Introduction
Lung cancer is the leading cause of cancer-related
death worldwide with non-small cell lung carcinoma (NSCLC)
accounting for ~80% of lung cancers (1). Activating mutations of the epidermal
growth factor receptor (EGFR) occur in 30–40% of the patients with
NSCLC in China, and are associated with poor prognosis (2). EGFR mutations result in constitutive
activation of the EGFR in the absence of the EGF ligand and
abnormal activation of downstream signaling pathways, including
mitogen-activated protein kinase (Mek)/extracellular signal
regulated kinase (ERK) and phosphatidylinositol-3 kinase (PI3K)
(3–5). Activation of these downstream
effectors upregulates Mcl-1, Bcl-XL and survivin, allowing cancer
cells to evade apoptosis (6–8).
EGFR inhibitors have recently been used clinically
to improve the poor prognosis of NSCLC with EGFR mutations. Almost
90% of these somatic activating mutations in EGFR consist of
in-frame deletions in exon 19 and L858R point mutations in exon 21
(9,10). Gefitinib, a synthetic
anilinoquinazoline, is an orally active and highly selective EGF
receptor inhibitor that blocks EGF receptor autophosphorylation and
subsequent signal transduction pathways implicated in the promotion
of cancer cell proliferation (11).
At present, gefitinib is applied to a number of human cancers and
benefits some patients during treatment (12). However, while most NSCLC patients
with EGFR mutations initially respond to EGFR-tyrosine kinase
inhibitors (TKIs), acquired resistance ultimately develops
(13).
One potential explanation for the acquired
resistance may be a secondary mutation in EGFR, EGFR T790M, which
occurs in ~50% of patients acquiring resistance to EGFR-TKIs
(14). Additionally, MET oncogene
amplification occurs in 20% of the patients with EGFR-TKI
resistance (15). Amplification of
MET was found to cause gefitinib resistance by driving ERBB3
(HER3)-dependent activation of PI3K, a pathway thought to be
specific to the EGFR/ERBB family receptors (16). Through genetic changes, cancer cells
acquire a survival advantage, such as resisting drug-induced
apoptosis, to decrease the sensitivity to drug therapy.
Nevertheless, this durable genetic resistance takes a relatively
long time to develop, whereas other temporary or weak types of
resistance mechanisms come into play earlier in treatment (17). There is evidence that the behavior
of carcinomas is influenced by crosstalk between tumor cells and
the host microenvironment (18,19).
Stromal cells reduce the sensitivity of cancer cells to
chemotherapy drugs (20,21), leading to the suggestion that
co-culture of cancer cells with stromal cells may cause reduced
gefitinib-induced apoptosis in cancer cells.
Since fibroblasts play a definitive role in tumor
progression and drug response (22,23),
in the present study we confirmed that fibroblasts efficiently
induced gefitinib resistance in the HCC827 cell line which
expresses EGFR exon 19 deletion mutations. In order to investigate
how the susceptibility of lung cancer cells with EGFR-activating
mutations to an EGFR-TKI could be affected by fibroblasts, we
performed bioinformatic analysis and found that Aurora-A kinase
(AURKA) overexpression played an important role in the reduced
apoptosis in HCC827 cells. Further investigations showed that the
p53 pathway may play a key role in the regulation of gefitinib
resistance.
Materials and methods
Cell lines and reagents
We purchased EGFR-mutant human lung adenocarcinoma
cell line HCC827 (del E746_A750) and human lung embryonic
fibroblast MRC-5 cells from the Cell Bank of the Chinese Academy of
Sciences. We maintained the cell lines in RPMI-1640 medium
containing 10% FBS (Gibco-BRL, Gaithersburg, MD, USA) at 37°C in a
humidified 5% CO2 atmosphere. To study the effect of
cancer-associated fibroblasts on gefitinib sensitivity of HCC827
cells, one patient with histologically proven lung cancer and who
underwent surgical resection in Changzheng Hospital, was enrolled.
We isolated and expanded cancer-associated fibroblasts (CAFs) as
previously described (24).
Briefly, we distributed small pieces of tumor tissue at the bottom
of 25 cm2 cell culture flasks that had been precoated
with 2 μl of RPMI-1640 medium supplemented with 50 IU/ml
penicillin, 50 μg/ml streptomycin, and 20% FBS. We incubated the
tissue cultures at 37°C in humidified air with 5% CO2,
and changed the media every 3–4 days. After 7–10 days, the cells
formed homogeneous monolayers morphologically consistent with
fibroblast-like cells. Immunoblots of vimentin and E-cadherin
confirmed the CAF cultures (data not shown).
We purchased gefitinib from Selleck Chemicals
(Houston, TX, USA) and E-cadherin, vimentin, EGFR, p-EGFR, AKT,
p-Akt, ERK, p-ERK, AURKA and GAPDH antibodies were from Epitomics
Inc. (Burlingame, CA, USA).
Co-culture and viability assays
For co-culture experiments, we cultured the HCC827
cells with or without gefitinib in the lower chamber of 24-well
Transwell plates (Corning Costar, Cambridge, MA, USA) and plated
MRC-5 cells or CAFs in the accompanying inserts (3-μm pore size).
We treated the co-cultured cells with the indicated gefitinib
concentrations for 48 h. At the assay end-point, we evaluated tumor
cell viability using MTT assay (Sigma-Aldrich, St. Louis, MO, USA).
We completed each experiment at least 3 times, each with triplicate
samples.
Microarray analysis of gene
expression
We used Affymetrix PrimeView Array chips (>60,000
probes interrogating 40,000 transcripts) to compare gene expression
patterns between cDNA reserve transcripts of mono-cultured HCC827
and MRC-5 cells co-cultured with HCC827 cells under gefitinib
treatment. We used the t-test to identify differences. We
considered P<0.05 as a significant difference and a cut-off line
of 1.1-fold change. We built gene co-expression networks based upon
the KEGG database by searching interactions among genes and
locating upstream and downstream genes (25).
Transfection of the AURKA shRNA plasmid
and AURKA overexpression plasmid
Dr Zhou Wang (Shanghai, China) kindly provided
pGenesil and the pEGFPC3 plasmid. We cloned oligonucleotides
containing the selected shRNA sequences into pGenesil. The
sequences of the clones are as follows: sense,
5′-GATCCGGGCTACAGCTCCAGTTGGA TTCAAGACGTCCAACTGGAGCTGTAGCCTTTTTA-3′
and antisense, 5′-AGCTTAAAAAGGCTACAGCTCCAGTT
GGACGTCTTGAATCCAACTGGAGCTGTAGCCCG-3′.
We transfected AURKA-shRNA and empty vectors into
the HCC827 cells using FuGENE HD transfection agent (Promega,
Madison, WI, USA) according to the manufacturer’s instructions.
We isolated total RNA from HCC827 cells and used a
reverse transcription system kit (Takara, Tokyo, Japan) to
synthesize the first-strand complementary DNA. We amplified the
open reading frame (ORF) of AURKA using PCR with the following
primer pairs: sense, 5′-ATGGACCGATCTAAAGA AAACTGCA-3′ and
antisense, 5′-TCTCCCCCTGCACGATT CCTAA-3′. We further ligated the
fragment containing the ORF of AURKA into the pEGFP-C3 vector, at
the HindIII/BamHI site and named the recombinant
plasmid p-EGFPC3-AURKA. We used FuGENE HD transfecting agent
(Promega) to infect the HCC827 cells with either the p-EGFPC3-AURKA
or the empty vectors.
Immunofluorescence analysis
Gefitinib-treated and untreated HCC827 cells grown
on glass coverslips were fixed with 4% PBS buffered formaldehyde
for 20 min, permeabilized with 0.05% Triton X-100 in PBS for 5 min,
and blocked. We washed the slides followed by incubation with the
rabbit anti-human AURKA antibody (1:200) for 2 h at 37°C. The
slides were washed with PBS, stained with Alexa Fluor 594
(red)-conjugated matched secondary antibody and DAPI (for nuclei),
and imaged with a fluorescence microscope (Olympus, Tokyo,
Japan).
Real-time quantitative reverse
transcription PCR
We validated the microarray results by qPCR. We used
TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) to
isolate total RNA from cells. We performed RT using a reverse
transcription system kit to synthesize the first-strand
complementary DNA. For PCR, we used SYBR-Green dye (Takara) with a
7900HT Detection system (Applied Biosystems, Foster City, CA, USA)
and normalized the results to GAPDH. We used the following primer
sets: AURKA forward, 5′-GAG GCCAATGCTCAGAGAAG-3′ and reverse,
5′-AGGGAGGT TAAGGCACACCT-3; p53 forward, 5′-GGCCCACTTCACC
GTACTAA-3′ and reverse, 5′-GTGGTTTCAAGGCCAGA TGT-3′; HDM2 forward,
5′-ACGACAAAGAAAACGCC ACA-3′ and reverse,
5′-ACCAGCATCAAGATCCGGAT-3′; GAPDH forward, 5′-AATTCCATGGCACCGTCA-3′
and reverse, 5′-TGGACTCCACGACGTACTCA-3′.
Western blotting
We harvested and lysed mono- and co-cultured tumor
cells treated and untreated with gefitinib for immunoblotting. We
resolved equal amounts of protein samples using 8% SDS-PAGE gels
and transferred the proteins to a nitrocellulose membrane. We
blocked the membrane with 5% nonfat milk and 0.05% Tween-20 for 1 h
at room temperature. We incubated the blots overnight at 4°C with
the respective primary antibodies. Following the washes, we
incubated the blots for 2 h at room temperature with the
HRP-conjugated antibodies. We used ECL Plus reagent (Thermo Fisher
Scientific, Waltham, MA, USA) to visualize immunoactivity. We
performed densitometric analysis of the bands using Image Lab
(Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
We compared differences by one-way ANOVA. We used
GraphPad Prism version 6.01 (GraphPad Software) for all statistical
analysis. We considered P<0.05 as a statistically significant
difference.
Results
Fibroblasts induce gefitinib resistance
in lung cancer cells
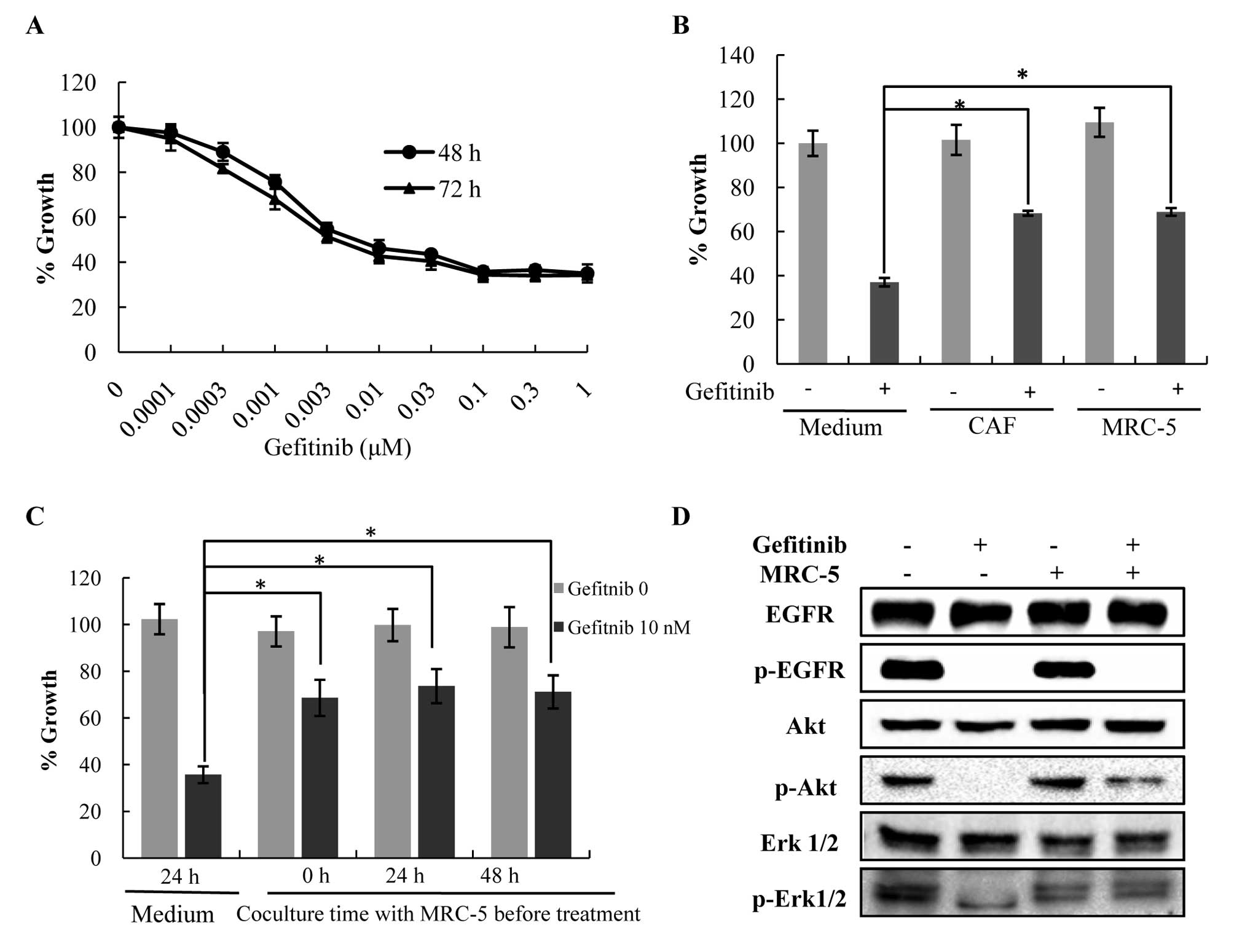
We examined the effect of co-culture with
fibroblasts on the protection of HCC827 cells from
gefitinib-induced apoptosis. HCC827 cells treated with 0.3 μM
gefitinib resulted in a maximal 60% decrease in baseline viability
at 48 and 72 h (Fig. 1A).
Therefore, we used this concentration for the remainder of the
experiments. Co-culture with MRC-5 cells protected HCC827 cells
from gefitinib-induced apoptosis (P<0.05) (Fig. 1B). In the absence of gefitinib,
MRC-5 cells did not promote the growth of HCC827 cells. Similar
results were obtained using a CAF cell line. We did not find
evidence of any increasing protective effect when cells were
exposed for longer time periods (24 or 48 h, Fig. 1C). These results suggest that
fibroblasts reduce gefitinib sensitivity in lung cancer cells with
EGFR-activating mutations, regardless of the state of the
fibroblasts. Moreover, we found that the expression levels of p-ERK
and p-Akt increased in the HCC827 cells co-cultured with MRC-5
cells compared to the mono-culture following treatment with
gefitinib, while the p-EGFR expression levels did not change.
(Fig. 1D). This revealed that the
MRC-5 cells prevented the reduction of p-ERK and p-Akt levels in a
p-EGFR-independent manner in the HCC827 cells in the presence of
gefitinib.
Gefitinib regulates expression of AURKA
in HCC827 cells
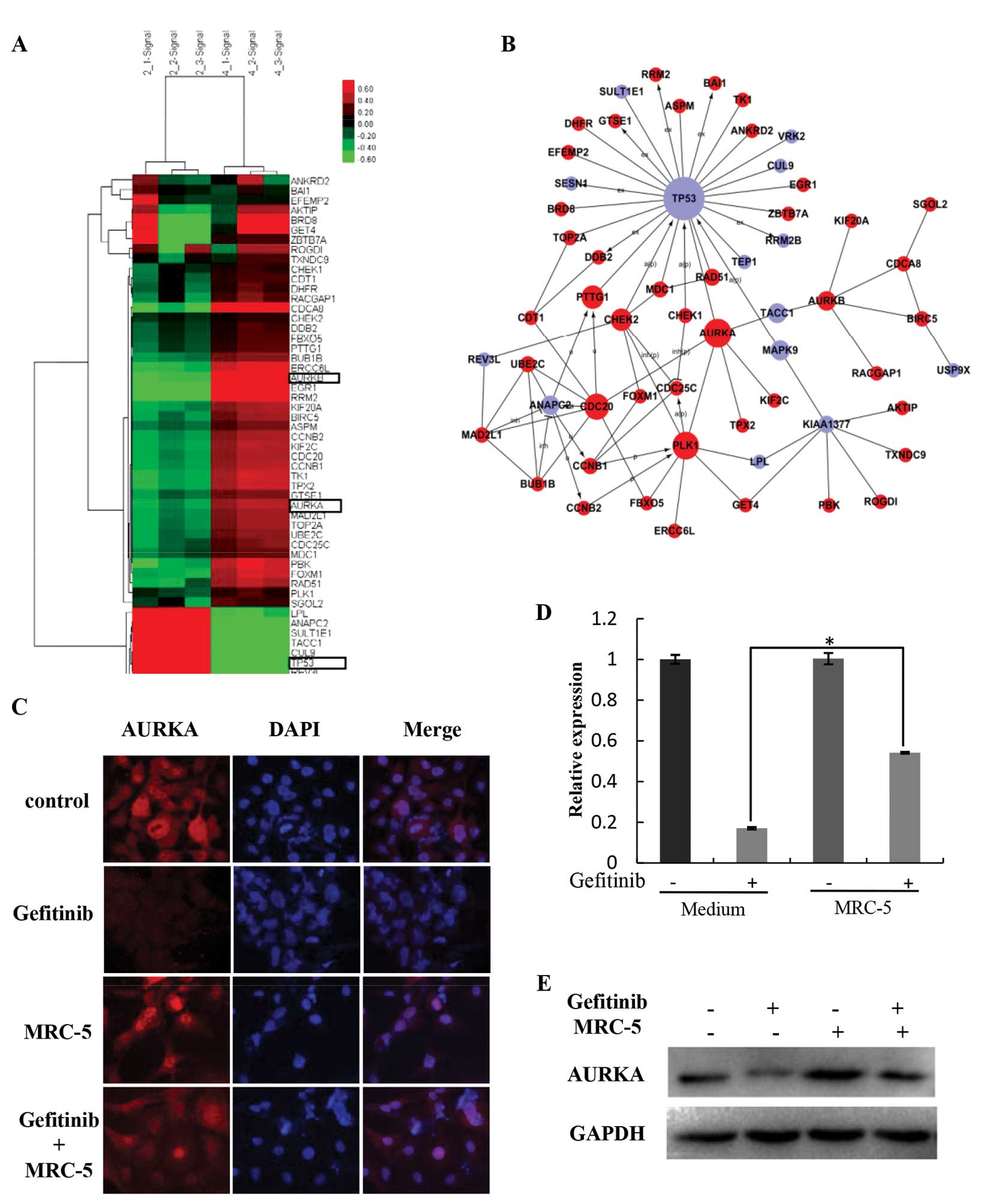
In order to further explore the mechanisms
underlying the different growth rates of HCC827 cells following
treatment with gefitinib between mono- and co-culture with MRC-5
cells, we compared global gene expression using an Affymetrix
PrimeView assay. The genes with significant change (P<0.05 and
false discovery rate <0.05) were selected. Cluster analysis
revealed an increased expression of AURKA and decreased expression
of p53 (Fig. 2A). A co-expression
network of altered gene expression was built, in which AURKA and
p53 were centrally located (Fig.
2B). Since studies indicate that AURKA is involved in the
regulation of resistance to various chemotherapeutic drugs, we
elected to further explore the role of AURKA in reduced gefitinib
sensitivity in HCC827 cells. We confirmed that in the presence of
gefitinib, HCC827 cells expressed higher levels of AURKA under
stromal co-cultures compared with the mono-culture cells (Fig. 2D and E). Immunofluorescence results
corroborated the PCR and western blotting results (Fig. 2C). Importantly, in the absence of
gefitinib, HCC827 cells co-cultured with fibroblasts expressed the
same level of AURKA when compared with this level in the HCC827
cells in the mono-culture. This suggests that AURKA actively
supports the survival of gefitinib-treated cells while being
co-cultured with MRC-5 cells.
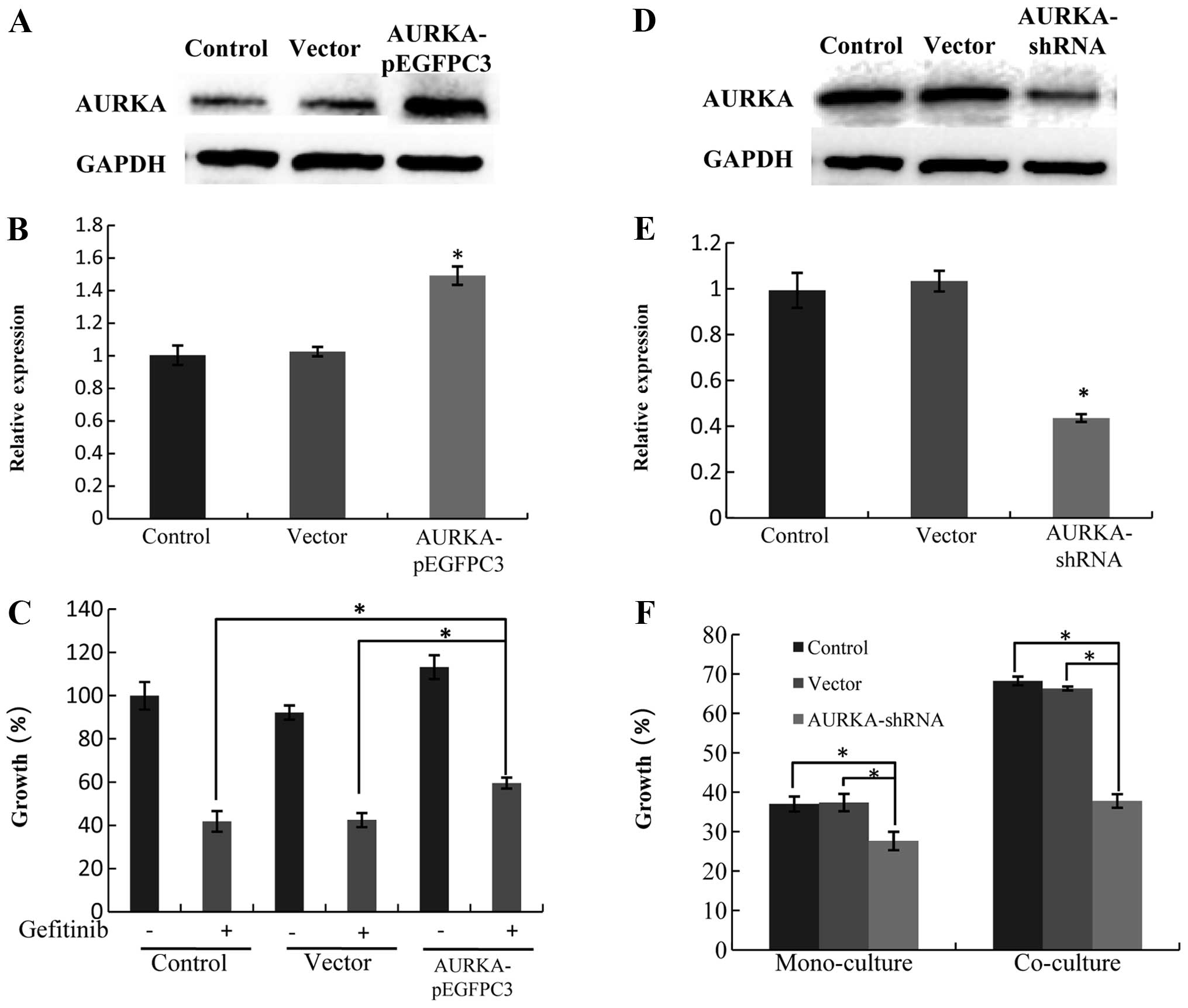
Overexpression of AURKA reduces gefitinib
sensitivity of HCC827 cells
We constructed an overexpression plasmid of AURKA to
investigate whether AURKA overexpression in HCC827 cells affects
the protective effect caused by MRC-5 cells against
gefitinib-induced apoptosis (Fig. 3A
and B). We found that AURKA overexpression reduced the
gefitinib sensitivity of HCC827 cells (Fig. 3C), suggesting that AURKA
upregulation in HCC827 cells may be the leading cause of resistance
to gefitinib when co-cultured with fibroblasts.
Inhibition of AURKA restores gefitinib
sensitivity in HCC827 cells co-cultured with MRC-5 cells
We constructed an AURKA-shRNA plasmid (Fig. 3D and E) to investigate whether AURKA
knockdown influences the gefitinib sensitivity of HCC827 cells. We
found that AURKA-shRNA abrogated the protective effect of MRC-5
cells against gefitinib-induced apoptosis (Fig. 3F).
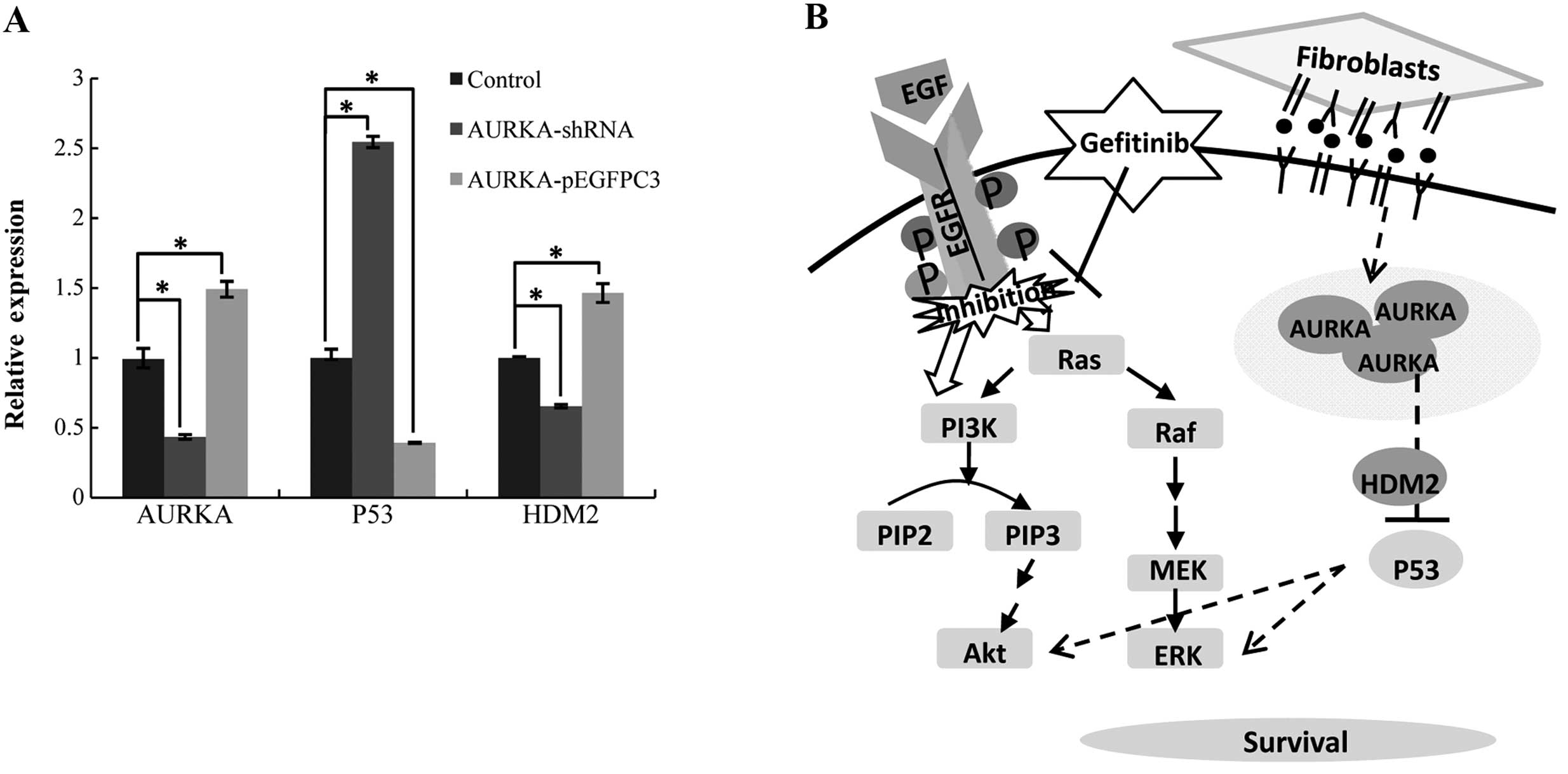
AURKA influences gefitinib sensitivity by
regulating p53 signaling
We investigated whether decreased expression of
AURKA affects the p53 signaling pathway. Inhibition of AURKA
upregulated expression of p53 and downregulated expression of HDM2
(Fig. 4A). Conversely,
overexpression of AURKA downregulated p53 expression and
upregulated HDM2 expression. It has been reported that AURKA
promotes tumor growth and cell survival through regulation of
HDM2-induced ubiquitination and inhibition of p53 (26). From the above results, we propose
that fibroblasts induce gefitinib resistance by regulating the
expression of AURKA, thus inhibiting the p53 signaling pathway
(Fig. 4B).
Discussion
In the present study, we confirmed that co-culture
with fibroblasts partially reduced the sensitivity of HCC827 cells
to gefitinib-induced apoptosis. Since NSCLC EGFR-TKI therapy blocks
the activation of EGFR, we evaluated expression levels of p-EGFR, a
molecular correlate of EGFR activity, and its downstream effectors,
p-ERK and p-Akt, in HCC827 cells in mono- and co-culture with MRC-5
cells. Co-culture with MRC-5 cells did not protect HCC827 cells
from gefitinib induced pEGFR inhibition but significantly increased
p-Akt and p-ERK activation compared with the mono-culture cells.
Without gefitinib, the activation of EGFR and its downstream
effectors in HCC827 cells did not change when co-cultured with
MRC-5 cells. Our data, along with previous research, demonstrating
that activation of MAPK pathways promote survival of cancer cells
(27) suggest that co-culture with
fibroblasts partially abrogates the gefitinib-induced inhibitory
effect of downstream molecules in an EGFR-independent manner in
HCC827 cells.
The tumor microenvironment is important for tumor
progression. Fibroblasts have been linked to several activities
that promote tumor progression, including angiogenesis, progressive
genetic instability, deregulation of antitumor immune responses,
enhanced metastasis, and enhanced growth (28,29).
Several prospective studies indicate that HGF secreted by
fibroblasts reduces gefitinib sensitivity in tumor cells (30), but the underlying mechanism by which
cancer cells survive, remains poorly understood.
Through bioinformatics analysis we found that
following treatment with gefitinib, AURKA expression was higher in
the HCC827 cells co-cultured with fibroblasts compared with the
mono-cultured cells. AURKA is a member of the Aurora kinase family
of serine/threonine kinases whose structures and functions are
highly conserved in different model organisms (31). They play an important role during
cell mitosis, such as promoting the cell to enter mitosis,
assisting in centrosome maturation and separation, assembling and
maintaining spindles, segregating chromosomes, and aiding in
cytokinesis (32,33). Previous studies showed that AURKA
phosphorylates p53 at Ser315, leading to its ubiquitination by Mdm2
and proteolysis. Knockdown of AURKA results in less phosphorylation
of p53 at Ser315, greater stability of p53, and cycle arrest at
G2-M (33). Overexpression of
Aurora kinase has been observed in some tumor cells and aberration
in Aurora kinases has proven to be involved in tumorigenesis
(34). It has been reported that
AURKA promotes tumor growth and cell survival through regulation of
HDM2-induced ubiquitination and inhibition of p53 (35). Consistent with this, we indentified
that the expression levels of p53 and HDM2 are regulated by AURKA
in HCC827 cells. Notably, the expression level of AURKA in
co-cultured HCC827 cells did not change compared with the
mono-cultured HCC827 cells in the absence of gefitinib, suggesting
that fibroblasts promoted tumor cell survival only in the presence
of gefitinib. We also found that AURKA inhibition alone led to
reduced survival in the HCC827 cells. AURKA plays an important role
in mediating gefitinib resistance as shown by the restoration of
gefitinib resistance of HCC827 cells co-cultured with fibroblasts
in the presence of AURKA-shRNA.
Taken together, our results provide genetic evidence
that the tumor microenvironment is a novel modulator of resistance
to gefitinib. Importantly, increased cell survival did not result
from reduced p-EGFR inhibition, but from downstream EGFR effectors.
Moreover, we also demonstrated that decreased drug sensitivity was
caused by increased expression of AURKA, which may rescue cells
from gefitinib-induced apoptosis by p53 signaling pathway
inhibition. In conclusion, our findings have important implications
in cancer-targeted therapy. AURKA inhibition enhances the efficacy
of EGFR-TKIs and reverses acquired resistance resulting from
fibroblasts in the microenvironment. Consequently, preventing AURKA
upregulation may provide valuable enhancement of the efficacy of
specific targeted therapy regimens.
Acknowledgements
This study was supported by the International
Science and Technology Cooperation Program of China
(S2014ZR0042).
References
|
1
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
2
|
Xue C, Hu Z, Jiang W, et al: National
survey of the medical treatment status for non-small cell lung
cancer (NSCLC) in China. Lung Cancer. 77:371–375. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yarden Y: The EGFR family and its ligands
in human cancer. Signaling mechanisms and therapeutic
opportunities. Eur J Cancer. 37:S3–S8. 2001. View Article : Google Scholar
|
|
4
|
Schlessinger J: Cell signaling by receptor
tyrosine kinases. Cell. 103:211–225. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhu H, Cao X, Ali-Osman F, Keir S and Lo
HW: EGFR and EGFRvIII interact with PUMA to inhibit mitochondrial
translocalization of PUMA and PUMA-mediated apoptosis independent
of EGFR kinase activity. Cancer Lett. 294:101–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leu CM, Chang C and Hu C: Epidermal growth
factor (EGF) suppresses staurosporine-induced apoptosis by inducing
mcl-1 via the mitogen-activated protein kinase pathway. Oncogene.
19:1665–1675. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SH, Li Y, Chen HG, Rong J and Ye S:
Activation of proteinase-activated receptor 2 prevents apoptosis of
lung cancer cells. Cancer Invest. 31:578–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Q and Greene MI: EGFR enhances
survivin expression through the phosphoinositide3 (PI-3) kinase
signaling pathway. Exp Mol Pathol. 79:100–107. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lynch TJ, Bell DW, Sordella R, et al:
Activating mutations in the epidermal growth factor receptor
underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paez JG, Jänne PA, Lee JC, et al: EGFR
mutations in lung cancer: correlation with clinical response to
gefitinib therapy. Science. 304:1497–1500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Woodburn JR: The epidermal growth factor
receptor and its inhibition in cancer therapy. Pharmacol Ther.
82:241–250. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wakeling AE, Guy SP, Woodburn JR, et al:
ZD1839 (Iressa): an orally active inhibitor of epidermal growth
factor signaling with potential for cancer therapy. Cancer Res.
62:5749–5754. 2002.PubMed/NCBI
|
|
13
|
Kobayashi S, Boggon TJ, Dayaram T, et al:
EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Balak MN, Gong Y, Riely GJ, et al: Novel
D761Y and common secondary T790M mutations in epidermal growth
factor receptor-mutant lung adenocarcinomas with acquired
resistance to kinase inhibitors. Clin Cancer Res. 12:6494–6501.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bean J, Brennan C, Shih JY, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baguley BC: Multiple drug resistance
mechanisms in cancer. Mol Biotechnol. 46:308–316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liotta LA and Kohn EC: The
microenvironment of the tumor-host interface. Nature. 411:375–379.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Witz IP: Tumor-microenvironment
interactions: dangerous liaisons. Adv Cancer Res. 100:203–229.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cukierman E and Bassi DE: The mesenchymal
tumor microenvironment: a drug-resistant niche. Cell Adh Migr.
6:285–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Flach EH, Rebecca VW, Herlyn M, Smalley KS
and Anderson AR: Fibroblasts contribute to melanoma tumor growth
and drug resistance. Mol Pharm. 8:2039–2049. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ostman A and Augsten M: Cancer-associated
fibroblasts and tumor growth - bystanders turning into key players.
Curr Opin Genet Dev. 19:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johansson AC, Ansell A, Jerhammar F, et
al: Cancer-associated fibroblasts induce matrix
metalloproteinase-mediated cetuximab resistance in head and neck
squamous cell carcinoma cells. Mol Cancer Res. 10:1158–1168. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jansen R, Greenbaum D and Gerstein M:
Relating whole-genome expression data with protein-protein
interactions. Genome Res. 12:37–46. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sehdev V, Katsha A, Arras J, et al: HDM2
regulation by AURKA promotes cell survival in gastric cancer. Clin
Cancer Res. 20:76–86. 2014. View Article : Google Scholar :
|
|
27
|
Zimmer S, Kahl P, Buhl TM, et al:
Epidermal growth factor receptor mutations in non-small cell lung
cancer influence downstream Akt, MAPK and Stat3 signaling. J Cancer
Res Clin Oncol. 135:723–730. 2009. View Article : Google Scholar
|
|
28
|
Ostman A and Augsten M: Cancer-associated
fibroblasts and tumor growth - bystanders turning into key players.
Curr Opin Genet Dev. 19:67–73. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhowmick NA, Neilson EG and Moses HL:
Stromal fibroblasts in cancer initiation and progression. Nature.
432:332–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Donev IS, Wang W, Yamada T, et al:
Transient PI3K inhibition induces apoptosis and overcomes
HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer.
Clin Cancer Res. 17:2260–2269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Giet R and Prigent C: Aurora/Ipl1p-related
kinases, a new oncogenic family of mitotic serine-threonine
kinases. J Cell Sci. 112:3591–3610. 1999.PubMed/NCBI
|
|
32
|
Gautschi O, Heighway J, Mack PC, Purnell
PR, Lara PN and Gandara DR: Aurora kinases as anticancer drug
targets. Clin Cancer Res. 14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nishimura Y, Endo T, Kano K and Naito K:
Porcine Aurora A accelerates Cyclin B and Mos synthesis and
promotes meiotic resumption of porcine oocytes. Anim Reprod Sci.
113:114–124. 2009. View Article : Google Scholar
|
|
34
|
Anand S, Penrhyn-Lowe S and Venkitaraman
AR: Aurora-A amplification overrides the mitotic spindle assembly
checkpoint, inducing resistance to taxol. Cancer Cell. 3:51–62.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Katayama H, Sasai K, Kawai H, et al:
Phosphorylation by aurora kinase A induces Mdm2-mediated
destabilization and inhibition of p53. Nat Genet. 36:55–62. 2004.
View Article : Google Scholar : PubMed/NCBI
|