Introduction
Oral squamous cell carcinoma (OSCC) is one of the
most common malignancies in the head and neck region. OSCC is
derived from oral mucosal epithelium and is characterized by its
strong local infiltration and cervical lymph node metastasis. Many
factors, including smoking, can lead to the tumorigenesis and
development of OSCC, which has a poor prognosis and low overall
survival rate (1,2). OSCC not only affects the lives of
late-stage patients but also affects their ability to chew and
swallow and their appearance. Although surgery, radiotherapy and
chemotherapy have made considerable progress in terms of treatment,
the prognosis of OSCC patients remains poor. Early detection and
radical treatment of tumors both present challenges and
opportunities for cancer researchers (3). The solution to this issue depends on a
comprehensive understanding of the molecular mechanisms of
tumorigenesis and development. Many research groups are actively
involved in the study of OSCC pathogenesis; however, the underlying
mechanisms of OSCC tumorigenesis and development have not been
fully elucidated (4–6). Therefore, further studies focusing on
the mechanisms of OSCC need to be performed to improve early
diagnosis, targeted therapy and prognosis.
Long non-coding RNAs (lncRNAs) are a group of
non-protein coding RNAs >200 nt (7). It is estimated that only 2% of the
human genome is transcribed into mRNAs, while 70–90% of the genome
is transcribed into lncRNAs (7).
lncRNAs play important roles in epigenetic modification,
transcription and post-transcriptional regulation, maintenance of
normal tissue development and differentiation (3,4).
Recently, increasing evidence has indicated that lncRNAs exert
vital roles in a number of biological processes, including cell
metabolism and immune response, through comprehensive mechanisms
(8). lncRNAs that affect
tumorigenesis and development are considered novel candidates for
targeted tumor therapy (9).
According to previous research, lncRNAs are closely correlated with
tumorigenesis and development of esophageal, liver, lung and breast
cancer (10–13). lncRNAs affect the expression of mRNA
by regulating the transcription and stability of their target genes
(14). Delineating the lncRNA-mRNA
coexpression network is an important method for analyzing the
functional and regulatory mechanisms of lncRNAs.
The aim of the present study was to identify
dysregulated lncRNAs and mRNAs in OSCC patients. The results of the
present study indicated that abnormal expression of lncRNAs may
contribute to the tumorigenesis and development of OSCC.
Furthermore, the present study provides new insight into the
molecular markers and therapeutic targets for OSCC.
Materials and methods
Samples
Seventy-two oral squamous cell carcinoma (OSCC)
tissues and paired adjacent normal tissues (excised 2 cm from the
tumor-free margin) were obtained from the Fourth Affiliated
Hospital of Hebei Medical University between January 2015 and
October 2016 and were pathologically confirmed to be OSCC (32 were
<50 and 40 were ≥50 years; 35 male and 37 female patients). The
present study was approved by the Ethics Committee of the Fourth
Affiliated Hospital of Hebei Medical University (Shijiazhuang,
China) and written informed consent was obtained from all subjects.
None of the patients received radiotherapy, chemotherapy or other
cancer treatment before tumor resection. Tumors were histologically
graded according to the World Health Organization (WHO) standards.
Classification of tumors was performed according to the TNM staging
revised by the International Union Against Cancer (UICC)
(https://www.uicc.org/).
All 72 patients were followed up by telephone and
outpatient methods after discharge with assessments of their
general condition and clinical symptoms and imaging examinations.
The starting point of follow-up was the date of surgery or
pathological biopsy, and the follow-up period ended on April 30,
2018. At the end of the follow-up period, 49 patients were still
alive, and 23 had died; and no patient was lost to follow-up.
Progression-free survival (PFS) was defined as the time between
diagnosis and progression of the disease, and overall survival (OS)
was defined as the time from diagnosis to death or last
follow-up.
Microarray assay
Of these samples, three tissue pairs including
tongue cancer (T1N2M0), gingival carcinoma (T2N0M0) and carcinoma
of the buccal mucosa (T3N1M0) were used for microarray analysis.
The microarray (SBC human 4*180K lncRNA array; Shanghai
Biotechnology Corp., Shanghai, China) used in the present study was
capable of detecting 77,103 lncRNAs and 18,853 mRNAs and covered
core databases, such as GENCODE v21 (https://www.gencodegenes.org), Lncipedia v3.1
(https://lncipedia.org), Ensembl (http://asia.ensembl.org) and Agilent_ncRNA (https://earray.chem.agilent.com).
Total RNA extraction
Total RNA was extracted from frozen samples by
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Quantitation was preformed using a NanoDrop 2000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.). Denaturing agarose gel electrophoresis was used to assess
the integrity of total RNA extracted from tissues.
cDNA synthesis, labeling and
hybridization
Qualified total RNA was used for the synthesis of
cDNA followed by fluorescent labeling according to the
manufacturer's instructions with the Agilent's Low Input Quick Amp
WT Labeling kit (Agilent Technologies, Santa Clara, CA, USA). The
labeled cRNA was purified with RNeasy Mini kit (Qiagen GmbH,
Hilden, Germany) and hybridization was performed at 65°C for 17
h.
Microarray data analysis
An Agilent Microarray Scanner (Agilent Technologies)
was used in the present study. Data were obtained using Feature
Extraction software 10.7 (Agilent Technologies). Raw data were
normalized by Quantile algorithm, Gene Spring Software 11.0
(Agilent Technologies). The lncRNAs and mRNAs were considered to be
differentially expressed when the fold change (FC) was >2
(P<0.05). A volcano plot was used to visualize differentially
expressed genes and was subsequently processed for hierarchical
clustering analysis using Gene Spring Software 11.0 (Agilent
Technologies). Finally, Pearson correlation coefficients between
differentially expressed lncRNAs and mRNAs were calculated, and
co-expression networks of lncRNAs and mRNAs were constructed.
Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway analysis
GO terms were used to annotate and classify gene
function. The differentially expressed genes were put into the
Database for Annotation, Visualization and Integrated Discovery
(DAVID; http://david.abcc.ncifcrf.gov/) v6.8, which utilizes
GO to identify the molecular function represented in the gene
profile. Furthermore, we used KEGG to analyze the potential
functions of these genes in metabolic pathways. P<0.05 was
recommended as a cut-off value.
Prediction of lncRNA target genes
Two independent algorithms were used to predict the
target genes of dysregulated lncRNAs. The first algorithm was
performed to predict cis-acting target genes using the
University of California Santa Cruz (UCSC; Santa Cruz, CA, USA)
genome browser (http://genome.ucsc.edu/). Genes transcribed within a
10-kb window upstream or downstream of lncRNAs were considered
cis target genes. The second algorithm predicted
trans-acting target genes using RNAplex 0.2 software
(http://www.bioinf.uni-leipzig.de/Software/RNAplex)
based on RNA duplex energy prediction and mRNA sequence
complementation according to the previous reference (15).
qRT-PCR
Differentially expressed lncRNAs from microarray
data were randomly selected for qRT-PCR. Reverse transcription of
total RNA was performed using PrimeScript RT reagent kit with gDNA
Eraser (Takara Biotechnology Co., Ltd., Dalian, China). SYBR-Green
qPCR Master Mix (Takara Biotechnology Co., Ltd.) was used for
qRT-PCR assay according to the manufacturer's protocol. The
thermocycling conditions were as follows: A denaturation step 10
min at 95°C, followed by 40 cycles of 15 sec at 95°C and 30 sec at
60°C (adjusted with the Tm of different lncRNAs), 30 sec at 72°C.
The housekeeping gene GAPDH was selected as an internal control.
The 2−ΔΔCq method was used to measure relative
expression levels, and each sample was analyzed at least in
triplicate (16). Specific primers
of each gene are listed in Table
I.
 | Table I.Primers used for qRT-PCR. |
Table I.
Primers used for qRT-PCR.
| lncRNA | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| lnc-MANSC4-8:1 |
AAGGAAAACAACAGAAGAACAC |
GCCAGCTTAAAGAGACAAATA |
| CXCR2P1 |
AGGGGAGTATGGGGAGTGATG |
GGGCCAAGGTGTTTTCTTTTTA |
| NRIR |
CCAAGAAAAGAGGGCTTAAAATGAA |
AAGGAGGTTAGAGGTGTCTGCTGC |
| lnc-CMPK2-1:3 |
TCAATAGAGAGGCAGACATACACA |
ACAAGAAACACAGCACTAACAACA |
| lnc-GLI3-4:1 |
GATGTGGTGGGTTCTCCAGTGTGA |
TTTCCATCTTGCCTTCATTGTTTT |
| NR_104048 |
AGTTTCCTTTTCATTGTTTTTTGC |
GATCCTGTTTGCTACTGCCAGA |
| MEG3 |
CTTTTCTGGGGGAATGGGG |
AGAGGGGTGGGAAGGGACT |
| lnc-WRN-10:1 |
ACATCAAGCTGTAACCAACCCAAC |
TGCCTCTTCATCCACACTACCAAA |
|
ENST00000583044 |
AAATAACCCTATCAATCACCAAG |
AGAGGAGAGAGATCAGGAAACC |
|
ENST00000527317 |
ACCAGAATGAGGTAAAAGAAGA |
TGAGAGTGTGTGAGAACAAAG |
| GAPDH |
ATCTTCCAGGAGCGAGATCCC |
TGAGTCCTTCCACGATACCAA |
Statistical analysis
Statistical analysis was performed using SPSS
statistical software package (version 22.0; IBM Corp., Armonk NY,
USA). Data are expressed as the mean ± standard deviation (SD).
Student's t-test was used to compare different groups. The
correlation between the lncRNA expression levels and the
clinicopathological factors was analyzed using the Chi-square
tests. Survival plots were generated by Kaplan-Meier analysis, and
the log-rank test was used to assess the significance of the
differences. P-values <0.05 were considered to indicate a
statistically significant result.
Results
Patient information
The clinical characteristics of the 72 patients are
summarized in Table II.
| Table II.Relationship between the expression
level of 4 critical node lncRNAs with the pathological
characteristics of the OSCC patients (N=72). |
Table II.
Relationship between the expression
level of 4 critical node lncRNAs with the pathological
characteristics of the OSCC patients (N=72).
|
|
|
ENST00000583044 | NR_104048 | lnc-WRN-10:1 |
ENST00000527317 |
|---|
|
|
|
|
|
|
|
|---|
| Clinical
characteristics | N 72 | High | Low | High | Low | High | Low | High | Low |
|---|
| Age (years) |
|
<50 | 32 | 14 | 18 | 15 | 17 | 14 | 18 | 15 | 17 |
|
≥50 | 40 | 16 | 24 | 13 | 27 | 12 | 28 | 15 | 25 |
|
P-value |
| 0.748 | 0.214 | 0.227 | 0.423 |
| Sex |
|
Male | 35 | 16 | 19 | 16 | 19 | 15 | 20 | 16 | 19 |
|
Female | 37 | 14 | 23 | 12 | 25 | 11 | 26 | 14 | 23 |
|
P-value |
| 0.498 | 0.248 | 0.246 | 0.498 |
| Smoking |
|
Yes | 42 | 15 | 27 | 16 | 26 | 14 | 28 | 18 | 24 |
| No | 30 | 15 | 15 | 12 | 18 | 12 | 18 | 12 | 18 |
|
P-value |
| 0.225 | 0.870 | 0.561 | 0.808 |
| Tumor location |
| Tongue
cancer | 30 | 13 | 17 | 12 | 18 | 11 | 19 | 16 | 14 |
|
Gingival carcinoma | 22 | 7 | 15 | 8 | 14 | 7 | 15 | 5 | 17 |
|
Carcinoma of the buccal
mucosa | 10 | 5 | 5 | 5 | 5 | 3 | 7 | 4 | 6 |
|
Others | 10 | 5 | 5 | 3 | 7 | 5 | 5 | 5 | 5 |
|
P-value |
| 0.686 | 0.819 | 0.758 | 0.156 |
| Clinical stage |
| T1 | 28 | 20 | 8 | 20 | 8 | 17 | 11 | 18 | 10 |
| T2 | 25 | 4 | 21 | 3 | 22 | 5 | 20 | 6 | 19 |
| T3 | 15 | 5 | 10 | 4 | 11 | 3 | 12 | 5 | 10 |
| T4 | 4 | 1 | 3 | 1 | 3 | 1 | 3 | 1 | 3 |
|
P-value |
| <0.001 | <0.001 | 0.007 | 0.019 |
| Lymphatic
metastasis |
|
Yes | 36 | 10 | 26 | 9 | 27 | 7 | 29 | 9 | 27 |
| No | 36 | 20 | 16 | 19 | 17 | 19 | 17 | 21 | 15 |
|
P-value |
| 0.017 | 0.016 | 0.003 | 0.004 |
| Distant
metastasis |
|
Yes | 18 | 3 | 15 | 3 | 15 | 3 | 15 | 2 | 16 |
| No | 54 | 27 | 27 | 25 | 29 | 23 | 31 | 28 | 26 |
|
P-value |
| 0.013 | 0.026 | 0.047 | 0.002 |
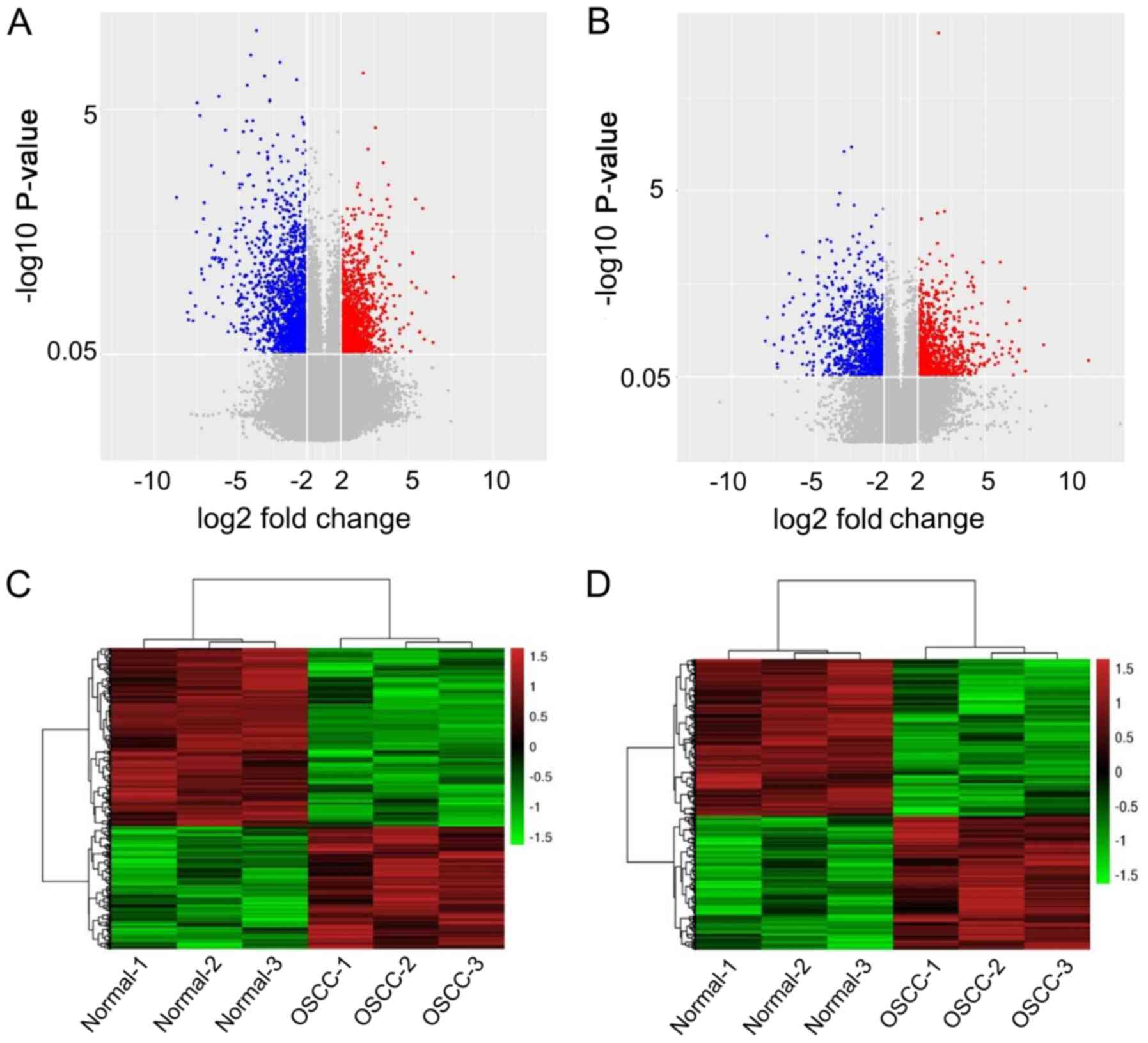
Volcano plot and hierarchical
clustering analysis
In the volcano plot, after normalization and
standardization, different signals were distributed in
corresponding regions. Gray signals indicated that the detected
genes did not meet the screening criteria. The results indicated
that there were many lncRNAs and mRNAs that were differentially
expressed between OSCC and normal tissues (Fig. 1A and B). Hierarchical clustering
analysis determined correlations among samples through grouping at
the gene level. In the hierarchical clustering analysis, each
column represented one sample, and each row represented one gene.
Hierarchical clustering analysis showed that the expression
profiles were significantly different between OSCC and paired
adjacent normal tissues (Fig. 1C and
D).
Differentially expressed lncRNAs and
mRNAs
To explore the role of lncRNAs in OSCC, we performed
a genome-wide analysis of lncRNA and mRNA expression in OSCC and
normal tissues. The results of the microarray assays showed that
2,294 differentially expressed lncRNAs (accounting for 2.9% of all
detectable lncRNAs) and 1,938 differentially expressed mRNAs
(accounting for 10.3% of all detectable mRNAs) were identified.
Furthermore, 933 lncRNAs and 891 mRNAs were upregulated and 1,361
lncRNAs and 1,047 mRNAs were downregulated. The most upregulated
lncRNA and mRNA were MANSC4-8:1 (FC=201.36) and MMP7 (FC=2167.59),
respectively. The most downregulated lncRNA and mRNA were NR_117092
(FC=418.62) and IL36A (FC=257.61), respectively. The top 20
dysregulated lncRNAs and mRNAs are summarized in Tables III and IV, respectively.
| Table III.Top 20 differentially expressed
lncRNAs between OSCC and paired adjacent normal tissues. |
Table III.
Top 20 differentially expressed
lncRNAs between OSCC and paired adjacent normal tissues.
|
| Upregulated
lncRNAs | Downregulated
lncRNAs |
|---|
|
|
|
|
|---|
| lncRNA | Source | Fold change | P-value | lncRNA | Source | Fold change | P-value |
|---|
| lnc-MANSC4-8:1 | Lncipedia | 201.357 | 0.003 | NR_117092 | RefSeq | 418.623 | 0.001 |
|
ENST00000520185 |
ENSEMBL_GENCODE | 86.973 | 0.033 | lnc-SPRR1B-1:1 | lncipedia | 266.212 | 0.012 |
| lnc-CXCR3-5:2 | Lncipedia | 64.771 | 0.006 |
ENST00000392385 |
ENSEMBL_GENCODE | 256.681 | 0.015 |
| NR_002712 | RefSeq | 61.104 | 0.030 |
lnc-TMPRSS11BNL-1:3 | lncipedia | 237.226 | 0.006 |
| lnc-IFI44-6:1 | Lncipedia | 57.542 | 0.001 | lnc-ANKS1A-1:1 | lncipedia | 212.475 | 0.016 |
| lnc-MANSC4-8:1 | Lncipedia | 51.022 | 0.023 |
ENST00000555864 |
ENSEMBL_GENCODE | 194.029 | 0.009 |
|
ENST00000522970 |
ENSEMBL_GENCODE | 50.388 | 0.023 | lnc-TPP2-7:2 | lncipedia | 185.490 | 0.001 |
| lnc-IFI44-5:1 | Lncipedia | 43.841 | 0.005 | NR_104048 | RefSeq | 180.413 | 8.02E-06 |
|
ENST00000562027 |
ENSEMBL_GENCODE | 42.530 | 0.001 | lnc-SPRR1A-2:1 | lncipedia | 160.518 | 1.26E-05 |
|
ENST00000455557 |
ENSEMBL_GENCODE | 38.310 | 0.001 |
ENST00000504297 |
ENSEMBL_GENCODE | 150.608 | 0.003 |
| lnc-GLI3-4:1 | Lncipedia | 37.568 | 0.001 | lnc-PROM2-1:2 | lncipedia | 150.001 | 0.003 |
|
ENST00000597169 |
ENSEMBL_GENCODE | 36.129 | 0.004 |
lnc-TMPRSS11BNL-1:2 | lncipedia | 138.905 | 0.001 |
| lnc-MSRB3-5:1 | Lncipedia | 31.778 | 0.018 | lnc-TPP2-7:2 | lncipedia | 135.176 | 0.004 |
|
ENST00000433410 |
ENSEMBL_GENCODE | 26.210 | 0.020 |
ENST00000614187 |
ENSEMBL_GENCODE | 134.054 | 0.014 |
|
ENST00000422194 |
ENSEMBL_GENCODE | 24.990 | 0.039 | lnc-KRT79-1:1 | lncipedia | 125.012 | 0.012 |
| NR_038369 | RefSeq | 22.397 | 0.021 | lnc-KRT79-2:1 | lncipedia | 105.665 | 0.002 |
| NR_110849 | RefSeq | 22.305 | 0.002 | NR_130720 | RefSeq | 104.857 | 0.022 |
|
ENST00000560994 |
ENSEMBL_GENCODE | 22.167 | 0.024 |
ENST00000516032 |
ENSEMBL_GENCODE | 101.046 | 7.09E-05 |
| NR_126359 | RefSeq | 18.934 | 0.015 |
ENST00000450445 |
ENSEMBL_GENCODE | 97.170 | 0.002 |
|
ENST00000365096 |
ENSEMBL_GENCODE | 18.736 | 0.048 | NR_104048 | RefSeq | 92.853 | 0.014 |
| Table IV.Top 20 differentially expressed mRNAs
between OSCC and paired adjacent normal tissues. |
Table IV.
Top 20 differentially expressed mRNAs
between OSCC and paired adjacent normal tissues.
|
| Upregulated
mRNAs | Downregulated
mRNAs |
|---|
|
|
|
|
|---|
| mRNA | Fold change | P-value | mRNA | Fold change | P-value |
|---|
| MMP7 | 2167.591 | 0.024 | IL36A | 257.616 | 0.010 |
| MMP10 | 344.744 | 0.012 | TMPRSS11B | 242.126 | 7.91E-05 |
| MMP7 | 163.871 | 0.038 | MAL | 237.820 | 0.003 |
| MMP13 | 161.637 | 0.001 | CRNN | 180.673 | 0.011 |
| PTHLH | 129.457 | 0.004 | SPINK7 | 159.889 | 0.027 |
| CSAG3 | 125.283 | 0.014 | KRT4 | 157.082 | 0.033 |
| CSAG3 | 124.324 | 0.015 | KRT13 | 129.559 | 0.015 |
| SLCO1B3 | 114.167 | 0.025 | SPINK7 | 127.208 | 0.009 |
| MMP12 | 99.936 | 0.046 | SPINK7 | 125.286 | 0.008 |
| MMP1 | 97.071 | 0.020 | FAM3B | 124.222 | 0.003 |
| IFIT2 | 94.992 | 0.010 | FAM3D | 120.592 | 0.002 |
| HOXD11 | 87.675 | 0.014 | TMPRSS11A | 97.086 | 0.001 |
| NLRP7 | 84.707 | 0.017 | KRT78 | 94.508 | 0.002 |
| CXCL11 | 81.590 | 0.002 | KRT13 | 86.058 | 0.016 |
| DNAH17 | 75.520 | 0.004 | ADH7 | 83.671 | 0.001 |
| PTHLH | 63.483 | 0.013 | CLCA4 | 81.754 | 0.008 |
| MMP1 | 62.481 | 0.018 | RHCG | 73.888 | 0.010 |
| INHBA | 58.435 | 0.003 | SERPINB11 | 63.682 | 0.006 |
| MMP11 | 54.858 | 0.009 | UPK1A | 62.443 | 0.001 |
| MMP3 | 53.994 | 0.026 | ADH7 | 57.245 | 0.005 |
Construction of the lncRNA-mRNA
co-expression network
Through construction of a co-expression network, we
identified 306 differentially expressed lncRNAs interacting with
other selected mRNAs and lncRNAs. According to the results,
ENST00000583044, NR_104048, lnc-WRN-10:1 and ENST00000527317 were
the four lncRNAs with the most frequent interactions (Fig. 2A and D). These four lncRNAs were
node genes for the entire network, with relationship coefficients
of 22, 22, 21 and 20. Other genes that did not directly interact
with these four node genes interacted with them via indirect means
through other relevant genes.
GO and KEGG pathway analyses
The differentially expressed mRNAs were processed
for GO annotation. The results showed that the differentially
expressed genes were enriched in molecular function (MF),
biological process (BP) and cellular component (CC). Furthermore,
three functions with the most enriched genes were interleukin-1
binding (GO:0019966; Ontology: molecular function; P=0.0003),
response to interferon-α (GO:0035455; Ontology: Biological process;
P=1.37E-10) and FHF complex (GO:0070695; Ontology: Cellular
component; P=0.0035).
The results of the KEGG analysis demonstrated that
differentially expressed mRNAs were mainly enriched in 38
biological pathways, including many cancer-related metabolic
pathways, e.g., ‘pathways in cancer’ (enriched with 36
differentially expressed genes), ‘bladder cancer’ (enriched with 8
differentially expressed genes), ‘metabolic pathways’ (enriched
with 109 differentially expressed genes), ‘pancreatic cancer’
(enriched with 11 differentially expressed genes) and ‘PPAR
signaling pathway’ (enriched with 11 differentially expressed
genes). The top 10 enriched GO and KEGG terms are summarized in the
Tables V and VI.
| Table V.Top 10 of GO enrichment in different
mRNAs. |
Table V.
Top 10 of GO enrichment in different
mRNAs.
| Description | Enrich factor | P-value |
|---|
| Interleukin-1
binding | 9.676 | 0.002 |
| Response to
interferon-α | 9.676 | 1.37E-10 |
| Establishment of
epithelial cell apical/basal polarity | 9.676 | 0.001 |
| Adenylate
cyclase-inhibiting dopamine receptor signaling pathway | 8.708 | 0.003 |
| Regulation of
synaptic vesicle priming | 8.708 | 0.003 |
| Oxidoreductase
activity | 8.708 | 0.003 |
| PML body
organization | 8.708 | 0.003 |
| Positive regulation
of gonadotropin secretion | 8.708 | 0.003 |
| Negative regulation
of viral-induced cytoplasmic pattern recognition receptor signaling
pathway | 8.708 | 0.003 |
| Negative regulation
of interferon-γ biosynthetic process | 8.708 | 0.003 |
| Table VI.Top 10 of pathway enrichment in
different mRNAs. |
Table VI.
Top 10 of pathway enrichment in
different mRNAs.
| Description | Enrich factor | P-value |
|---|
| Steroid
biosynthesis | 5.383 | 4.60E-05 |
| Glycosaminoglycan
biosynthesis | 5.230 | 2.00E-05 |
| Synthesis and
degradation of ketone bodies | 4.262 | 0.028 |
| Biosynthesis of
unsaturated fatty acids | 4.262 | 0.001 |
| Terpenoid backbone
biosynthesis | 3.409 | 0.025 |
| Arginine and
proline metabolism | 3.315 | 4.62E-05 |
| α-Linolenic acid
metabolism | 3.196 | 0.017 |
| Thyroid cancer | 3.086 | 0.006 |
| ECM-receptor
interaction | 2.858 | 2.14E-05 |
| Tryptophan
metabolism | 2.740 | 0.004 |
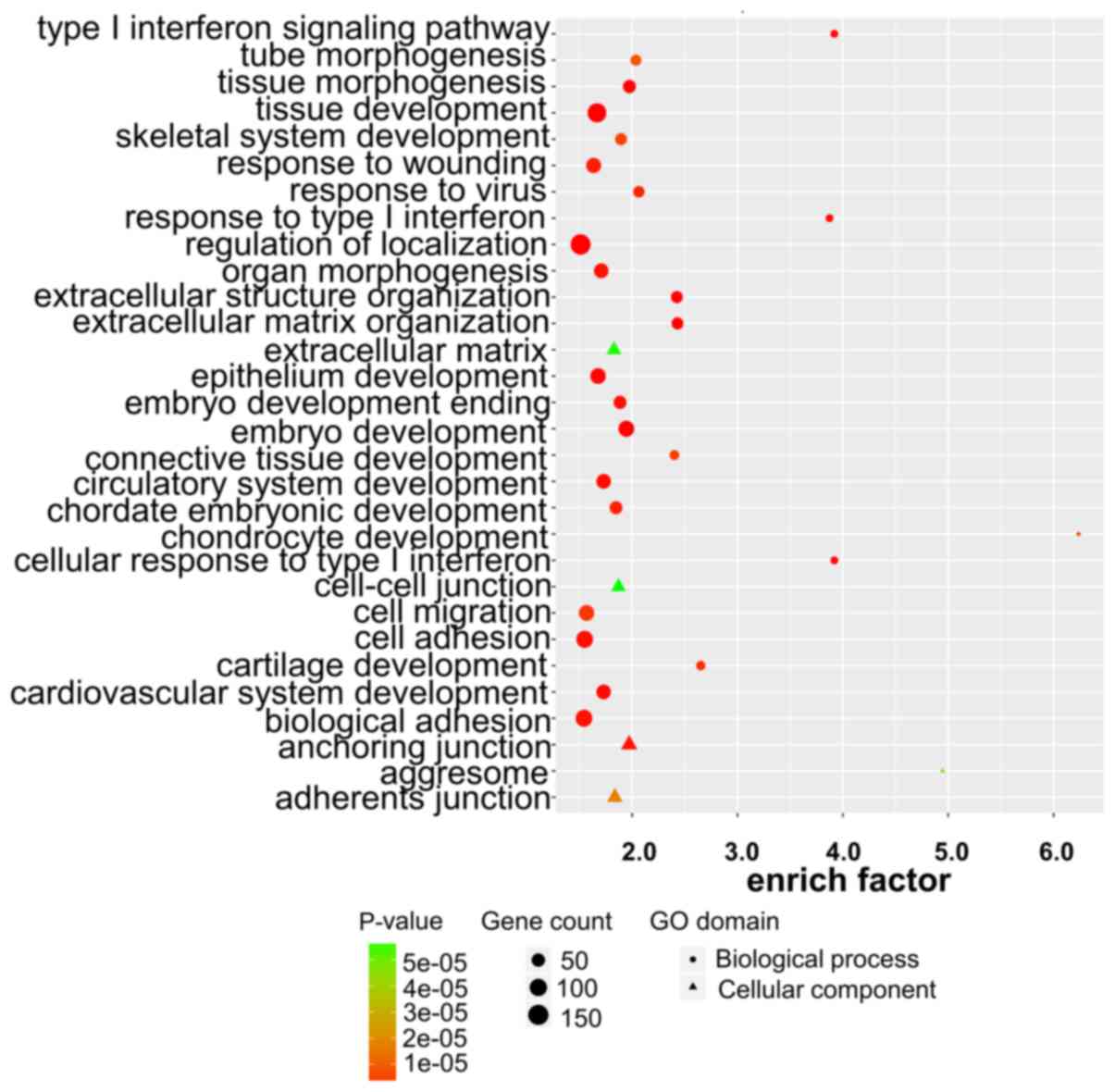
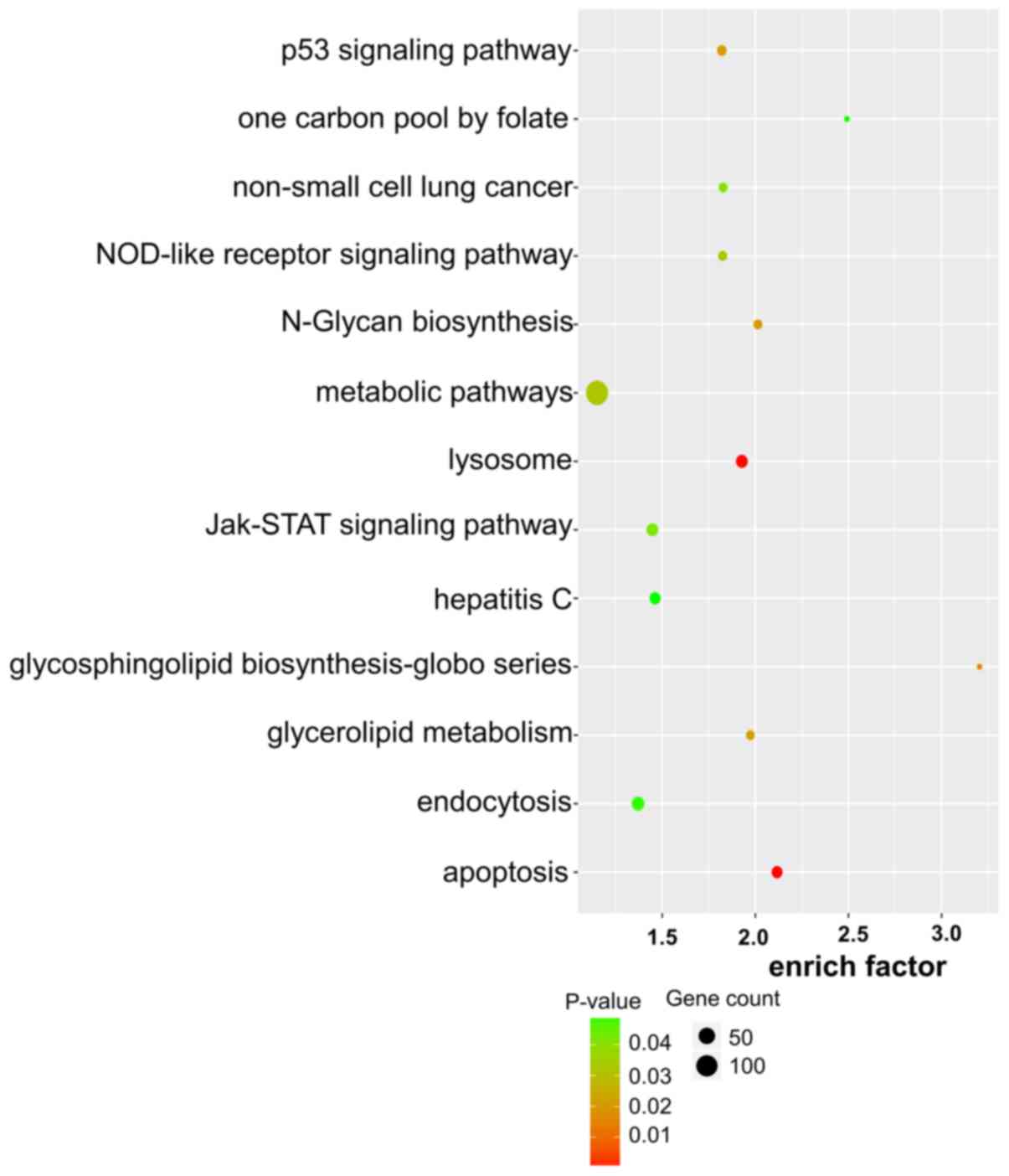
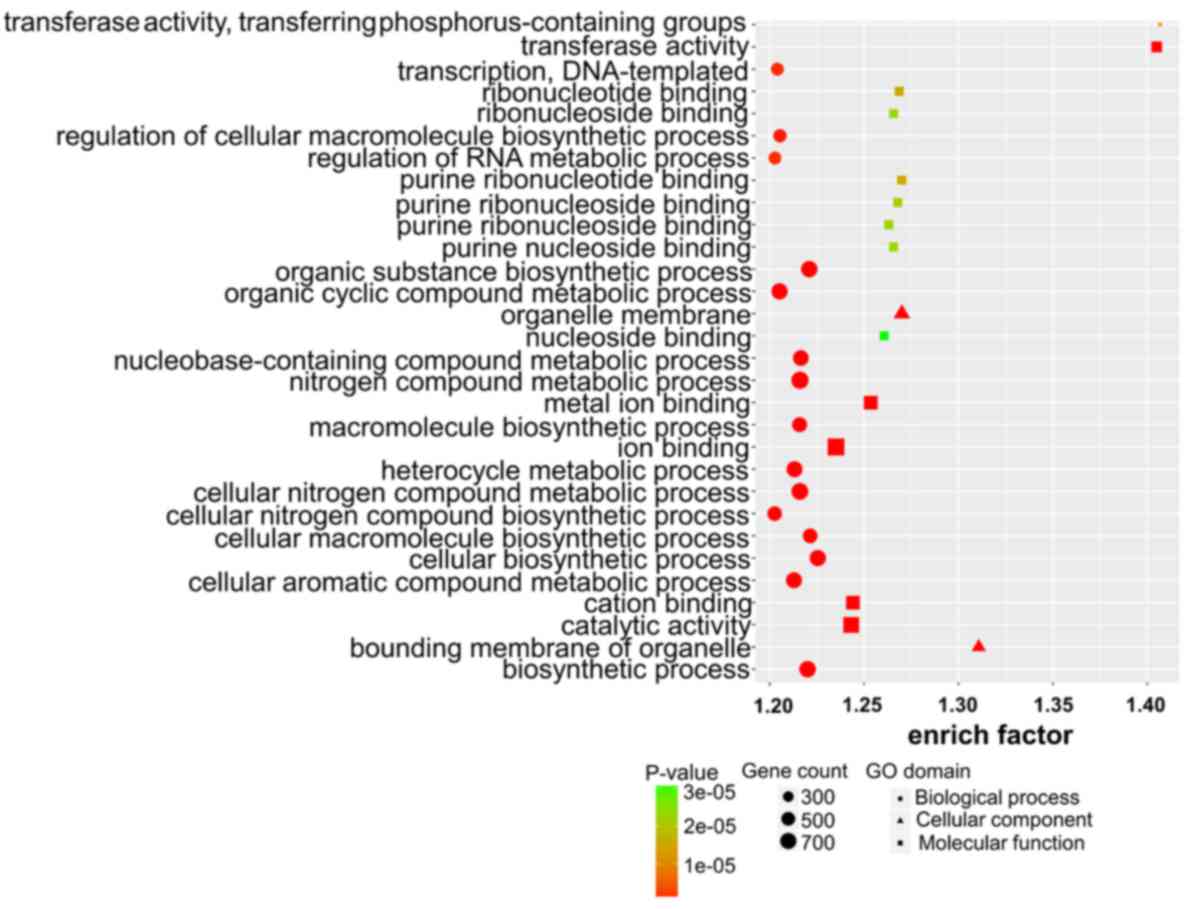
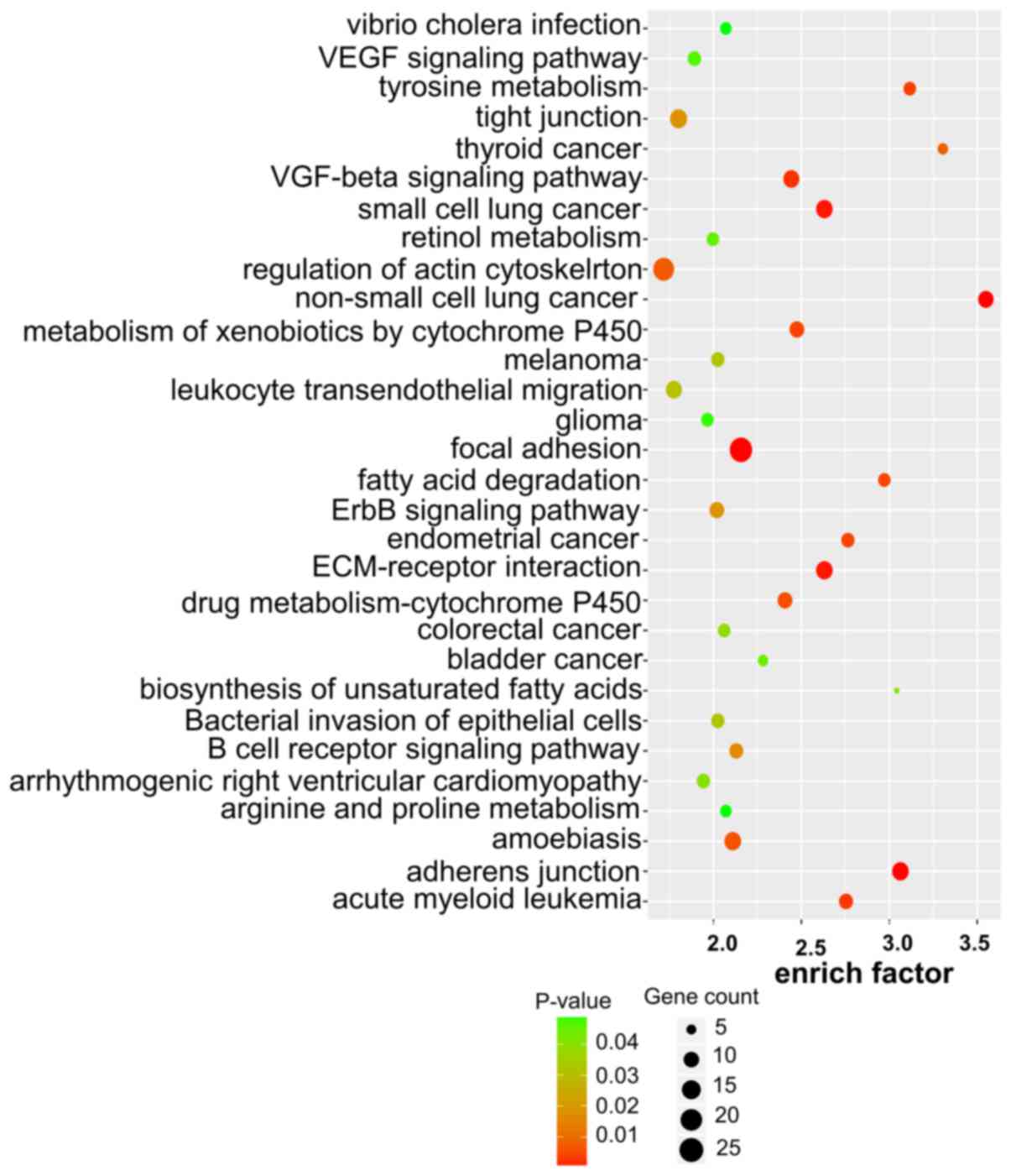
Target gene prediction, GO analysis
and KEGG pathway enrichment of lncRNAs
To explore the role of dysregulated lncRNAs in
OSCC-related gene regulation and metabolic pathways, the target
genes of lncRNAs were predicted using two independent algorithms.
The results revealed that 1,470 dysregulated lncRNAs were
identified to have cis or trans target genes,
including 1,356 lncRNAs targeting 1,250 cis-genes, 370
lncRNAs targeting 2,454 trans-genes and 256 lncRNAs
targeting both cis and trans target genes. We listed
the top 30 terms of GO and KEGG enrichment using cis and
trans methods (Figs.
3–6).
In the GO analysis, target genes with MF were mainly
enriched in molecular binding, e.g., ‘anion binding’ and ‘ion
binding’. Target genes with CC functions were mainly enriched in
cell organ and cell membrane, e.g., ‘aggresome’ and ‘organelle
membrane’. Target genes with the function of BP were mainly
enriched in the synthesis process, e.g., ‘cellular biosynthetic
process’ and ‘organic substance biosynthetic processs’ (Figs. 3 and 4).
In the KEGG pathway enrichment, the target genes
were enriched in 43 pathways. Among them, the cancer pathways were
the main pathways. Target genes were also involved in other
pathways, e.g., ‘biosynthesis’, ‘metabolism’ and ‘signal pathway’
(Figs. 5 and 6). These enriched metabolic pathways may
be the key pathways involved in the regulation of tumorigenesis and
development of OSCC by lncRNAs.
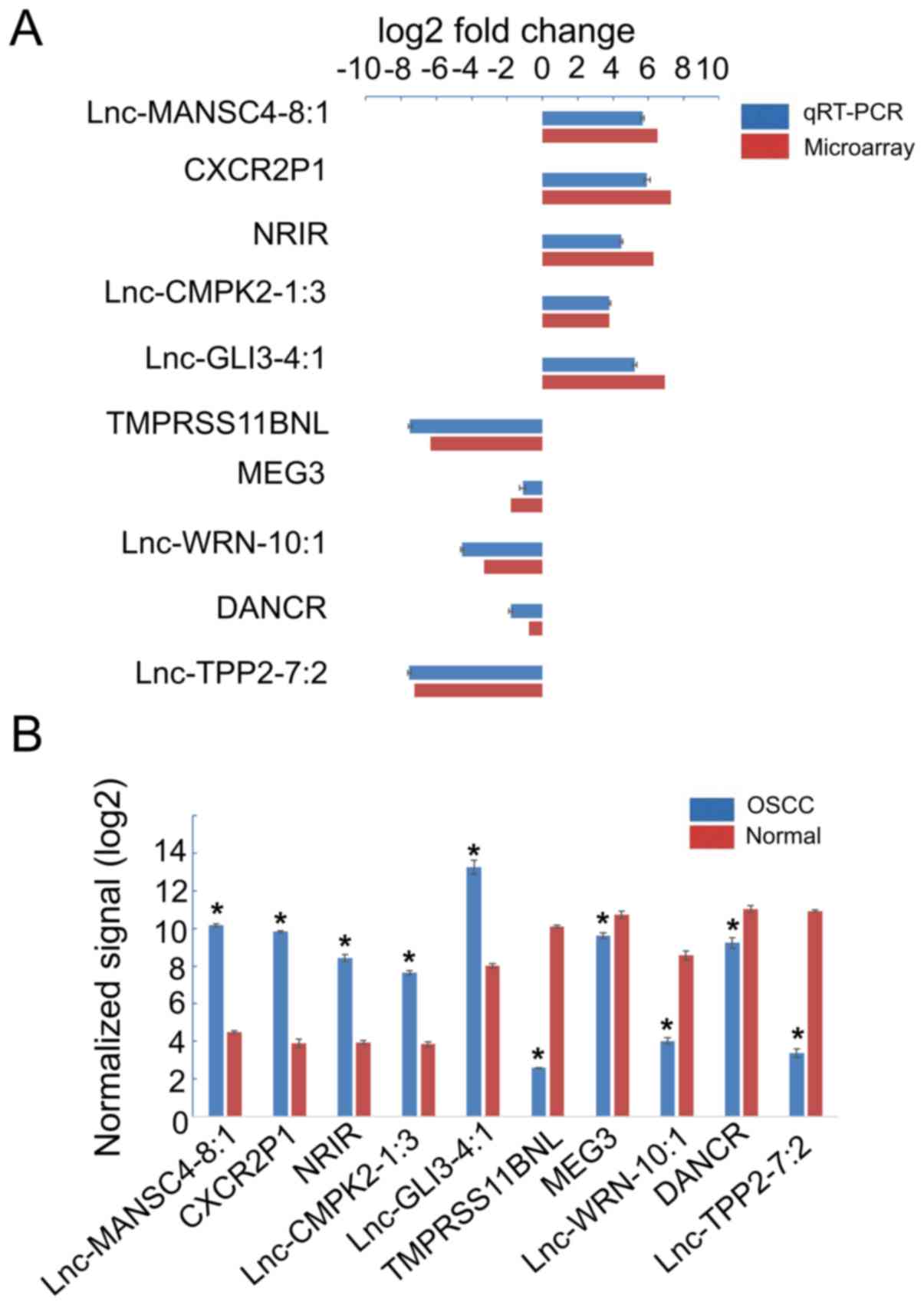
Validation by qRT-PCR
To verify the results of the microarray, 10
differentially expressed lncRNAs were randomly selected and
assessed by qRT-PCR in 72 patient tissues. The data indicated that
lnc-MANSC4-8:1, CXCR2P1, NRIR, lnc-CMPK2-1:3 and lnc-GLI3-4:1 were
significantly upregulated, while TMPRSS11BNL, MEG3, lnc-WRN-10:1,
DANCR and lnc-TPP2-7:2 were significantly downregulated in OSCC
(P<0.05, Fig. 7). The trend of
dysregulated lncRNAs detected by qRT-PCR was consistent with those
of the microarray assay.
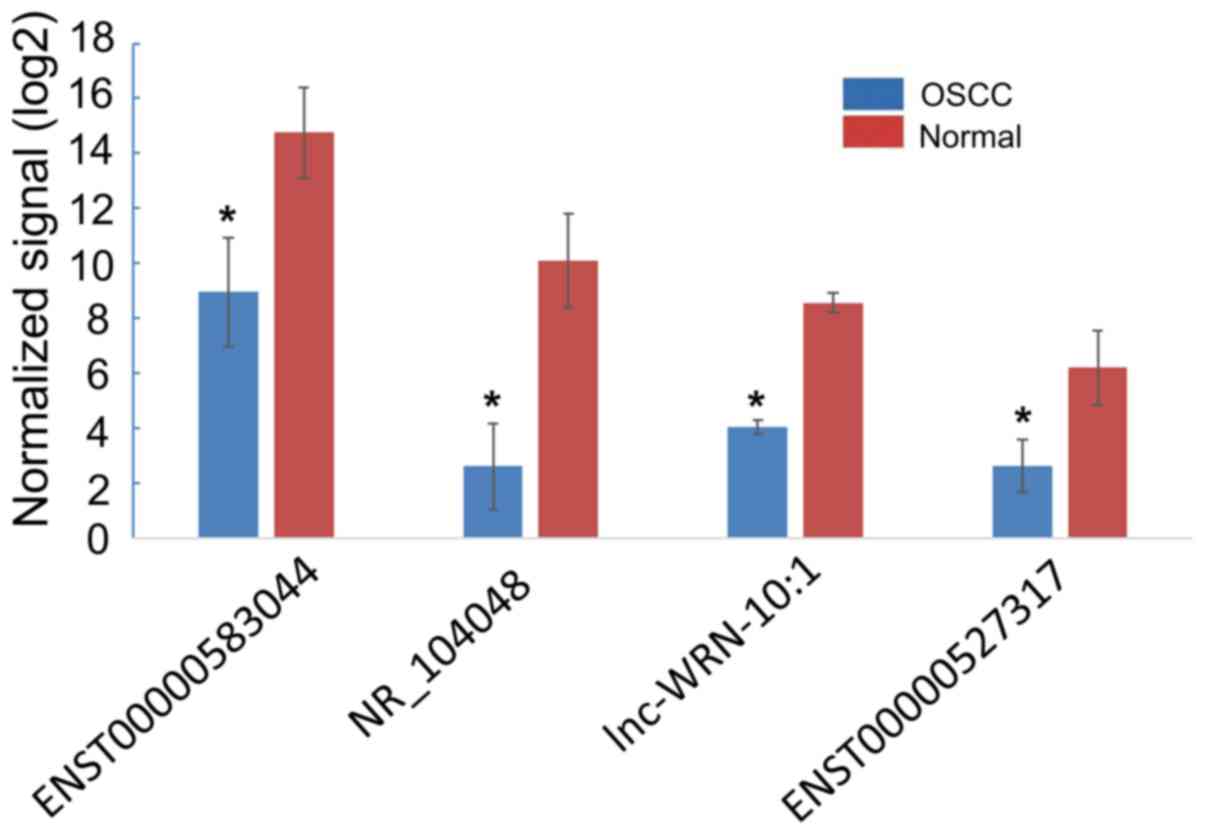
Relationship between the expression of
ENST00000583044, NR_104048, lnc-WRN-10:1, ENST00000527317 and the
clinicopathological features of OSCC patients
In 72 cases of OSCC, the expression levels of
ENST00000583044, NR_104048, lnc-WRN-10:1 and ENST00000527317 were
significantly lower than those in normal tissues (P<0.05)
(Fig. 8). According to the average
expression levels of ENST00000583044, NR_104048, lnc-WRN-10:1 and
ENST00000527317 in OSCC, patients with OSCC were divided into a
high ENST00000583044 expression group (≥3.15) (n=30) and a low
ENST00000583044 expression group (<3.15) (n=42); a high
NR_104048 expression group (≥2.99) (n=28) and a low NR_104048
expression group (<2.99) (n=44); a high lnc-WRN-10:1 expression
group (≥3.35) (n=26) and a low lnc-WRN-10:1 expression group
(<3.35) (n=46); and a high ENST00000527317 expression group
(≥2.91) (n=30) and a low ENST00000527317 expression group
(<2.91) (n=42). We then analyzed the relationships among the
expression of ENST00000583044, NR_104048, lnc-WRN-10:1,
ENST00000527317 and age, sex, smoking status, tumor location,
clinical stage, lymphatic metastasis, distant metastasis and
survival status. The present study demonstrated that the expression
of ENST00000583044, NR_104048, lnc-WRN-10:1 and ENST00000527317 was
significantly correlated with clinical stage, lymphatic metastasis,
distant metastasis and survival status (P<0.05). However, no
significant associations were detected among the expression of
ENST00000583044, NR_104048, lnc-WRN-10:1 and ENST00000527317 and
age, sex, smoking and tumor location (P>0.05, Table II).
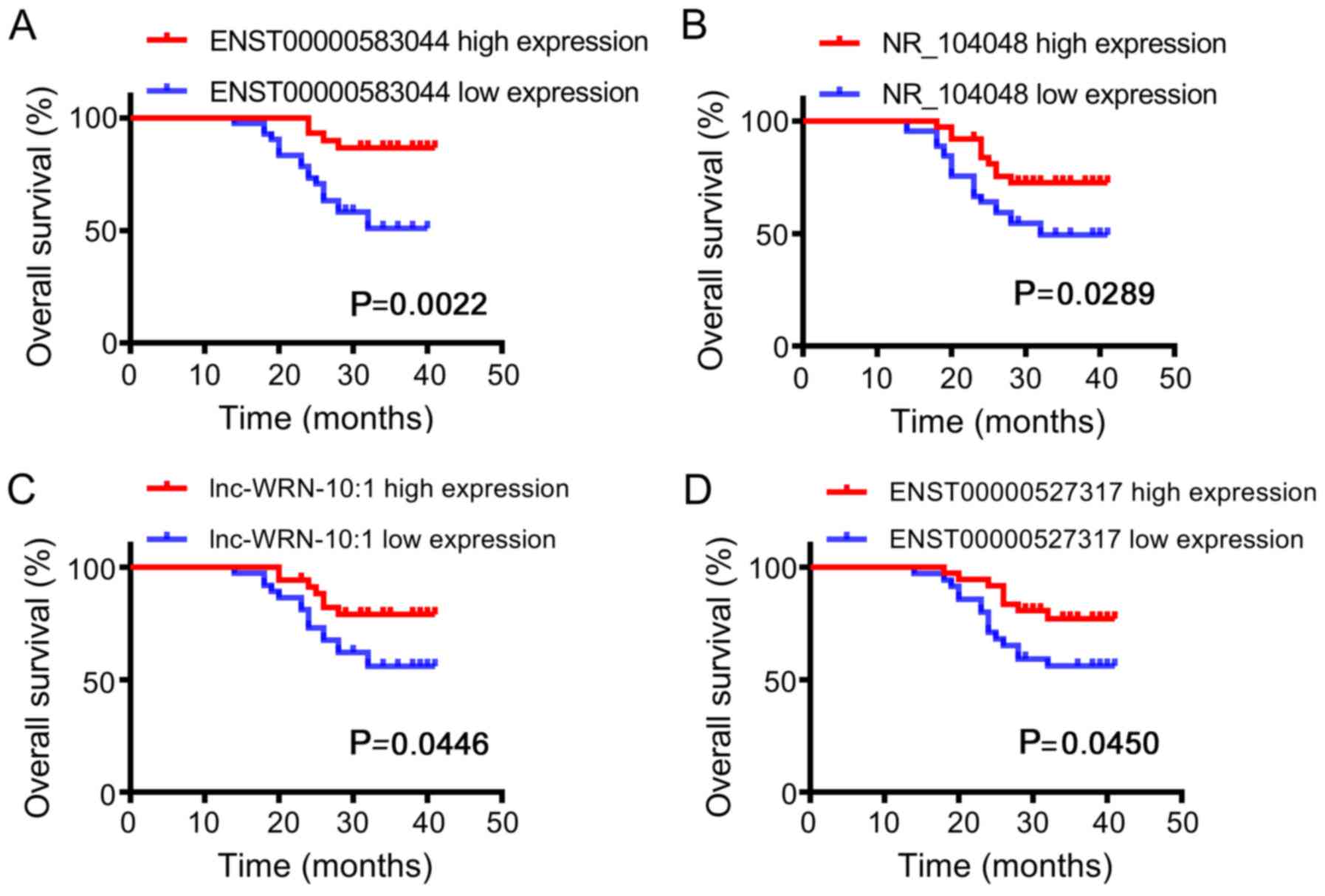
Kaplan-Meier analysis demonstrated that the median
OS for patients with low expression of ENST00000583044, NR_104048,
lnc-WRN-10:1 and ENST00000527317 was significantly lower than that
in patients with high expression of these factors (P<0.05)
(Fig. 9). Furthermore, the median
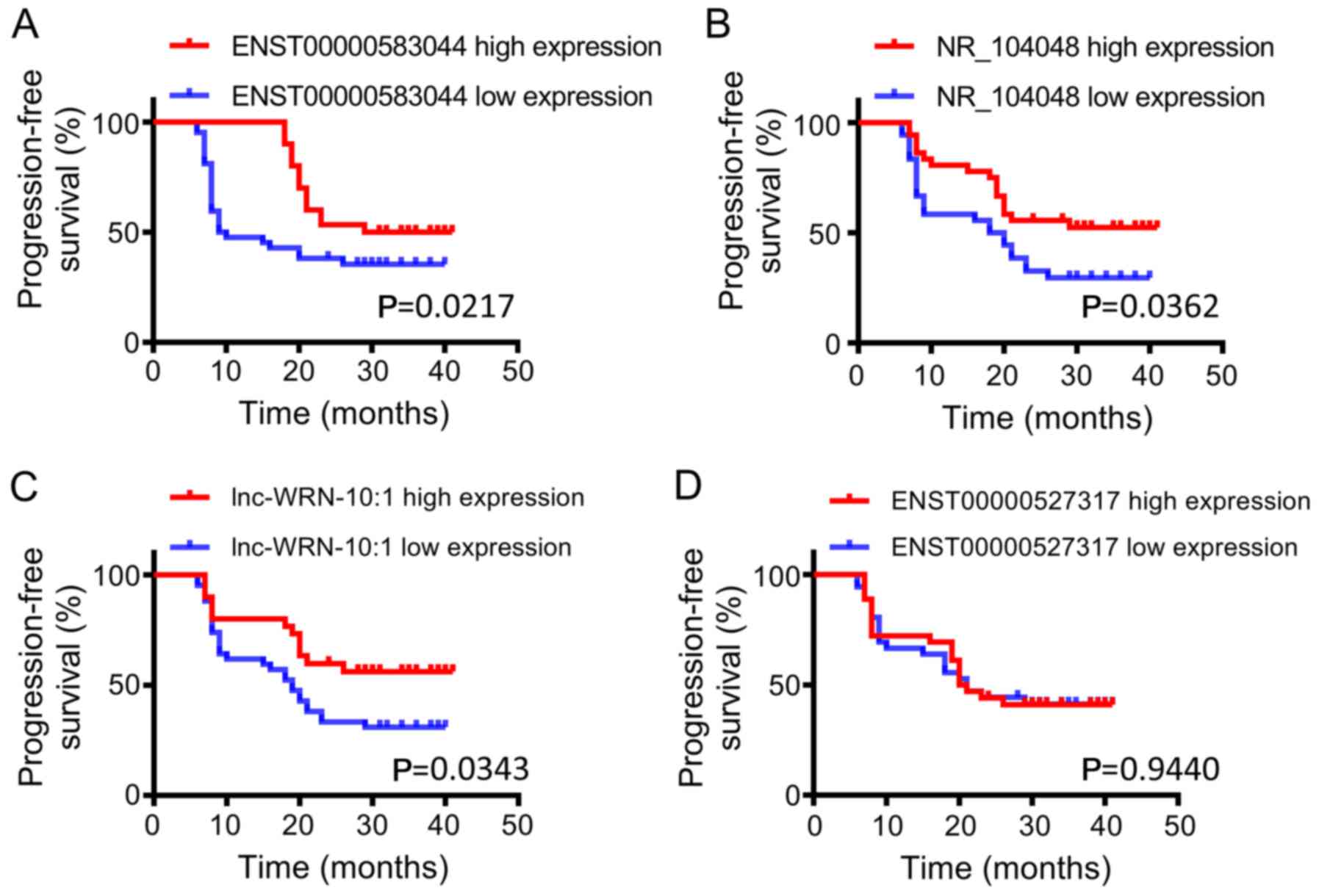
PFS for patients with high expression of ENST00000583044, NR_104048
and lnc-WRN-10:1 was significantly higher than that in patients
with low expression (P<0.05) (Fig.
10).
Discussion
Tumorigenesis and development of oral squamous cell
carcinoma (OSCC) consists of a complex process, and the underlying
mechanisms remain poorly understood. The aim of the present study
was to explore the relationship between lncRNAs and OSCC. lncRNAs
were initially considered ‘noise’ without any biological functions
in the human genome. They are now known to play important roles in
gene expression, and their differential expression may affect
corresponding functional performance (4). Subsequently, many studies have
demonstrated that lncRNAs are involved in many important regulatory
processes, including X-chromosome silencing, genomic imprinting,
chromatin modification, transcriptional activation, transcriptional
interference and intranuclear transport (17,18).
In the present study, we used an expression profile microarray to
identify differentially expressed genes in OSCC at the whole genome
level. The results revealed a large number of differentially
expressed lncRNAs and mRNAs, some of which may be important genes
involved in tumorigenesis and the development of OSCC. For example,
MALAT1 promotes the invasion and metastasis of lung cancer
(19). MEG3 expression levels were
highly correlated with invasion and metastasis of gastric cancer
(20,21). GAS5 indicates a poor prognosis in
ovarian cancer (22). Feng et
al showed that lncRNAs were abnormally expressed in OSCC and
metastatic tissue samples (5).
Recent studies have shown that HOTAIR is highly expressed in OSCC
and is associated with the biological behavior of tumor invasion
and metastasis (6,23).
In view of the complex transcriptional regulatory
mechanisms of lncRNAs and their ability to form a variety of
secondary functional structures, their biological functions cannot
be predicted based solely on nucleic acid sequence (24,25).
lncRNA loci are often located in intronic regions of the coding
gene and thus may affect expression of its adjacent genes (26–28).
To investigate the relationship between lncRNAs and mRNAs,
coexpression networks of dysregulated lncRNAs and mRNAs were
constructed based on differential expression. Differentially
expressed genes were assigned into subnetworks associated with
phenotypic functions to roughly deduce the function of lncRNAs in
this subnetwork and predict possible regulatory mechanisms. We
identified 4 lncRNAs with high correlations with other genes.
ENST00000583044, NR_104048, lnc-WRN-10:1 and ENST00000527317, which
were found in the present study, belonged to downregulated lncRNAs
and have never previously been reported in OSCC or any other solid
tumors. Our data are the first to reveal these four critical node
genes. We speculate that these results may depend on tumor
heterogeneity. As with any cancer, OSCC is also highly
heterogeneous and is characterized by different genetic
backgrounds, different pathological types, different
differentiation states, different gene mutation and transcriptional
patterns and proteome expression profiles (29,30).
We further examined the expression of these 4 genes in 72 patients
with OSCC and normal tissues and analyzed their relationship with
the clinicopathological features and prognosis. We found that these
4 genes were downregulated in OSCC. Moreover, their expression was
not correlated with age, sex, smoking, or tumor location but was
related to clinical stage, lymphatic metastasis, distant metastasis
and survival status. Furthermore, low expression levels of these 4
lncRNAs contributed to poor median PFS and OS.
In addition, by constructing a co-expression network
of differentially expressed genes, we found that some lncRNAs,
e.g., NR_002812, regulate many genes, including SP100 and B2M.
Previous studies have shown that the SP100 protein is involved in
viral infection, virus-related protein interaction and
self-ubiquitination regulation, and plays an important role in
interferon and p53 signaling pathways (31,32).
p53 protein inhibits the growth and invasion of oral malignancy by
regulating the phosphorylation of AKT (25,33,34).
Although the specific mechanism and related signaling pathways need
to be further studied, they can be used as molecular markers for
early diagnosis, treatment and prognostic evaluation of OSCC.
Since lncRNAs do not encode protein, we studied the
mechanism of pathogenesis from another angle by enriching the
biological function of differentially expressed mRNAs. In the
present study, GO and KEGG pathway analyses were performed to
examine the biological function of dysregulated genes (35,36).
GO functional annotations indicated that the differentially
expressed genes were enriched in different BP, CC and MF
categories. KEGG enrichment identified many metabolic pathways
associated with cancer, e.g., ‘metabolic pathways’ and ‘pathways in
cancer’, demonstrating that OSCC is associated with cell structure
changes, metabolic process disorders, tumor suppressor genes and
oncogene signaling pathway abnormalities and was a consequence of
multiple intracellular and external factors. These results further
verified the results of the microarray assay. lncRNAs can guide
gene expression in either a cis or trans manner. In
the present study, two independent algorithms were used to predict
cis and trans target genes of differentially
expressed lncRNAs in OSCC. Through GO functional annotation and
KEGG pathway analysis, we found that these target genes regulate
relevant OSCC proteins and affect OSCC tumorigenesis and
development via their functions in organ, molecular binding,
metabolism and cancer pathways. Target prediction of lncRNAs
provides important information for further study of potential
functional lncRNAs and target genes in OSCC.
The present study also has several limitations
including its small sample size. In the future, our results need to
be validated in large-scale samples.
In conclusion, in the present study, 2,294
dysregulated lncRNAs and 1,938 dysregulated mRNAs were identified
by a microarray assay. We explored 4 critical lncRNAs nodes, which
may play an important role in the pathogenesis of OSCC. GO and
pathway analyses indicated that the functions and enriched pathways
of many dysregulated genes were associated with cancer. The
potential target genes of dysregulated lncRNAs were enriched in 43
KEGG pathways, and cancer pathways were the primary enrichment
pathways. These results provide new insight into the molecular
markers and therapeutic targets for OSCC.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (no. 81372150).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
YLQ, YHL and BHL acquired the data and created a
draft of the manuscript; JDB, WJW, MH and PK conducted the
experiments and collected the data; YLQ and YHL interpreted the
data, performed the statistical analysis and analyzed the results;
BHL revised and approved the final version of the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the Fourth Affiliated Hospital of Hebei Medical
University (Shijiazhuang, China) and written informed consent was
obtained from all subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Poveda-Roda R, Bagán JV, Jiménez-Soriano
Y, Margaix-Muñoz M and Sarrión-Pérez G: Changes in smoking habit
among patients with a history of oral squamous cell carcinoma
(OSCC). Med Oral Patol Oral Cir Bucal. 15:e721–e726. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Messadi DV: Diagnostic aids for detection
of oral precancerous conditions. Int J Oral Sci. 5:59–65. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cui Z, Ren S, Lu J, Wang F, Xu W, Sun Y,
Wei M, Chen J, Gao X, Xu C, et al: The prostate cancer-up-regulated
long noncoding RNA PlncRNA-1 modulates apoptosis and proliferation
through reciprocal regulation of androgen receptor. Urol Oncol.
31:1117–1123. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou X, Liu S, Cai G, Kong L, Zhang T, Ren
Y, Wu Y, Mei M, Zhang L and Wang X: Long non coding RNA MALAT1
promotes tumor growth and metastasis by inducing
epithelial-mesenchymal transition in oral squamous cell carcinoma.
Sci Rep. 5:159722015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feng L, Houck JR, Lohavanichbutr P and
Chen C: Transcriptome analysis reveals differentially expressed
lncRNAs between oral squamous cell carcinoma and healthy oral
mucosa. Oncotarget. 8:31521–31531. 2017.PubMed/NCBI
|
|
6
|
Wu Y, Zhang L, Zhang L, Wang Y, Li H, Ren
X, Wei F, Yu W, Liu T, Wang X, et al: Long non-coding RNA HOTAIR
promotes tumor cell invasion and metastasis by recruiting EZH2 and
repressing E-cadherin in oral squamous cell carcinoma. Int J Oncol.
46:2586–2594. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eades G, Zhang YS, Li QL, Xia JX, Yao Y
and Zhou Q: Long non-coding RNAs in stem cells and cancer. World J
Clin Oncol. 5:134–141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang L, Xu HG and Lu C: A novel long
non-coding RNA T-ALL-R-LncR1 knockdown and Par-4 cooperate to
induce cellular apoptosis in T-cell acute lymphoblastic leukemia
cells. Leuk Lymphoma. 55:1373–1382. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zang W, Wang T, Huang J, Li M, Wang Y, Du
Y, Chen X and Zhao G: Long noncoding RNA PEG10 regulates
proliferation and invasion of esophageal cancer cells. Cancer Gene
Ther. 22:138–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, He L, Du Y, Zhu P, Huang G, Luo J,
Yan X, Ye B, Li C, Xia P, et al: The long noncoding RNA
lncTCF7 promotes self-renewal of human liver cancer stem
cells through activation of Wnt signaling. Cell Stem Cell.
16:413–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Loewen G, Jayawickramarajah J, Zhuo Y and
Shan B: Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol.
7:902014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi SJ, Wang LJ, Yu B, Li YH, Jin Y and
Bai XZ: LncRNA-ATB promotes trastuzumab resistance and
invasion-metastasis cascade in breast cancer. Oncotarget.
6:11652–11663. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yan P, Luo S, Lu JY and Shen X: Cis- and
trans-acting lncRNAs in pluripotency and reprogramming. Curr Opin
Genet Dev. 46:170–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tafer H and Hofacker IL: RNAplex: A fast
tool for RNA-RNA interaction search. Bioinformatics. 24:2657–2663.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu DX, Zhu W, Fang C, Fan L, Zou ZJ, Wang
YH, Liu P, Hong M, Miao KR, Liu P, et al: miR-181a/b significantly
enhances drug sensitivity in chronic lymphocytic leukemia cells via
targeting multiple anti-apoptosis genes. Carcinogenesis.
33:1294–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kornienko AE, Guenzl PM, Barlow DP and
Pauler FM: Gene regulation by the act of long non-coding RNA
transcription. BMC Biol. 11:592013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gutschner T, Hämmerle M, Eissmann M, Hsu
J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al:
The noncoding RNA MALAT1 is a critical regulator of the metastasis
phenotype of lung cancer cells. Cancer Res. 73:1180–1189. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–R53.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista
DL, Gejman R, Ansell PJ, Zhao J, Weng C and Klibanski A: Activation
of p53 by MEG3 non-coding RNA. J Biol Chem. 282:24731–24742. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma N, Li S, Zhang Q, Wang H, Qin H and
Wang S: Long non-coding RNA GAS5 inhibits ovarian cancer cell
proliferation via the control of microRNA-21 and SPRY2 expression.
Exp Ther Med. 16:73–82. 2018.PubMed/NCBI
|
|
23
|
Lu MY, Liao YW, Chen PY, Hsieh PL, Fang
CY, Wu CY, Yen ML, Peng BY, Wang DP, Cheng HC, et al: Targeting
LncRNA HOTAIR suppresses cancer stemness and metastasis in oral
carcinomas stem cells through modulation of EMT. Oncotarget.
8:98542–98552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang B, Arun G, Mao YS, Lazar Z, Hung G,
Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, et al: The lncRNA
Malat1 is dispensable for mouse development but its
transcription plays a cis-regulatory role in the adult. Cell
Rep. 2:111–123. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang G, Li Z, Zhao Q, Zhu Y, Zhao C, Li X,
Ma Z, Li X and Zhang Y: LincRNA-p21 enhances the sensitivity of
radiotherapy for human colorectal cancer by targeting the
Wnt/beta-catenin signaling pathway. Oncol Rep. 31:1839–1845. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roy R, Singh R, Chattopadhyay E, Ray A,
Sarkar N, Aich R, Paul RR, Pal M and Roy B: MicroRNA and target
gene expression based clustering of oral cancer, precancer and
normal tissues. Gene. 593:58–63. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zaid KW, Nhar BM, Ghadeer Alanazi SM,
Murad R, Domani A and Alhafi AJ: Lack of effects of recombinant
human bone morphogenetic protein2 on angiogenesis in oral squamous
cell carcinoma induced in the syrian hamster cheek Pouch. Asian Pac
J Cancer Prev. 17:3527–3531. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is
associated with meningioma pathogenesis and progression. Cancer
Res. 70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sasahira T and Kirita T: Hallmarks of
cancer-related newly prognostic factors of oral squamous cell
carcinoma. Int J Mol Sci. 19:E24132018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lala M, Chirovsky D, Cheng JD and Mayawala
K: Clinical outcomes with therapies for previously treated
recurrent/metastatic head-and-neck squamous cell carcinoma (R/M
HNSCC): A systematic literature review. Oral Oncol. 84:108–120.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wurdak M, Schneider M, Iftner T and
Stubenrauch F: The contribution of SP100 to cottontail rabbit
papillomavirus transcription and replication. J Gen Virol. Jan
24–2018.(Epub ahead of print). doi: 10.1099/jgv.0.001012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Berscheminski J, Brun J, Speiseder T,
Wimmer P, Ip WH, Terzic M, Dobner T and Schreiner S: Schreiner,
Sp100A is a tumor suppressor that activates p53-dependent
transcription and counteracts E1A/E1B-55K-mediated transformation.
Oncogene. 35:3178–3189. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim T, Veronese A, Pichiorri F, Lee TJ,
Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al:
p53 regulates epithelial-mesenchymal transition through microRNAs
targeting ZEB1 and ZEB2. J Exp Med. 208:875–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan BS, Tiong KH, Choo HL, Chung FF, Hii
LW, Tan SH, Yap IK, Pani S, Khor NT, Wong SF, et al: Mutant
p53-R273H mediates cancer cell survival and anoikis resistance
through AKT-dependent suppression of BCL2-modifying factor (BMF).
Cell Death Dis. 6:e18262015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng W, Gao W and Feng J: Long noncoding
RNA HULC is a novel biomarker of poor prognosis in patients with
pancreatic cancer. Med Oncol. 31:3462014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao Z, Bai J, Wu A, Wang Y, Zhang J, Wang
Z, Li Y, Xu J and Li X: Co-LncRNA: Investigating the lncRNA
combinatorial effects in GO annotations and KEGG pathways based on
human RNA-Seq data. Database. 2015:bav0822015. View Article : Google Scholar : PubMed/NCBI
|