGG-02 cells. Expression of PDGFs and PDGFRs
has been observed even in low-grade gliomas, which suggests that
this pathway possibly represents an early oncogenic event in
gliomagenesis (27,28). According to previous studies, PDGFRα
is overexpressed in GBM cells, whereas PDGFRβ is detected in
adjacent vascular cells (27).
However, a recent study interrogated the proposed role of PDGFRs in
intratumoral GBM heterogeneity (29). Kim et al found that
expression of PDGFRβ, but not PDGFRα, was strongly associated with
the glioblastoma stem cell (GSC) phenotype. Moreover, targeting
PDGFRβ decreased GSC self-renewal, survival, tumor growth and
invasion. Using in silico survival analysis, the authors
noted that PDGFRβ indicated a poor prognosis, whereas PDGFRα was a
positive prognostic marker. Together, these data support our
observation in HGG-02 cells. Higher expression and signaling
activity of PDGFRα, instead of PDGFRβ, may indicate the presence of
a limited number of GSCs in the HGG-02 cell line when compared with
the GM7 reference cell line derived from an aggressive tumor type.
While our data stress the importance of PDGF signaling in GBM with
respect to the distinct effects of PDGFRα and PDGFRβ in
gliomagenesis, we strongly encourage further research of these RTKs
in GBM biology.
We identified increased signaling through the p38
and JNK pathways in HGG-02 cells. It is known that p38 kinase
activation is associated with anti-proliferative functions and
plays a role in the induction of apoptosis (30). Moreover, recent studies have shown
that p38 kinase activation is necessary for GBM cell death
initiated by various anticancer agents (31–33).
Furthermore, we detected increased signaling through the JNK
kinases, which are also involved in apoptosis, and their activation
is required for UV-induced apoptosis (30,34).
As previously noted, the JNK pathway can also mediate TrkA-induced
autophagic cell death (25,26). Overall, the increased activation of
the p38 and JNK pathways, with regard to anti-proliferative and
apoptotic functions, may represent the less aggressive
characteristics of the HGG-02 cell line.
Although gene expression profiling detected changes
in the expression of 118 genes, analysis of the most upregulated
and downregulated genes in the HGG-02 cells revealed a specific
expression profile of HGG-02 cells that may reflect the
non-aggressive phenotype of this tumor. Seven of the 17 highly
upregulated genes in the HGG-02 cells represent well accepted or
potential tumor suppressors, including CDKN2A (35,36),
TFAP2C (37), IGFBP3 (38), CDKN2B (36), FRZB (39), SRPX (40) and Siva1 (41). However, the downregulated genes
involved genes associated with the ECM, including COL6A3, COL1A1,
DCN, FN1 and FBN1. These genes have been proposed to play an
important role in controlling and facilitating the motility of
glioma cells (42). For example,
COL6A3 was found to correlate with glioma grade (43). Apart from the ECM-related genes, we
detected significantly lower expression of GDF15 in the HGG-02
cells. Recent studies revealed that GDF15 contributes to the
proliferation and immune escape of malignant gliomas (44,45).
This finding is in agreement with its low expression in the HGG-02
cell line that was derived from the unusually non-aggressive
GBM.
In conclusion, the detailed analysis of the HGG-02
cell line confirmed the unusually reduced EGFR signaling and
indicated several other targets that may contribute to the
non-aggressive phenotype of the respective tumor. As suggested by
our results, TAM receptors, Trk receptors and PDGFRs need to be
further investigated since these proteins may play an important
role in GBM biology, gliomagenesis and patient outcome.
Acknowledgements
The present study was supported by the European
Regional Development Fund and the State Budget of the Czech
Republic (RECAMO, CZ.1.05/2.1.00/03.0101) and OP VK
CZ.1.07/2.3.00/20.0183.
References
|
1
|
Linggi B and Carpenter G: ErbB receptors:
new insights on mechanisms and biology. Trends Cell Biol.
16:649–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yarden Y and Sliwkowski MX: Untangling the
ErbB signalling network. Nat Rev Mol Cell Biol. 2:127–137. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ohgaki H and Kleihues P: Population-based
studies on incidence, survival rates, and genetic alterations in
astrocytic and oligodendroglial gliomas. J Neuropathol Exp Neurol.
64:479–489. 2005.PubMed/NCBI
|
|
4
|
Ohgaki H and Kleihues P: Genetic pathways
to primary and secondary glioblastoma. Am J Pathol. 170:1445–1453.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hatanpaa KJ, Burma S, Zhao D and Habib AA:
Epidermal growth factor receptor in glioma: signal transduction,
neuropathology, imaging, and radioresistance. Neoplasia.
12:675–684. 2010.PubMed/NCBI
|
|
6
|
Lopez-Gines C, Gil-Benso R, Ferrer-Luna R,
et al: New pattern of EGFR amplification in glioblastoma and
the relationship of gene copy number with gene expression profile.
Mod Pathol. 23:856–865. 2010.
|
|
7
|
Bredel M, Scholtens DM, Yadav AK, et al:
NFKBIA deletion in glioblastomas. N Engl J Med. 364:627–637.
2011. View Article : Google Scholar
|
|
8
|
Veselska R, Skoda J, Loja T, et al: An
unusual loss of EGFR gene copy in glioblastoma multiforme in
a child: a case report and analysis of a successfully derived
HGG-02 cell line. Childs Nerv Syst. 26:841–846. 2010.
|
|
9
|
Loja T, Chlapek P, Kuglik P, Pesakova M,
Oltova A, Cejpek P and Veselska R: Characterization of a GM7
glioblastoma cell line showing CD133 positivity and both
cytoplasmic and nuclear localization of nestin. Oncol Rep.
21:119–127. 2009.PubMed/NCBI
|
|
10
|
Clark PA, Iida M, Treisman DM, et al:
Activation of multiple ERBB family receptors mediates glioblastoma
cancer stem-like cell resistance to EGFR-targeted inhibition.
Neoplasia. 14:420–428. 2012.PubMed/NCBI
|
|
11
|
Gry M, Rimini R, Strömberg S, Asplund A,
Pontén F, Uhlén M and Nilsson P: Correlations between RNA and
protein expression profiles in 23 human cell lines. BMC Genomics.
10:3652009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang P, Steck PA and Yung WK: The
autocrine loop of TGF-α/EGFR and brain tumors. J Neurooncol.
35:303–314. 1997.
|
|
13
|
Ekstrand AJ, James CD, Cavenee WK, Seliger
B, Pettersson RF and Collins VP: Genes for epidermal growth factor
receptor, transforming growth factor α, and epidermal growth factor
and their expression in human gliomas in vivo. Cancer Res.
51:2164–2172. 1991.
|
|
14
|
Stommel JM, Kimmelman AC, Ying H, et al:
Coactivation of receptor tyrosine kinases affects the response of
tumor cells to targeted therapies. Science. 318:287–290. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hutterer M, Knyazev P, Abate A, et al: Axl
and growth arrest-specific gene 6 are frequently overexpressed in
human gliomas and predict poor prognosis in patients with
glioblastoma multiforme. Clin Cancer Res. 14:130–138. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Linger RM, Keating AK, Earp HS and Graham
DK: TAM receptor tyrosine kinases: biologic functions, signaling,
and potential therapeutic targeting in human cancer. Adv Cancer
Res. 100:35–83. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rogers AE, Le JP, Sather S, Pernu BM,
Graham DK, Pierce AM and Keating AK: Mer receptor tyrosine kinase
inhibition impedes glioblastoma multiforme migration and alters
cellular morphology. Oncogene. 31:4171–4181. 2012. View Article : Google Scholar
|
|
18
|
Wang Y, Moncayo G, Morin P Jr, et al: Mer
receptor tyrosine kinase promotes invasion and survival in
glioblastoma multiforme. Oncogene. 32:872–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Velpula KK, Dasari VR, Asuthkar S,
Gorantla B and Tsung AJ: EGFR and c-Met cross talk in glioblastoma
and its regulation by human cord blood stem cells. Transl Oncol.
5:379–392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kong DS, Song SY, Kim DH, et al:
Prognostic significance of c-Met expression in glioblastomas.
Cancer. 115:140–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Joo KM, Jin J, Kim E, et al: MET signaling
regulates glioblastoma stem cells. Cancer Res. 72:3828–3838. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eckerich C, Schulte A, Martens T, Zapf S,
Westphal M and Lamszus K: RON receptor tyrosine kinase in human
gliomas: expression, function, and identification of a novel
soluble splice variant. J Neurochem. 109:969–980. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harel L, Costa B and Fainzilber M: On the
death Trk. Dev Neurobiol. 70:298–303. 2010.PubMed/NCBI
|
|
24
|
Wadhwa S, Nag TC, Jindal A, Kushwaha R,
Mahapatra AK and Sarkar C: Expression of the neurotrophin receptors
Trk A and Trk B in adult human astrocytoma and glioblastoma. J
Biosci. 28:181–188. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hansen K, Wagner B, Hamel W, Schweizer M,
Haag F, Westphal M and Lamszus K: Autophagic cell death induced by
TrkA receptor activation in human glioblastoma cells. J Neurochem.
103:259–275. 2007.PubMed/NCBI
|
|
26
|
Dadakhujaev S, Noh HS, Jung EJ, Hah YS,
Kim CJ and Kim DR: The reduced catalase expression in TrkA-induced
cells leads to autophagic cell death via ROS accumulation. Exp Cell
Res. 314:3094–3106. 2008. View Article : Google Scholar
|
|
27
|
Hermanson M, Funa K, Hartman M,
Claesson-Welsh L, Heldin CH, Westermark B and Nistér M:
Platelet-derived growth factor and its receptors in human glioma
tissue: expression of messenger RNA and protein suggests the
presence of autocrine and paracrine loops. Cancer Res.
52:3213–3219. 1992.
|
|
28
|
Maher EA, Furnari FB, Bachoo RM, Rowitch
DH, Louis DN, Cavenee WK and DePinho RA: Malignant glioma: genetics
and biology of a grave matter. Genes Dev. 15:1311–1333. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim Y, Kim E, Wu Q, et al:
Platelet-derived growth factor receptors differentially inform
intertumoral and intratumoral heterogeneity. Genes Dev.
26:1247–1262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wagner EF and Nebreda AR: Signal
integration by JNK and p38 MAPK pathways in cancer development. Nat
Rev Cancer. 9:537–549. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Amantini C, Mosca M, Nabissi M, et al:
Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1
vanilloid receptor and requires p38 MAPK activation. J Neurochem.
102:977–990. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yao YQ, Ding X, Jia YC, Huang CX, Wang YZ
and Xu YH: Anti-tumor effect of β-elemene in glioblastoma cells
depends on p38 MAPK activation. Cancer Lett. 264:127–134. 2008.
|
|
33
|
Das A, Banik NL and Ray SK: Flavonoids
activated caspases for apoptosis in human glioblastoma T98G and
U87MG cells but not in human normal astrocytes. Cancer.
116:164–176. 2010.PubMed/NCBI
|
|
34
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu W, Lv G, Li Y, Li L and Wang B:
Downregulation of CDKN2A and suppression of cyclin D1 gene
expressions in malignant gliomas. J Exp Clin Cancer Res. 30:762011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feng J, Kim ST, Liu W, et al: An
integrated analysis of germline and somatic, genetic and epigenetic
alterations at 9p21.3 in glioblastoma. Cancer. 118:232–240. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gabriely G, Yi M, Narayan RS, et al: Human
glioma growth is controlled by microRNA-10b. Cancer Res.
71:3563–3572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu C, Liu X, Wang Y, et al: Insulin-like
factor binding protein-3 promotes the G1 cell cycle arrest in
several cancer cell lines. Gene. 512:127–133. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zi X, Guo Y, Simoneau AR, Hope C, Xie J,
Holcombe RF and Hoang BH: Expression of Frzb/secreted
Frizzled-related protein 3, a secreted Wnt antagonist, in human
androgen-independent prostate cancer PC-3 cells suppresses tumor
growth and cellular invasiveness. Cancer Res. 65:9762–9770. 2005.
View Article : Google Scholar
|
|
40
|
Tambe Y, Isono T, Haraguchi S,
Yoshioka-Yamashita A, Yutsudo M and Inoue H: A novel apoptotic
pathway induced by the drs tumor suppressor gene. Oncogene.
23:2977–2987. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li N, Jiang P, Du W, et al: Siva1
suppresses epithelial-mesenchymal transition and metastasis of
tumor cells by inhibiting stathmin and stabilizing microtubules.
Proc Natl Acad Sci USA. 108:12851–12856. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mahesparan R, Tysnes BB, Read TA, Enger
PO, Bjerkvig R and Lund-Johansen M: Extracellular matrix-induced
cell migration from glioblastoma biopsy specimens in vitro. Acta
Neuropathol. 97:231–239. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fujita A, Sato JR, Festa F, et al:
Identification of COL6A1 as a differentially expressed gene
in human astrocytomas. Genet Mol Res. 7:371–378. 2008.
|
|
44
|
Wu A, Wei J, Kong LY, et al: Glioma cancer
stem cells induce immunosuppressive macrophages/microglia. Neuro
Oncol. 12:1113–1125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Roth P, Junker M, Tritschler I, et al:
GDF-15 contributes to proliferation and immune escape of malignant
gliomas. Clin Cancer Res. 16:3851–3859. 2010. View Article : Google Scholar : PubMed/NCBI
|