Introduction
Lung cancer is the leading cause of cancer-related
deaths worldwide. The majority of non-small cell lung cancer
(NSCLC) patients are in the advanced stage of the disease at the
time of diagnosis. Systemic platinum-based chemotherapy is usually
the treatment of choice for advanced lung cancer (1). Unfortunately, conventional
chemotherapy regimens are hampered by limited efficacy, significant
toxicity and a high relapse rate, indicating an urgent need for
developing alternative therapeutic approaches such as targeted
therapy. The development of tyrosine kinase inhibitors targeting
mutant epidermal growth factor receptor (EGFR) is a good example of
lung cancer targeted therapy (2).
However, the identification of novel targets is still required in
the future development of lung cancer therapy.
Cullin 4A (Cul4A) is a member of the evolutionarily
conserved cullin protein family, which is related to the ubiquitin
proteosome pathway. Overexpression of Cul4A has been observed in
breast (3,4), mesothelioma (5) and liver cancers (6). Cul4A has also been implicated in the
ubiquitination and proteolysis of tumor suppressors, including p53
(7), NF2 (8), RASSF1A (9) and p21 (10,11),
to promote oncogenesis. Our previous study demonstrated that the
expression of Cul4A causes ubiquitination and destablization of p21
and p27. Consequently, downregulation of Cul4A expression resulted
in G0/G1 arrest; and overexpression, increased growth of
mesothelioma cells (5). Cul4A is
also a critical gene for the survival of hematopoietic cells and
development and growth of cancer cells (12). In a transgenic mouse study,
conditional expression of Cul4A resulted in the formation of lung
tumors (11,13). Since Cul4A is overexpressed in
cancers and is associated with oncogenesis, Cul4A can be a
candidate for targeted therapy.
Transforming growth factor β-induced, 68-kDa protein
(TGFBI) is known as keratoepithelin or βIg-h3, and contains four
conserved fasciclin-1 (FAS1) domains and a carboxyl-terminal
Arg-Gly-Asp (RGD) integrin-binding sequence. TGFBI mediates
integrin binding to extracellular matrix proteins such as collagen,
laminin and fibronectin (14). Loss
of TGFBI expression has been reported in several types of cancers
including lung cancer (15), and it
has been suggested to act as a tumor-suppressor gene (16). Additionally, overexpression of TGFBI
in lung cancer cells increased the sensitivity to gemcitabine
(17), and overexpression of TGFBI
has also been reported to be associated with a better response to
gemcitabine chemotherapy in lung cancer cells and patients.
Although the Cul4A-DDB1 E3 ligase complex has been reported to
ubiquitinate and degrade many tumor-suppressor proteins, it is
unclear whether Cul4A causes ubiquitination of TGFBI to decrease
its stability.
The present study aimed to investigate the
association of TGFBI and Cul4A and the mechanism by which Cul4A
regulates TGFBI. In addition, we also evaluated the therapeutic
value of Cul4A RNAi using adenoviral transfection of Cul4A RNAi in
a nude mouse xenograft model.
Materials and methods
Cell culture
The NSCLC cell lines A549 (ATCC CCL-185) and H460
(ATCC HTB-177) were purchased from the American Type Culture
Collection (ATCC, Manassas, VA, USA). Both A549 and H460 cell lines
were grown in complete RPMI-1640 growth medium supplemented with
10% fetal calf serum, 10 U/ml penicillin and 10 µg/ml
streptomycin at 37°C and in 5% CO2.
Retroviral production and
transduction
The pBabe-puro retroviral vector (Addgene,
Cambridge, MA, USA) was used to transduce the Cul4A gene, and the
pSUPER-retro-puro vector (Oligoengine, Seattle, WA, USA) was used
to express Cul4A shRNA. The resultant pBabe Cul4A vector expressing
the Cul4A-myc-His fusion protein and the pSuper Cul4A vector
expressing Cul4A shRNA were constructed as described previously
(5). Retroviral vectors were
transfected into the HEK 293 Phoenix ampho packaging cells (ATCC)
using Fu-GENE6 transfection reagent (Roche, Lewes, UK). After
transfection for 48 h, the supernatant was filtered using a 0.45-mm
syringe filter. Retroviral infection was performed by adding the
filtered supernatant to lung cancer cell lines cultured on 10-cm
dishes with 50% confluency in the presence of 8 µg/ml of
Polybrene (Sigma-Aldrich, St. Louis, MO, USA). Six hours after
infection, the culture medium was replaced with fresh medium. The
infected cells were allowed to recover for 48 h. Infected cells
were selected by adding 1 mg/ml puromycin (Sigma-Aldrich) for 48 h
and then maintained in complete medium with 0.5 mg/ml puromycin.
Empty retroviral-infected stable cell lines were also produced
following the above protocols (5).
Western blot analysis
Whole protein was extracted using Mammalian Protein
Extraction Reagent (M-PER) from the cell lines added with
Phosphatase Inhibitor Cocktail Set II (Calbiochem, San Diego, CA,
USA) and Complete Protease Inhibitor Cocktails (Roche) according to
the manufacturer's protocols. The proteins were separated on 4–15%
gradient SDS-polyacrylamide gels and transferred to Immobilon-P
membranes (Millipore, Billerica, MA, USA). The following primary
antibodies were used: anti-Cul4A (Abcam, Cambridge, MA, USA),
anti-β-actin (Sigma Chemical, St. Louis, MO, USA), anti-TGFBI
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and anti-HA
(Cell Signaling, Danvers, MA, USA). After being incubated with the
appropriate secondary antibodies, the antigen-antibody complexes
were detected using an ECL blotting analysis system (Amersham
Pharmacia Biotech, Piscataway, NJ, USA). Densitometry of western
blot analysis was determined by ImageJ (v1.44m for Windows;
National Institutes of Health).
Semi-quantitative reverse
transcription-PCR (RT-PCR) analysis
RNA was extracted using the RNeasy Mini kit (Qiagen,
Hilden, Germany) from cell pellets according to the manufacturer's
instructions. Total RNA was then transcripted to cDNA using the
iScript™ cDNA synthesis kit (Bio-Rad Laboratories, Munich,
Germany). Reverse-transcribed cDNA (2 µl) was subjected to
with a total volume of 20 µl and the Bio-Rad CFX96™
quantitative PCR system (Bio-Rad Laboratories). The following
primers were used for PCR: Cul4A, 5′-GCACTGGAGCGAGTACATCA-3′
(sense) and 5′-CACATGCTTTGCGATCAGTT-3′ (antisense); TGFBI,
5′-AGCCCTGCCACCAAGAGAA-3′ (sense) and 5′-CTCCGCTAACCAGGATTTCATC-3′
(antisense); GAPDH, 5′-CATCCATGACAACTTTGGTATCGT-3′ (sense) and
5′-CAGTCTTCTGGGTGGCAGTGA-3′ (antisense). Amplification conditions
were as follows: 1 cycle of 95°C for 5 min and 30 cycles of 95°C
for 20 sec, 58°C for 30 sec, 72°C for 30 sec and 1 cycle of 72°C
for 5 min and then maintained at 4°C.
Immunofluorescence
For immunofluorescence microscopy, the cells were
grown on coverslips, fixed in cold methanol for 10 min at −20°C,
and blocked with 2% bovine serum albumin for 30 min. The cells were
incubated with the primary anti-TGFBI (Santa Cruz Biotechnology,
Inc.) antibody in 2% bovine serum albumin for 1 h at room
temperature. The cells were washed with PBS and subsequently
incubated with FITC-conjugated secondary antibodies in 2% bovine
serum albumin for 1 h at room temperature. After washing with PBS,
the cells were counterstained with DAPI and mounted in Vectashield
(Vector Laboratories, Burlingame, CA, USA). Images were acquired
using a TCS SP5 confocal microscope (Leica, Wetzlar, Germany). The
intensity of TGFBI was quantified by MetaMorph®
Microscopy Automation & Image Analysis Software (Molecular
Devices, Sunnyvale, CA, USA).
Cell viability assay
H460 and A549 stable cells (1×104/ml)
were cultured in 6-well plates for 48 h and then treated with the
indicated concentration of gemcitabine for 72 or 96 h as indicated.
Then, cells were trypsinized. The number of viable cells was
counted by trypan blue dye exclusion using a hemocytometer. The
result was expressed as a percentage, relative to the empty
virus-transfected control groups. The half maximal inhibitory
concentration (IC50) value was determined using GraphPad
Prism® log (inhibitor) vs. response (variable slope)
software (version 6; La Jolla, CA, USA).
Transfection of small interfering RNA
(siRNA) and vectors
Pre-designed and validated TGFBI and universal
negative control siRNA were purchased from Santa Cruz
Biotechnology, Inc. Transfection was performed using Lipofectamine™
RNAiMAX transfection reagent (Invitrogen Life Technologies,
Carlsbad, CA, USA) according to the manufacturer's manual. The
cells were plated in 6-well plates in antibiotic-free media, and
transfection was performed with cells at 80% confluency with a
final concentration of 50 nM for each siRNA. At 96 h after
transfection, the cells were treated with the indicated
concentration of gemcitabine for 72 h and then the number of viable
cells was counted.
The pCMV6-TGFBI-GFP (OriGene, Rockville, MD, USA)
and empty pCDNA3 (Invitrogen Life Technologies) vectors were
transfected with OmniFect™ transfection reagent (TransOMIC,
Huntsville, AL, USA), according to the manufacturer's manual. The
cells were plated in 6-well plates in antibiotic-free media, and
transfection was performed with cells at 80% confluency with a
final concentration of 0.5 µg for each vector. At 96 h after
transfection, the cells were treated with the indicated
concentration of gemcitabine for 72 h and then the number of viable
cells was counted.
Protein degradation assay
A protein degradation assay was used to evaluate the
effects of Cul4A on the decay of TGFBI in lung cancer cells. Lung
cancer cells that were retrovirally transfected with Cul4A shRNA
and Cul4A were plated on 6-cm culture dishes. At 80% confluency,
the cells were exposed to 20 mg/ml of cycloheximide. Then, the
cells were harvested at the indicated time points. Total cellular
proteins were extracted and analyzed by western blot analysis using
β-actin as a loading control.
Co-immunoprecipitation assay
293T cells were transiently co-transfected with the
pBabe-Cul4A-myc-his and pCMV6-TGFBI-GFP (OriGene) vectors with
Lipofectamine 2000 transfection reagent (Invitrogen Life
Technologies). Twenty-four hours after transfection, the cells were
treated with 10 mM of MG132 (Sigma-Aldrich) and then harvested in
NP-40 lysis buffer [150 mM NaCl, 50 mM Tris (pH 8.0), 1% NP40],
protease inhibitor and phosphatase inhibitor cocktail (Roche).
Immunoprecipitation was performed by the Catch and Release v2.0
reversible immunoprecipitation system (Millipore) according to the
manufacturer's protocols. Anti-GFP (OriGene) and anti-Myc tag (Cell
Signaling, Danvers, MA, USA) antibodies were used for
immunoprecipitation, respectively.
In vivo ubiquitination assay
293T cells were cotransfected in combination with
pBabe-Cul4A-myc-his and pCMV6-TGFBI-GFP (OriGene) with or without
pRK5-HA-Ubiquitin-WT (Addgene). All cells were treated with 10
µM of MG132 for 24 h prior to being lysed. Anti-GFP antibody
was used for immunoprecipitation. Anti-HA antibody was used for
western blot analysis.
Adenovirus expressing Cul4A RNAi
An adenovirus expressing Cul4A RNAi was generated
with BLOCK-iT™ adenoviral RNAi expression system (Invitrogen, Life
Technologies) according to the instructions from the manufacturer's
manual. Briefly, double-stranded oligonucleotides that encode Cul4A
siRNA were cloned into the pENTR™/U6 vector. The following Cul4A
siRNAi sequences were used for cloning: 5′-GGUUUAUCCACGGUAAAGA-3′
(5) and 5′-GCAGAACUGAUCGCAAAGCAU-3′
(18). The resulting clones were
verified by DNA sequencing. This study used the pAd/BLOCK-iT™-DEST
vector and a pENTR™/U6 entry clone in an LR recombination reaction
to generate an expression clone containing the U6 RNAi cassette of
Cul4A. After digesting the DNA with PacI, the
pAd/BLOCK-iT™-DEST expression construct was used to transfect the
293A cell line to produce an adenoviral stock. An empty virus was
also generated using the same method. The adenoviral stock was
amplified and titered using the Adeno-X™ Rapid Titer kit (Clontech
Laboratories, Inc., Mountain View, CA, USA). Knockdown of Cul4A was
confirmed by immunoblotting using the empty virus-transfected group
as the control. The adenovirus was used for the xenograft
study.
Nude mouse xenograft models
After approval from the Institutional Animal Care
and Use Committee at Chang Gung Memorial Hospital, Chiayi, Taiwan,
female Balb/c athymic nude mice of 5–6 weeks of age were housed
under specific pathogen-free conditions. H460 or A549 lung cancer
cells (1×106) in 100 µl of serum-free RPMI-1640
medium and 20% Matrigel (BD Biosciences, San Jose, CA, USA) were
injected subcutaneously into the flank areas of the mice. When
tumors reached the mean size of ~50–100 mm3, the mice
were divided randomly into groups of five and injected with either
AdCul4A or AdEV viruses with or without gemcitabine, at 120 mg/kg
intraperitoneally (i.p.) weekly for three weeks. An intratumoral
injection of 2×109 plaque forming units (PFU) of the
AdCul4A virus or AdEV control virus was performed at days 0, 7 and
14 from the day of injection. Tumors were measured with a caliper
twice a week for 6 weeks, and the tumor volume was calculated by
the formula (L × W2/2), where L represents the largest
tumor diameter and W represents the smallest tumor diameter. Then,
the mice were sacrificed, and tumors were excised for further
studies at the indicated times.
Statistical analysis
The Student's t-test was used to compare variables
in the different groups studied. Statistical analysis was carried
out using SPSS (version 10.0; SPSS, Inc., Chicago, IL, USA).
Significance was defined as P<0.05 with two-sided analysis.
Results
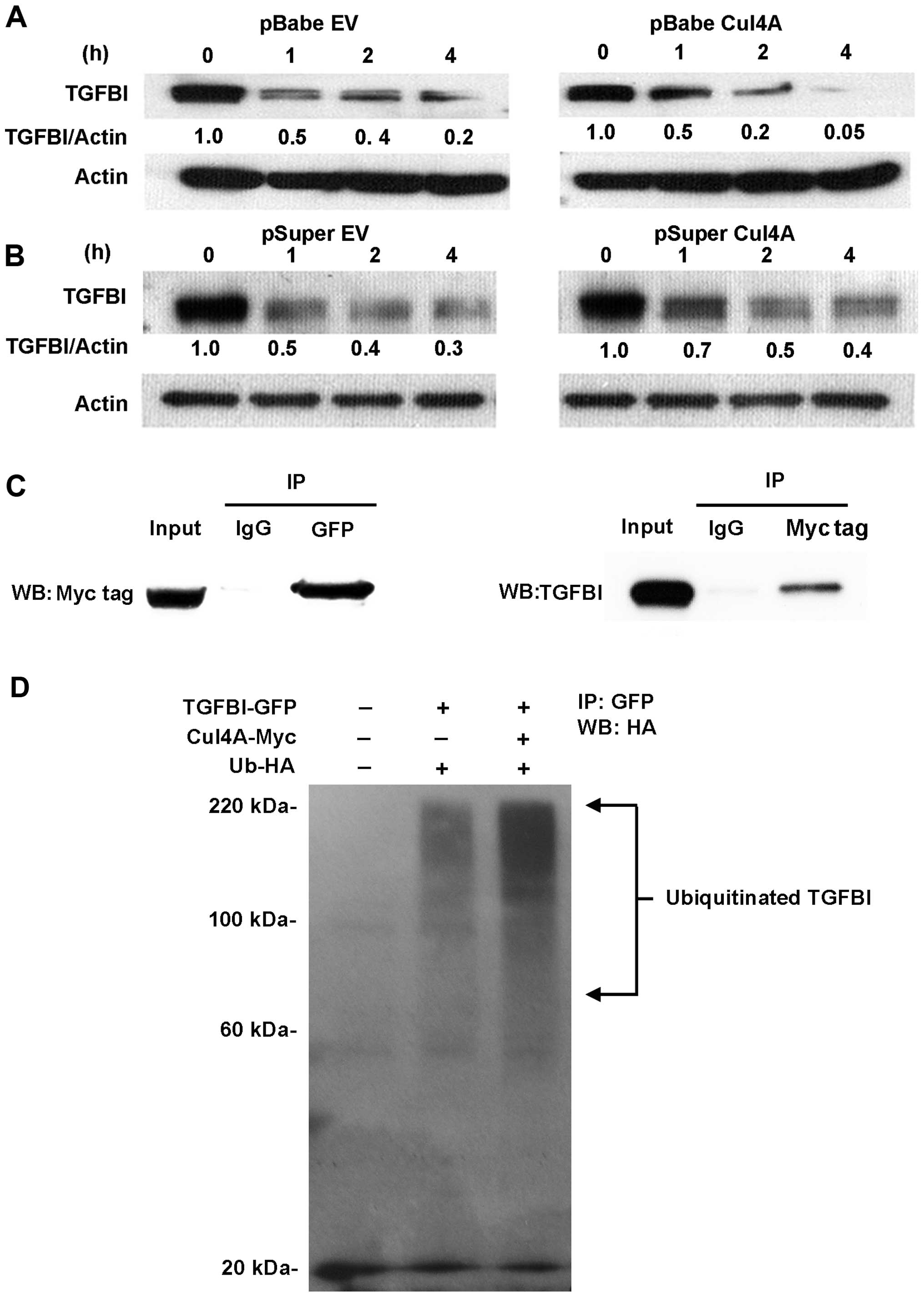
Overexpression of Cul4A downregulates
TGFBI expression in lung cancer cells
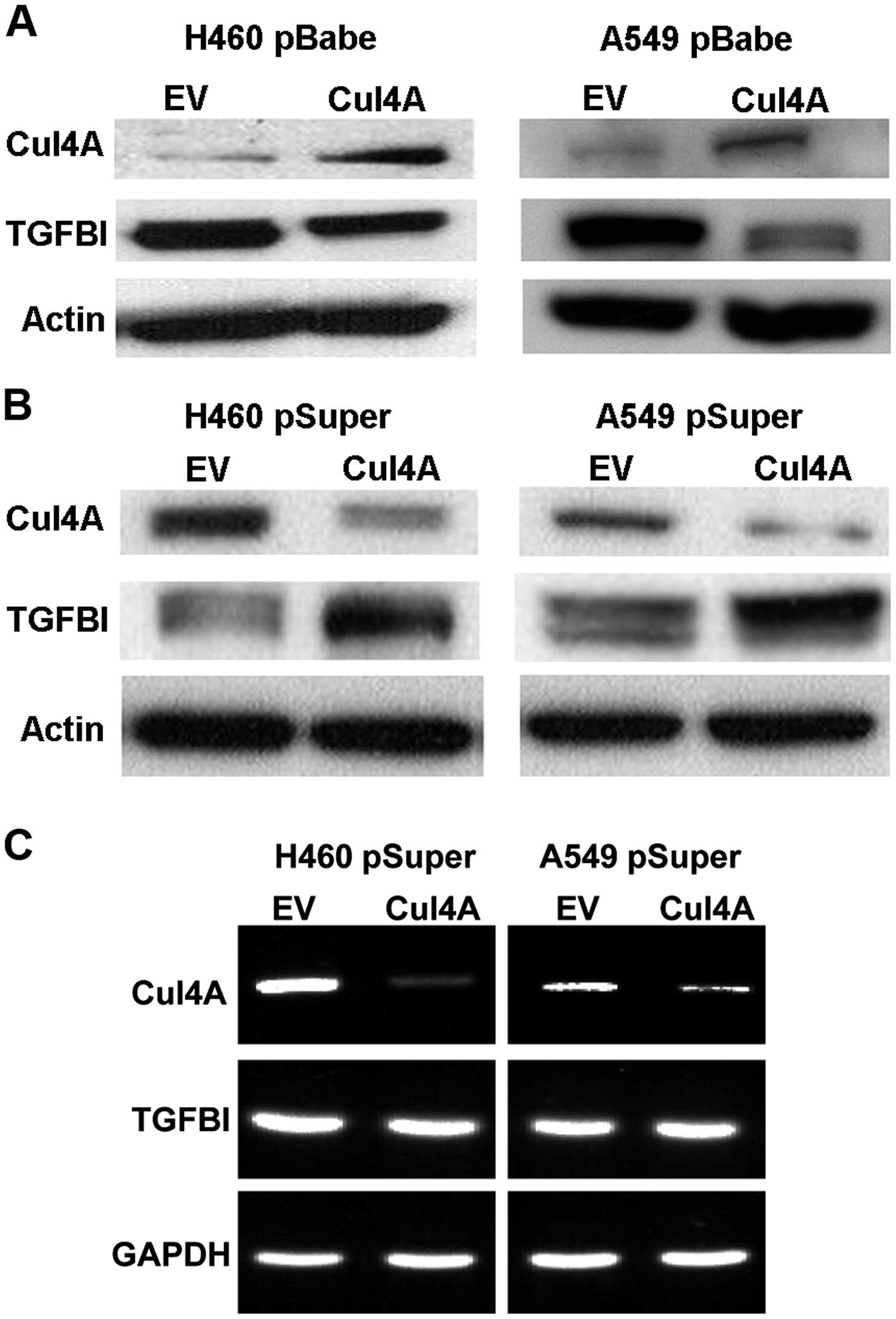
To study the correlation between Cul4A expression
and TGFBI in lung cancer cells, we established stable cell lines
expressing Cul4A using retroviral transfection with the pBabe-puro
vector in H460 and A549 (H460 pBabeCul4A and A549 pBabeCul4A) lung
cancer cells. In the stable cell lines overexpressing Cul4A,
downregulation of TGFBI was observed when compared to the
expression level in the empty vector-transfected stable cells (H460
pBabeEV and A549 pBabeEV) (Fig.
1A).
Downregulation of Cul4A upregulates TGFBI
expression in lung cancer cells
To further study the association of Cul4A and TGFBI
expression in lung cancer cells, knockdown of Cul4A expression in
the H460 and A549 lung cancer cells was performed using retroviral
transfection of Cul4A-specific shRNA with the pSuper-puro vector.
In the Cul4A-knockdown H460 and A549 (H460 pSuperCul4A and A549
pSuperCul4A) lung cancer cells, upregulation of TGFBI was observed
compared to the expression level in the empty vector-transfected
stable cells (H460 pSuperEV and A549 pSuperEV) (Fig. 1B). To further evaluate whether
upregulation of TGFBI is associated with increased transcription of
TGFBI mRNA, RT-PCR was performed to evaluate the expression of
Cul4A and TGFBI in the Cul4A-knockdown H460 and A549 lung cancer
cells. The expression of TGFBI mRNA was not changed in both the
Cul4A-knockdown lung cancer cell lines (Fig. 1C). Thus, Cul4A may not regulate
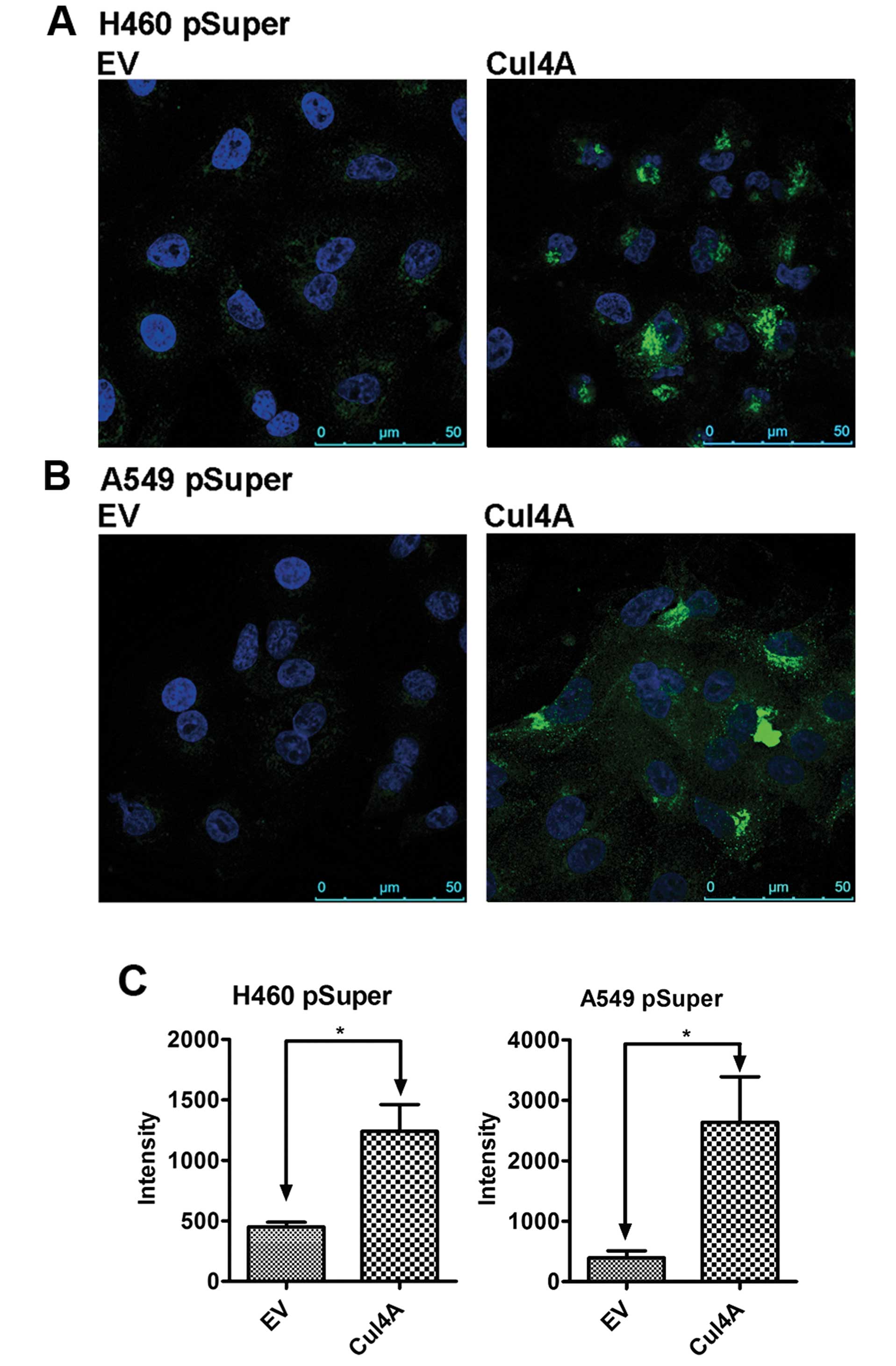
TGFBI through transcriptional regulation of mRNA. Immunofluorescent
analysis was also performed to evaluate the expression of TGFBI in
the Cul4A-knockdown H460 (Fig. 2A)
and A549 (Fig. 2B) (H460
pSuperCul4A and A549 pSuperCul4A) lung cancer cells. Upregulation
of TGFBI was also observed in these cells when compared to the
expression level in the empty vector-transfected stable cells (H460
pSuperEV and A549 pSuperEV), and TGFBI was mainly expressed in the
cytoplasm and on the surface of the lung cancer cells studied.
Downregulation of Cul4A increases
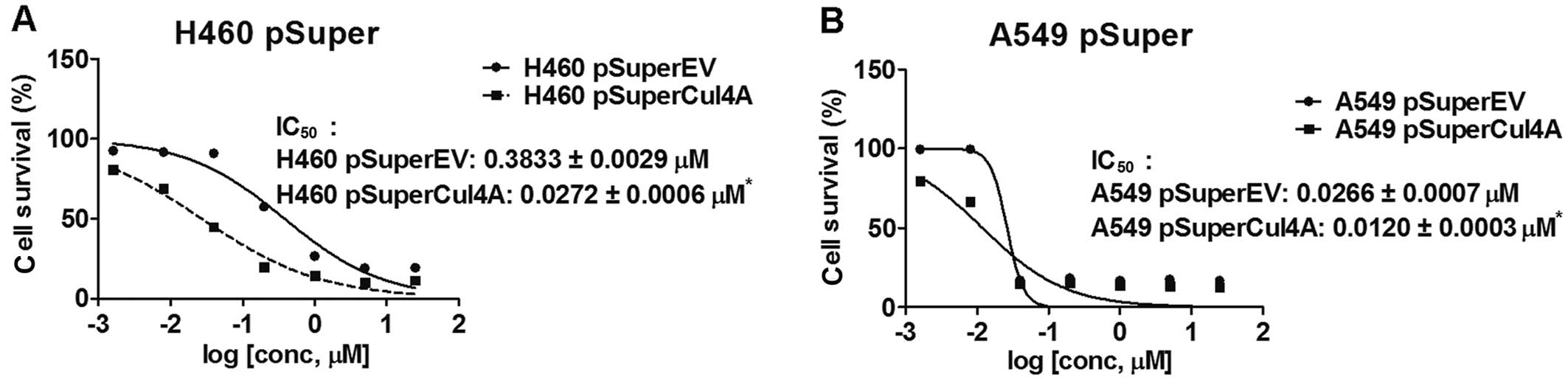
chemosensitivity to gemcitabine in lung cancer cells
We next studied the chemo-sensitivity to gemcitabine
in the Cul4A-knockdown H460 and A549 lung cancer cells. We treated
Cul4A-knockdown (H460 pSuperCul4A and A549 pSuperCul4A) and empty
vector-transfected (H460 pSuperEV and A549 pSuperEV) lung cancer
cells with the indicated concentrations of gemcitabine for 96 h.
Viable cells were counted and normal-ized to the cell number in the
empty vector-transfected groups without treatment. IC50
values were determined in the H460 pSuperCul4A (0.0272±0.0006
µM), A549 pSuperCul4A (0.0120±0.0003 µM), H460
pSuperEV (0.3833±0.0029 µM) and A549 pSuperEV (0.0266±0.0007
µM) lung cancer cells. Significantly lower IC50
values were observed in the Cul4A-knockdown (H460 pSuperCul4A and
A549 pSuper-Cul4A) cells when compared to the corresponding empty
vector-transfected (H460 pSuperEV and A549 pSuperEV) lung cancer
cells (Fig. 3A and B).
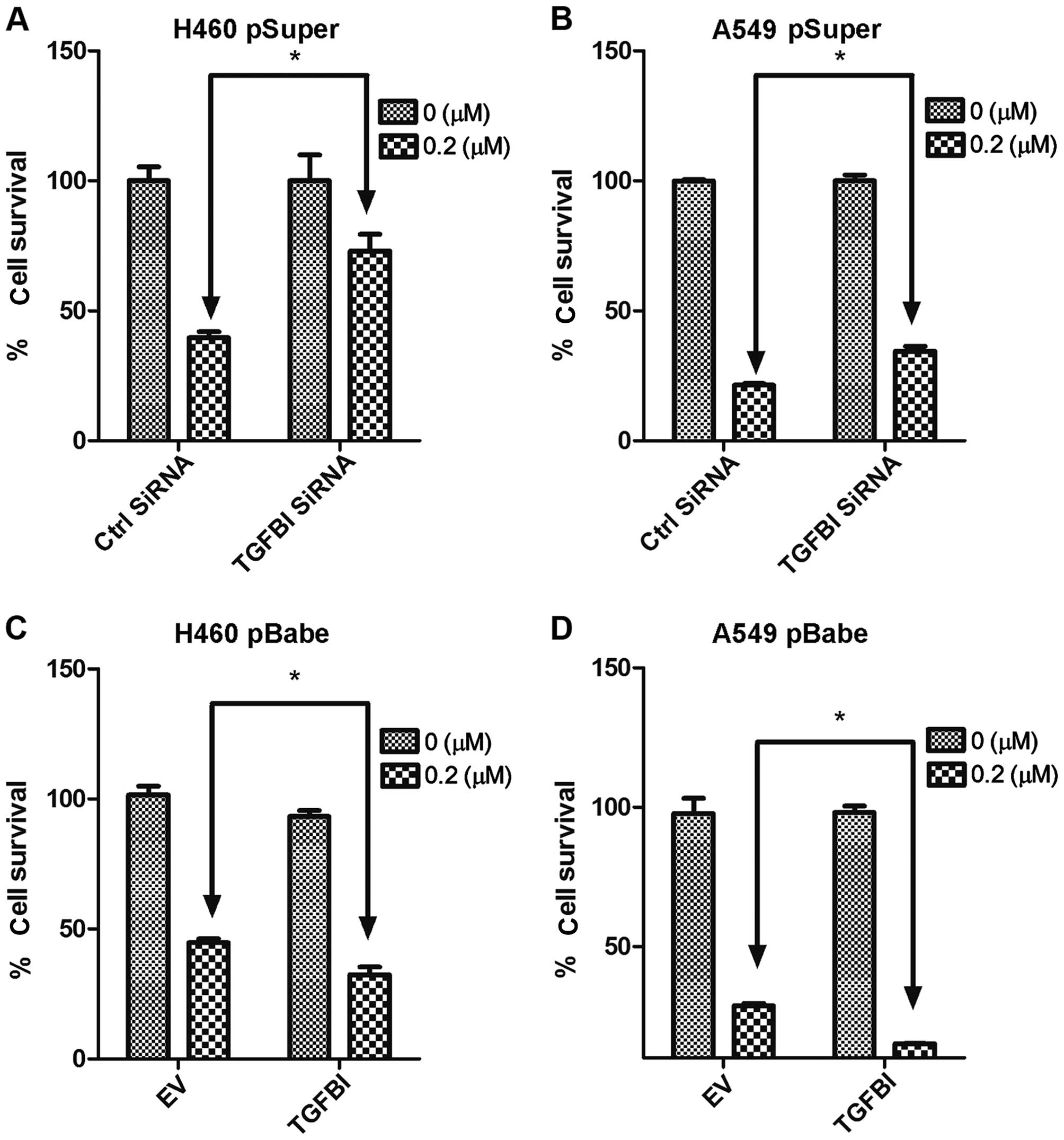
Downregulation of TGFBI decreases
chemosensitivity to gemcitabine in Cul4A-knockdown lung cancer
cells
To further evaluation the effects of TGFBI on
chemosensitivity to gemcitabine, we downregulated TGFBI expression
in the Cul4A-knockdown H460 pSuperCul4A and A549 pSuper-Cul4A lung
cancer cells. All groups of cells were treated with 0.2 µM
of gemcitabine for 72 h. Compared to the control siRNA-treated
groups, significantly decreased chemosensitivity to gemcitabine was
observed in the TGFBI siRNA-treated H460 pSuperCul4A (Fig. 4A) and A549 pSuperCul4A (Fig. 4B) lung cancer cells.
Overexpression of TGFBI increases
chemosensitivity to gemcitabine in Cul4A-overexpressing lung cancer
cells
The effects of TGFBI overexpression on
chemosensitivity to gemcitabine were also studied. We overexpressed
TGFBI in the H460 pBabeCul4A and A549 pBabeCul4A lung cancer cells.
All groups of cells were treated with 0.2 µM of gemcitabine
for 72 h. Compared to the empty vector-transfected groups,
significantly increased chemosensitivity to gemcitabine was
observed in the TGFBI-overexpressing H460 pBabeCul4A (Fig. 4C) and A549 pBabeCul4A (Fig. 4D) lung cancer cells.
Cul4A regulates TGFBI stability through
protein degradation and ubiquitination
Since Cul4A-DDB1 E3 ligase complex has been reported
to ubiquitinate and degrade many tumor-suppressor proteins, we thus
studied whether Cul4A regulates TGFBI stability. A protein
degradation assay was performed using the Cul4A-overexpressing H460
pBabeCul4A lung cancer cells. Compared to the empty
vector-transfected H460 pBabeEV lung cancer cells, increased
degradation of TGFBI was noted in the H460 pBabeCul4A lung cancer
cells (Fig. 5A), which indicated
that overexpression of Cul4A is related to increased degradation of
TGFBI. A protein degradation assay was also performed using
Cul4A-knockdown H460 pSuperCul4A lung cancer cells. Compared to the
empty vector-transfected H460 pSuperEV lung cancer cells, decreased
degradation of TGFBI was noted in the H460 pSuperCul4A lung cancer
cells (Fig. 5B), which indicated
that downregulation of Cul4A is related to decreased degradation of
TGFBI.
The interaction between Cul4A and TGFBI was also
evaluated in our study. Reciprocal immunoprecipitation of
Myc-tagged Cul4A and GFP-tagged TGFBI proteins was performed.
Direct and preferential binding between the Cul4A and TGFBI
proteins was noted (Fig. 5C). Since
Cul4A exerts its functions through forming the multifunctional
ubiquitin-protein ligase E3 complex by interacting with ROC1 and
DDB1, we further performed in vivo ubiquitination assays to
determine whether Cul4A is a TGFBI E3 ligase. 293T cells were
cotransfected with HA-tagged ubiquitin and Myc-tagged Cul4A in
combination with GFP-tagged TGFBI. Our results showed that TGFBI
was ubiquitinated by Cul4A (Fig.
5D).
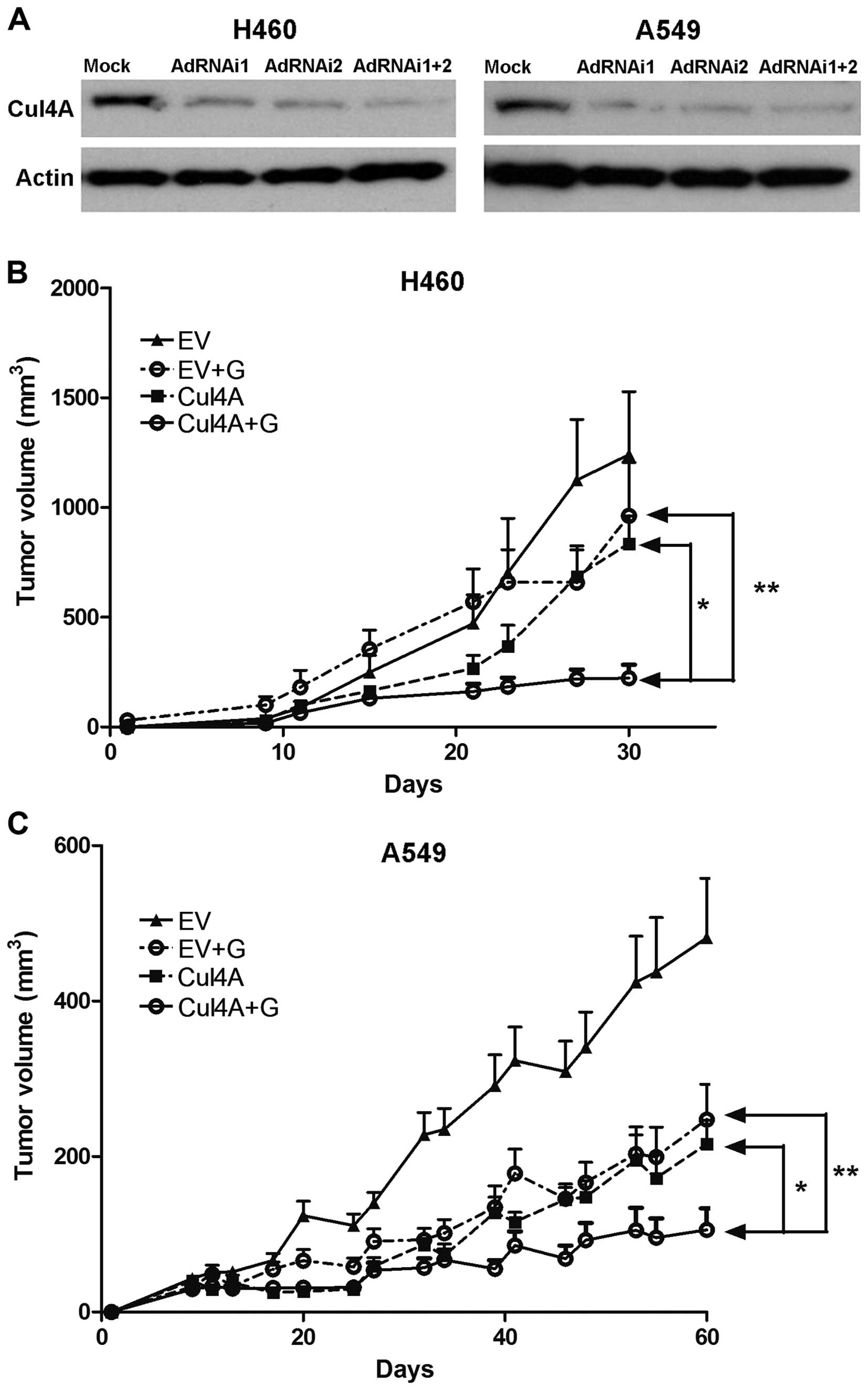
Adenovirus-mediated transfection of Cul4A
RNAi inhibits tumor growth in lung cancer xenograft models
Since the susceptibility of lung cancer cells to
gemcitabine was elevated after the knockdown of Cul4A in the stable
lung cancer cell lines (Fig. 3A and
B), we further studied the therapeutic effects of Cul4A RNAi
and the synergistic effects of Cul4A RNAi with gemcitabine in nude
mouse xenograft models. Transient transfection of cells with Cul4A
RNAi was performed in this model. Adenoviruses expressing Cul4A
RNAi (AdRNAi1 and AdRNAi2) targeting different regions of the Cul4A
gene were generated. Compared to the empty virus-transfected cells,
the combination of both AdRNAi1 and AdRNAi2 viruses resulted in
downregulation of Cul4A in the H460 and A549 lung cancer cells
(Fig. 6A). Nude mouse xenografts
derived from the H460 and A549 lung cancer cells were then
established. Compared to the empty virus combined with gemcitabine
and the Cul4A RNAi-transfected groups, significantly decreased
tumor growth was observed in the groups which combined Cul4A RNAi
and gemcitabine in the H460 (Fig.
6B) and A549 (Fig. 6C) lung
cancer xenograft models.
Discussion
In the present study, we observed that knockdown of
Cul4A enhanced chemosensitivity to gemcitabine in lung cancer
cells. Upregulation of TGFBI was noted in the Cul4A-knockdown lung
cancer cells and downregulation of TGFBI was noted in the
Cul4A-overexpressing lung cancer cells. Further knockdown of TGFBI
expression in the Cul4A-knockdown lung cancer cells decreased
chemosensitivity. In contrast, overexpression of TGFBI in the
Cul4A-overexpressing lung cancer cells increased chemosensitivity.
Thus, the mechanisms by which Cul4A affects chemosensitivity to
gemcitabine in lung cancer cells may be through regulation of TGFBI
protein.
In addition, Cul4A knockdown significantly
upregulated TGFBI protein levels and decreased its degradation,
whereas overexpression of Cul4A markedly enhanced TGFBI protein
ubiquitination and increased degradation of TGFBI protein. We also
observed that Cul4A regulated TGFBI protein stability through
direct interaction and ubiquitin mediated proteolysis. Thus, TGFBI
is a target of the Cul4A E3 ligase complex. Cul4A has been reported
to play a role in the ubiquitination and proteolysis of some
well-defined tumor suppressors, including p53 (7), NF2 (8), RASSF1A (9), p27 (19), DDB2 (20) and p21 (10,11).
To the best of our knowledge, the association of Cul4A and TGFBI
has not been previously reported.
Knockdown of Cul4A has been reported to increase
chemo-sensitivity to cisplatin in lung cancer cells (13). Overexpression of Cul4A was also
reported to participate in multiple drug resistance (MDR) in breast
cancer cells through upregulation of MDR1/P-gp expression at both
the transcription and protein levels, and knockdown of Cul4A
increased chemosensitivity to P-gp substrate drugs, paclitaxel,
vincristine and adriamycin in breast cancer cells (21). In the present study, we observed
that knockdown of Cul4A is related to chemosensitivity to
gemcitabine through upregulation of TGFBI in lung cancer cells for
the first time. TGFBI-mediated chemosensitivity to gemcitabine has
been reported to be related to proteolytic fragments derived from
TGFBI-induced cell death through binding to the αvβ3 integrin on
the surface of NSCLC cells and the subsequent activation of
caspase-8 and caspase-3/7 signaling and resultant apoptosis of
NSCLC cells (17). Gemcitabine is
one of the most widely used drugs for the treatment of NSCLC.
Cisplatin plus gemcitabine regimen has become a commonly used
combination for advanced NSCLC (22) and maintenance therapy (23). When used as a first-line
monotherapy, the objective response rate to gemcitabine is 16–22%
(24). Thus, developing novel
adjuvant chemotherapy in combination with gemcitabine chemotherapy
is urgent. Our study may provide the rationale to improve the
efficacy of gemcitabine chemotherapy as well as other chemotherapy
drugs in NSCLC in combination with anti-Cul4A targeted therapy in
the future.
Moreover, we also established an animal model to
demonstrate the therapeutic value of Cul4A RNAi. We observed that
tumor size was significantly decreased in the group of mice treated
with Cul4A RNAi and gemcitabine. To date, there is no
Cul4A-specific small compound reported. RNAi is a potential new
class of pharmaceutical drugs. RNAi-based therapeutics can offer a
powerful method for rapidly identifying specific and potent
inhibitors of disease targets from all molecular classes. The broad
potential application of RNAi therapeutics has been demonstrated by
numerous proof-of-concept studies in animal models of human
diseases (25). The use of
adenoviral vectors is considered to be a very powerful tool in
cancer gene therapy as they have been shown to transduce genes
efficiently into many types of cancer cells (26). In the present study, we designed
recombinant adenoviral vectors encoding siRNAs against Cul4A and
investigated their efficacy in regard to the suppression of Cul4A
expression in cancer cells and consequent antitumor potential.
Through in vivo studies, our results support the feasibility
of adenoviral-mediated transfer of Cul4A RNAi for treatment of
NSCLC and other cancers in the future.
In summary, the present study showed that knockdown
of Cul4A is associated with increased sensitivity to gemcitabine
through upregulation of TGFBI in NSCLC cells. Cul4A regulates TGFBI
through direct interaction and then ubiquitin-mediated protein
degradation. In nude mouse xeno-graft models, adenoviral-mediated
transfer of Cul4A RNAi in combination with gemcitabine chemotherapy
inhibited NSCLC tumor growth. Therefore, combination of Cul4A RNAi
with chemotherapy may provide a new approach to lung cancer
treatment.
Acknowledgments
The present study was supported by grants from the
Chang Gung Memorial Hospital (CMRPG6E0081, NMRPD1B1331 and
NMRPD1B1331) and the National Science Council, Taiwan
(101-2314-B-182-086-MY2). We would like to acknowledge the Leica
SP5II confocal microscope service provided by the Expensive
Advanced Instrument Core Laboratory, Department of Medical Research
and Development, Chang Gung Memorial Hospital at Chiayi.
References
|
1
|
Azzoli CG, Baker S Jr, Temin S, Pao W,
Aliff T, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, et
al American Society of Clinical Oncology: American Society of
Clinical Oncology Clinical Practice Guideline update on
chemotherapy for stage IV non-small-cell lung cancer. J Clin Oncol.
27:6251–6266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maemondo M, Inoue A, Kobayashi K, Sugawara
S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I,
et al North-East Japan Study Group: Gefitinib or chemotherapy for
non-small-cell lung cancer with mutated EGFR. N Engl J Med.
362:2380–2388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abba MC, Fabris VT, Hu Y, Kittrell FS, Cai
WW, Donehower LA, Sahin A, Medina D and Aldaz CM: Identification of
novel amplification gene targets in mouse and human breast cancer
at a syntenic cluster mapping to mouse ch8A1 and human ch13q34.
Cancer Res. 67:4104–4112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Melchor L, Saucedo-Cuevas LP, Muñoz-Repeto
I, Rodríguez-Pinilla SM, Honrado E, Campoverde A, Palacios J,
Nathanson KL, García MJ and Benítez J: Comprehensive
characterization of the DNA amplification at 13q34 in human breast
cancer reveals TFDP1 and CUL4A as likely candidate target genes.
Breast Cancer Res. 11:R862009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hung MS, Mao JH, Xu Z, Yang CT, Yu JS,
Harvard C, Lin YC, Bravo DT, Jablons DM and You L: Cul4A is an
oncogene in malignant pleural mesothelioma. J Cell Mol Med.
15:350–358. 2011. View Article : Google Scholar
|
|
6
|
Yasui K, Arii S, Zhao C, Imoto I, Ueda M,
Nagai H, Emi M and Inazawa J: TFDP1, CUL4A, and CDC16 identified as
targets for amplification at 13q34 in hepatocellular carcinomas.
Hepatology. 35:1476–1484. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nag A, Bagchi S and Raychaudhuri P: Cul4A
physically associates with MDM2 and participates in the proteolysis
of p53. Cancer Res. 64:8152–8155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang J and Chen J: VprBP targets Merlin
to the Roc1-Cul4A-DDB1 E3 ligase complex for degradation. Oncogene.
27:4056–4064. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiang X, Gu X, Liu Y, Ding F, Gu X, Huan
Y, Ren L and Wang Y: Molecular cloning and expression analysis of
evolutionarily conserved stathmin from Gekko japonicus spinal cord.
Indian J Biochem Biophys. 46:289–293. 2009.PubMed/NCBI
|
|
10
|
Nishitani H, Shiomi Y, Iida H, Michishita
M, Takami T and Tsurimoto T: CDK inhibitor p21 is degraded by a
PCNA coupled Cul4 DDB1Cdt2 pathway during S phase and after UV
irradiation. J Biol Chem. 43:29045–29052. 2008. View Article : Google Scholar
|
|
11
|
Li T, Hung MS, Wang Y, Mao JH, Tan JL,
Jahan K, Roos H, Xu Z, Jablons DM and You L: Transgenic mice for
cre-inducible overexpression of the Cul4A gene. Genesis.
49:134–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miranda-Carboni GA, Krum SA, Yee K, Nava
M, Deng QE, Pervin S, Collado-Hidalgo A, Galic Z, Zack JA, Nakayama
K, et al: A functional link between Wnt signaling and
SKP2-independent p27 turnover in mammary tumors. Genes Dev.
22:3121–3134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang YL, Hung MS, Wang Y, Ni J, Mao JH,
Hsieh D, Au A, Kumar A, Quigley D, Fang LT, et al: Lung
tumourigenesis in a conditional Cul4A transgenic mouse model. J
Pathol. 233:113–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim JE, Kim SJ, Lee BH, Park RW, Kim KS
and Kim IS: Identification of motifs for cell adhesion within the
repeated domains of transforming growth factor-beta-induced gene,
betaig-h3. J Biol Chem. 275:30907–30915. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhao Y, El-Gabry M and Hei TK: Loss of
Betaig-h3 protein is frequent in primary lung carcinoma and related
to tumorigenic phenotype in lung cancer cells. Mol Carcinog.
45:84–92. 2006. View
Article : Google Scholar
|
|
16
|
Zhao YL, Piao CQ and Hei TK:
Downregulation of Betaig-h3 gene is causally linked to tumorigenic
phenotype in asbestos treated immortalized human bronchial
epithelial cells. Oncogene. 21:7471–7477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Irigoyen M, Pajares MJ, Agorreta J,
Ponz-Sarvisé M, Salvo E, Lozano MD, Pío R, Gil-Bazo I and Rouzaut
A: TGFBI expression is associated with a better response to
chemotherapy in NSCLC. Mol Cancer. 9:1302010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang L, Rong R, Sheikh MS and Huang Y:
Cullin-4A DNA damage-binding protein 1 E3 ligase complex targets
tumor suppressor RASSF1A for degradation during mitosis. J Biol
Chem. 286:6971–6978. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bondar T, Kalinina A, Khair L, Kopanja D,
Nag A, Bagchi S and Raychaudhuri P: Cul4A and DDB1 associate with
Skp2 to target p27Kip1 for proteolysis involving the COP9
signalosome. Mol Cell Biol. 26:2531–2539. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
El-Mahdy MA, Zhu Q, Wang QE, Wani G,
Praetorius-Ibba M and Wani AA: Cullin 4A-mediated proteolysis of
DDB2 protein at DNA damage sites regulates in vivo lesion
recognition by XPC. J Biol Chem. 281:13404–13411. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Ma G, Wang Q, Wen M, Xu Y, He X,
Zhang P, Wang Y, Yang T, Zhan P, et al: Involvement of CUL4A in
regulation of multidrug resistance to P-gp substrate drugs in
breast cancer cells. Molecules. 19:159–176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Inal A, Kos FT, Algin E, Yildiz R,
Dikiltas M, Unek IT, Colak D, Elkiran ET, Helvaci K, Geredeli C, et
al: Gemcitabine alone versus combination of gemcitabine and
cisplatin for the treatment of patients with locally advanced
and/or metastatic pancreatic carcinoma: A retrospective analysis of
multicenter study. Neoplasma. 59:297–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brodowicz T, Krzakowski M, Zwitter M,
Tzekova V, Ramlau R, Ghilezan N, Ciuleanu T, Cucevic B, Gyurkovits
K, Ulsperger E, et al Central European Cooperative Oncology Group
CECOG: Cisplatin and gemcitabine first-line chemotherapy followed
by maintenance gemcitabine or best supportive care in advanced
non-small cell lung cancer: A phase III trial. Lung Cancer.
52:155–163. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ricci S, Antonuzzo A, Galli L, Tibaldi C,
Bertuccelli M, Lopes Pegna A, Petruzzelli S, Algeri R, Bonifazi V,
Fioretto ML, et al: Gemcitabine monotherapy in elderly patients
with advanced non-small cell lung cancer: A multicenter phase II
study. Lung Cancer. 27:75–80. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bumcrot D, Manoharan M, Koteliansky V and
Sah DW: RNAi therapeutics: A potential new class of pharmaceutical
drugs. Nat Chem Biol. 2:711–719. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Uchida H, Tanaka T, Sasaki K, Kato K,
Dehari H, Ito Y, Kobune M, Miyagishi M, Taira K, Tahara H, et al:
Adenovirus-mediated transfer of siRNA against survivin induced
apoptosis and attenuated tumor cell growth in vitro and in vivo.
Mol Ther. 10:162–171. 2004. View Article : Google Scholar : PubMed/NCBI
|