Introduction
Salivary gland tumor (SGT) is one of the most
pathologically heterogeneous and least studied tumor types
(1). Currently, the molecular
mechanisms that cause SGT pathogenesis remain poorly understood.
Although rare, the limited treatment options available for patients
with SGT make this disease a significant health problem (2–4). SGT
is classified into >20 distinct histological subtypes (5). The most common benign SGT is salivary
pleomorphic adenoma, and the most common malignant SGTs are
salivary adenoid cystic carcinoma (SACC) and mucoepidermoid
carcinoma (5).
The incidence of SACC is ~4.5 cases/106
individuals and accounts for 10% of all SGTs (6–8). SACC
is characterized by indolent, yet progressive, growth, perineural
invasion and a high rate of distant metastasis (8). SACC presents with three different
histological growth patterns, i.e. cribriform, tubular and solid
patterns. Among them, the solid-pattern histological type
represents the most aggressive form of SACC. Solid-pattern SACCs
are typically poorly differentiated, higher-grade malignancy with
increased rates of metastasis, all of which lead to shorter
disease-specific survival times (9). Although the 5-year survival rate is
~70% for patients with SACC, the survival rates decrease to 40% at
10 years and 25% at 15 years owing to frequent local recurrence and
distant metastasis (3–8). Treatment options for SACC are
currently limited, with the standard treatment regimen being local
resection with radiation therapy (10). Chemotherapy has also been used in
isolated cases, but has failed to improve overall patient survival
(10). An improved understanding of
the molecular mechanisms of SACC pathogenesis should allow the
development of novel and more effective treatment options for
patients with SACC (11).
Significant progress has been achieved in the
molecular profiling of human SGT, particularly with the application
of exome sequencing (12–17). In addition to the commonly observed
chromosome translocations (e.g. nuclear factor IB and MYB)
(18), the phosphoinositide
3-kinase (PI3K)/phosphatase and tensin homolog (PTEN) signaling
pathway has been noted to be frequently altered in SGTs (12–14).
Three independent studies identified PTEN mutations in human SGTs,
specifically in SACC, salivary ductal carcinoma and carcinoma ex
pleomorphic adenoma, but not in mucoepidemoid carcinoma or acinic
cell carcinomas (12,19,20).
In addition, genetic loss or decreased PTEN expression has been
identified in human SGTs (21–24).
Previous studies, including ours, have indicated that loss or
decreased expression of PTEN (ranging between 20 and 47%) occurs in
several human SGT subtypes (21–24).
To investigate the molecular consequences of
decreased PTEN expression or PTEN gene loss, we previously
performed a gene expression microarray in human SACC cell lines
with ectopically induced PTEN knockdown (24). In the present study, the gene
expression profile associated with PTEN knockdown was analyzed
further and enrichment of genes associated with
epithelial-mesenchymal transition (EMT) was identified. Among the
EMT genes associated with PTEN downregulation, WD repeat-containing
protein 66 (WDR66) was selected for further characterization of its
functions in the mediation of EMT, maintenance of cancer stem cells
(CSCs). Specifically, basic cellular phenotype implications,
including cellular proliferation, migration and invasion, of WDR66
were also assessed in the context of PTEN downregulation. Finally,
the clinical relevance of the results was validated as WDR66 gene
expression was identified to be inversely correlated with PTEN
expression in SACC cell lines and tumor samples from SACC patients,
and its expression level was revealed to be negatively associated
with the overall survival of patients with SACC.
Materials and methods
Patients and collection of
tissues
A total of 20 normal salivary glands (SGs) and 46
SACC tumor tissues, which were surgically resected between January
2001 and December 2013 at Dalian Medical University (Dalian,
China), were included in the present study. All patient samples
were collected under the approved guidelines provided by the
Institutional Review Boards at Dalian Medical University. All
collected tissues were clinically examined by oral pathologists and
informed consent forms were obtained from all patients. The
demographic and clinical pathological information for each patient
is listed in Table I.
 | Table I.Clinicopathological characteristics
of patients. |
Table I.
Clinicopathological characteristics
of patients.
| Factor | n |
|---|
| SACC | 46 |
|
Tubular | 12 |
|
Cribriform | 14 |
|
Solid | 20 |
| NSG | 20 |
| Age, years |
|
|
<60 | 35 |
|
≥60 | 31 |
| Sex |
|
|
Male | 32 |
|
Female | 34 |
| Tumor site |
|
| Major
salivary glands | 32 |
| Minor
salivary glands | 14 |
Cell culture, treatment and
transfection
The human SACC cell line SACC83 was generously
provided by Dr Shenglin Li (Department of Oral and Maxillofacial
Surgery, Peking University School and Hospital of Stomatology,
Beijing, China). Cells were subcultured in RPMI-1640 medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.) and 1X penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). All cell cultures were maintained at 37°C in a
humidified incubator containing 5% CO2. To inhibit the
PI3K pathway, SACC cells were treated with 1 µM GDC-0941 (LC
Laboratories, Woburn, MA, USA) or vehicle (dimethylsulfoxide)
control for 4 h. To knock down the expression of WDR66, SACC83
cells were transfected with the WDR66 short interfering RNA
(siRNA) or control siRNA using Lipofectamine™ 3000 Transfection
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Three siRNAs against WDR66 were
tested. The sequence of these three siRNAs were: #1918,
5′-GCGGUGUACCACUUAACAATT-3′; #1566, 5′-GCUGGUUGUCUGGGACAUATT-3′;
and #969, 5′-GCAGAGAGUUCUUCUGUAUTT-3′. The sequence for the
negative control siRNA was 5′-UUCUCCGAACGUGUCACGUTT-3′. PTEN
knockdown was achieved by transfecting with PTEN siRNA
(5′-GAUCUUGACAAAGCAAAUATT-3′). At between 48 and 72 h
post-transfection, SACC83 cells were harvested and the knockdown
efficiencies were determined using the reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded (FFPE) slides of
SACC biopsies (4-mm thick) from patients and normal HSG tissue
sections were used for IHC staining according to a protocol
described previously (25).
Antibody binding was visualized using a Vectastain®
Avidin-Biotin Complex kit (cat. no. PK-4001 [rabbit immunoglobulin
G (IgG)] or PK-4002 (mouse IgG); Vector Laboratories, Inc.,
Burlingame, CA, USA) and a Diaminobenzidine kit (cat. no. DAB-1031,
Fuzhou Maixin Biotech Co., Ltd., Fuzhou, China), according to the
manufacturer's protocol, and counterstained with hematoxylin.
Images of the slides were captured using a BX43 light microscope
(Olympus Corporation, Tokyo, Japan) controlled with CellSens
software (version 1.12; Olympus Corporation). The primary
antibodies used for IHC were mouse anti-WDR66 (1:200; cat. no.
ab83694; Abcam, Cambridge, UK) and mouse anti-PTEN (1:200; cat. no.
18-0652; Zymed; Thermo Fisher Scientific, Inc.). The quantitative
analysis of IHC staining followed the criteria we have described
previously (24). The staining
index was determined as follows: 0–2, low (−); 3–6, medium (+);
7–9, high (++) expression.
Double immunofluorescence (IF)
staining
Double IF was performed as described previously
(26) using mouse anti-WDR66 (as
aforementioned) and rabbit anti-PTEN (1:100; cat. no. 60300;
ProteinTech Group, Inc., Chicago, IL, USA) primary antibodies, and
Alexa Fluor 488-conjugated goat anti-rabbit IgG [heavy and light
chains (H+L)] (1:500; cat. no. ab181448; Abcam, UK) and Alexa Fluor
594-conjugated goat anti-mouse IgG (H+L) (1:500; cat. no. ab150120;
Abcam, UK) secondary antibodies. Nuclei were labeled with DAPI
(1:200; cat. no. C1005; Beyotime Institute of Biotechnology,
Haimen, China).
RNA extraction and RT-qPCR
Total RNA was isolated with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Total RNA samples were
reverse-transcribed into cDNA using the PrimeScript™ RT reagent kit
with gDNA Eraser (cat. no. DRR047A; Takara Bio, Inc., Otsu, Japan).
qPCR was performed using a Thermal Cycler Dice Real-Time system
(TP800; Takara Bio, Inc.) using SYBR Premix Ex Taq II kit (cat. no.
DRR820A; Takara Bio, Inc.), according to the manufacturer's
protocol. GAPDH served as an internal control gene. All primers
were synthesized by Takara Bio, Inc., and the sequences are
presented in Table II.
| Table II.Primer sequences for the quantitative
polymerase chain reaction. |
Table II.
Primer sequences for the quantitative
polymerase chain reaction.
| Gene | Forward | Reverse |
|---|
| WDR66 |
5′-TTATGGTTCCCCTATTGAGC-3′ |
5′-GTCTTATGTGGATTGCCGTC-3′ |
| PTEN |
5′-GAGCGTGCAGATAATGACAAGGAAT-3′ |
5′-GGATTTGACGGCTCCTCTACTGTTT-3′ |
| CDH1 |
5′-TGCCCAGAAAATGAAAAAGG-3′ |
5′-GTGTATGTGGCAATGCGTTC-3′ |
| VIM |
5′-AATGGCTCGTCACCTTCGTGAAT-3′ | 5′-
CAGATTAGTTTCCCTCAGGTTCAG-3′ |
| ALDH1 |
5′-GCACGCCAGACTTACCTGTCCTA-3′ |
5′-GGCCTTCACTGCCTTGTCAAC-3′ |
| OCT4 |
5′-GTGCCGTGAAGCTGGAGAA-3′ |
5′-TGGTCGTTTGGCTGAATACCTT-3′ |
| NANOG |
5′-CCTGTGATTTGTGGGCCTGA-3′ |
5′-CTCTGCAGAAGTGGGTTGTTTG-3′ |
| SOX2 |
5′-CCAAGATGCACAACTCGGAGA-3′ |
5′-CCGGTATTTATAATCCGGGTGCT-3′ |
| ZEB1 |
5′-TCCATGCTTAAGAGCGCTAGCT-3′ |
5′-GTATCTTGTCTTTCATCCTGATTTCCA-3′ |
| ZEB2 |
5′-TTCCTGGGCTACGACCATACC-3′ |
5′-CAAGCAATTCTCCCTGAAATCC-3′ |
| TGFB1 |
5′-CGCCAGAGTGGTTATCTTTTGA-3′ |
5′-CGGTAGTGAACCCGTTGATGT-3′ |
| GAPDH |
5′-GCACCGTCAAGGCTGAGAAC-3′ |
5′-TGGTGAAGACGCCAGTGGA-3′ |
Western blot analysis
SACC cells were harvested and lysed in
radioimmunoprecipitation buffer containing protease/phosphatase
inhibitor cocktail (cat. no. KGP250, Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China). Total proteins (40 µg) were separated by
SDS/PAGE (10% gels) prior to transfer onto nitrocellulose
membranes, which were blocked with fat-free milk powder (5%)
diluted in Tris-buffered saline containing 0.1% Tween-20 at room
temperature for 1 h then incubated with primary antibody at 4°C
overnight followed by horseradish peroxidase-conjugated secondary
antibodies [HRP-conjugated goat anti-rabbit IgG (H+L); 1:2,000;
cat. no. ABC-AS014; and HRP-conjugated goat anti-mouse IgG (H+L);
1:2,000, cat. no. ABC-AS003; both from ABclonal Biotech Co., Ltd.,
Woburn, MA, USA] for 2 h at room temperature. A Prime Western
Blotting Detection Reagent enhanced chemiluminescence kit (cat. no.
K-12045-D10; Advansta Inc., San Jose, CA, USA) was used to detect
the signals.
Primary antibodies used for the western blotting
were: Mouse Anti-WDR66 (1:500; as aforementioned), rabbit
anti-epithelial (E-)cadherin (1:2,000; cat. no. 20874; ProteinTech
Group, Inc.), rabbit anti-vimentin (1:2,000; cat. no. 10366;
ProteinTech Group, Inc.), rabbit anti-aldehyde dehydrogenase 1
(ALDH1; 1:400; cat. no. 15910; ProteinTech Group, Inc.), rabbit
anti-Oct3/4 (1:500; cat. no. 11263; ProteinTech Group, Inc.),
rabbit anti-Nanog (1:2,000; cat. no. 14295; ProteinTech Group,
Inc.), rabbit anti-sex-determining region Y box 2 (Sox2; 1:1,000;
cat. no. 3579; Cell Signaling Technology, Inc., Danvers, MA, USA),
mouse anti-vinculin (1:10,000; cat. no. ab73412; Abcam) and mouse
anti-GAPDH (1:1,000; cat. no. AG019; Beyotime Institute of
Biotechnology).
Co-immunoprecipitation (IP) assay
SACC cells were harvested and were lysed with
ice-cold IP buffer. The cell lysates were incubated with Protein
G-conjugated sepharose beads at 4°C for 2 h and centrifuged at
1,000 × g for 5 min at 4°C. The extract was incubated with
pre-coupled antibodies bound to Protein G-conjugated beads at 4°C
overnight. Samples were resolved by SDS-PAGE (10% gels) and
transferred onto polyvinylidene fluoride membranes. Primary
antibodies against PTEN (1:100; as aforementioned) and WDR66
(1:100; as aforementioned) were used for IP.
Proliferation assay
Cell proliferation was evaluated using a Cell
Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan). Briefly, cells were seeded in a 96-well plate at
a density of 0.5×104 cells/well in RPMI-1640 with 10%
FBS. Cells were harvested from day 1 to day 6. CCK-8 reagent was
added to each well and incubated at 37°C for 4 h before each time
point of harvest. The absorbance values were determined at a
wavelength of 450 nm.
Migration and invasion assay
Cells at 6×104 cells/well in serum-free
RPMI-1640 medium were added to the upper chamber of a 6.5-mm
Transwell with 8.0-µm pore polyester membrane inserts (Corning
Incorporated, Corning, NY, USA) and the inserts were placed in
24-well plates with RPMI-1640 containing 10% FBS in the lower
chamber. For invasion assays, the Transwell membranes were
pre-coated with 50 µg/ml Matrigel solution (BD Biosciences, San
Jose, CA, USA), whereas Matrigel was not used for cell migration
assays. After 48 h of incubation at 37°C, cells that had invaded to
the reverse side of the Transwell membrane were fixed with methanol
and stained with 0.1% crystal violet at room temperature for 20
min. For each filter, five randomly selected fields were analyzed
under an inverted light microscope at ×20 magnification.
Wound healing assay
SACC cells (~1×106) were seeded onto
6-well plates at 37°C for 24 h. The plated cells were then
scratched with a 200 µl sterile pipette tip. Images of the wounded
areas were captured under a microscope at ×10 magnification after
0, 12 and 24 h of incubation at 37°C, and the distances between the
wound edges were determined at each time point.
TCGA database search
To investigate the gene status of WDR66 in human
cancer, a cross-cancer genomic analysis of WDR66 was performed
using The Cancer Genome Atlas (TCGA) database (www.cbioportal.org/public-portal) using
WDR66 as a key word.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism (version 6; GraphPad Software, Inc., La Jolla, CA,
USA) and SPSS software (version 19.0; IBM Corp., Armonk, NY, USA).
All data were acquired from at least three independent experiments
and are presented as the mean ± standard error of the mean. For two
group-designed experiments, comparisons were performed using
unpaired Student's t-test; for multiple comparisons, analysis of
variance (ANOVA) with two-way ANOVA followed by Dunn's post hoc
test or two-way repeated-measures ANOVA followed by Bonferroni's
post hoc test was performed. Additionally, the correlations of PTEN
and WDR66 were assessed using Spearman rank correlation
coefficients. Survival curves were estimated using the Kaplan-Meier
method and differences in survival distributions were evaluated by
the log-rank test. P<0.05 was considered to indicate a
statistically significant difference.
Results
WDR66 is identified as one of the
candidates for EMT in the context of PTEN deficiency
We previously reported a gene expression profile by
microarray analysis using human SACC83 cells with ectopic knockdown
of PTEN (24). Initial
review of the microarray data revealed a total of 244 mRNAs to be
differentially expressed (fold change >3) between SACC83 cells
and SACC83 cells with PTEN knockdown (24). Following reanalysis of these
microarray data for the present study, EMT genes were identified to
be particularly enriched in SACC83 cells with PTEN knockdown
(Fig. 1A). Selected genes were
validated by RT-qPCR. Decreases in CDH1 (E-cadherin) and
increase in VIM (vimentin) RNA expression levels were
determined in SACC83 cells with PTEN knockdown (Fig. 1B), suggesting that loss of PTEN may
promote EMT in SACC cells. In addition, the transcription factor of
EMT, zinc finger E-box-binding homeobox 1 (27), and a confirmed EMT promoter,
transforming growth factor β1 (28), were also validated to reveal
increased expression levels in SACC83 cells with PTEN
knockdown (Fig. 1B). Among the
validated genes, WDR66 (Fig. 1A
and B) was selected for further functional investigation as it
was identified to be the least-studied EMT gene candidate in the
context of cancer development and maintenance.
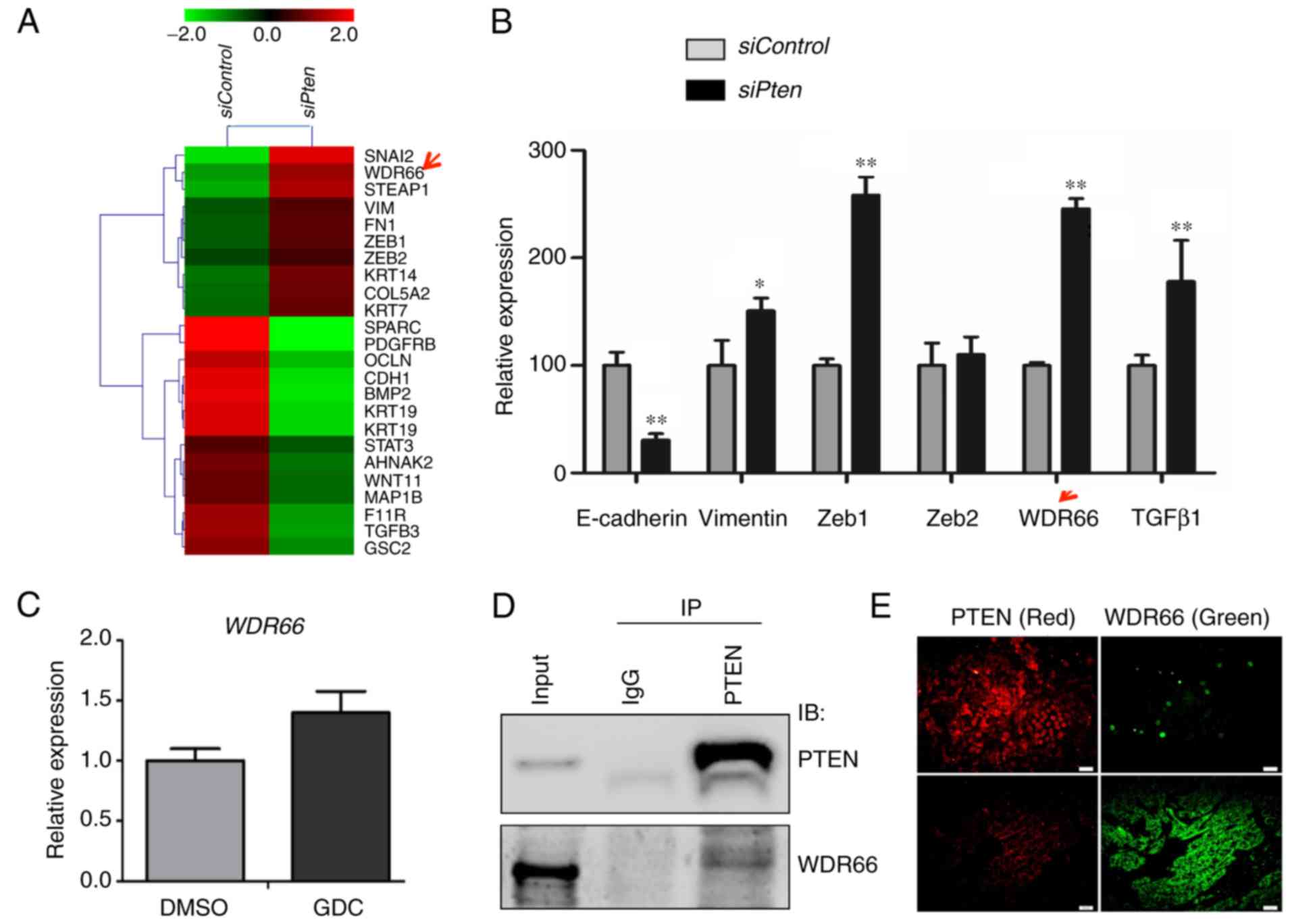 | Figure 1.WDR66 is identified as one of
candidates for EMT in the context of PTEN deficiency. (A) Heatmap
of the EMT-associated genes in the microarray data of the SACC83
cell with PTEN knocked down (siPten) in comparison with
control (siControl). Red represents an increase; green represents a
decrease. (B) Validation of the expression of EMT-associated genes
from the microarray data using RT-qPCR. *P<0.05; **P<0.01 vs.
Control (independent sample t-test). (C) Expression of WDR66
following treatment with a phosphoinositide 3-kinase inhibitor,
GDC, determined using RT-qPCR. (D) Co-IP of PTEN and WDR66 protein
in the SACC83 cell line. (E) Double immunofluorescence staining of
PTEN (red) and WDR66 (green) in tissues of human salivary adenoid
cystic carcinoma samples. Scale bar, 100 µm. WDR66, WD
repeat-containing protein 66; EMT, epithelial-mesenchymal
transition; PTEN, phosphatase and tensin homolog; si, short
interfering RNA; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; GDC, GDC-0941; DMSO, dimethylsulfoxide;
IP, immunoprecipitation; IB, immunoblot; E-cadherin, epithelial
cadherin; Zeb, zinc finger E-box-binding homeobox; TGFβ1,
transforming growth factor β1; IgG, immunoglobulin G. |
To investigate whether the inverse association
between PTEN and WDR66 is due to PTEN's regulation of the PI3K
signaling pathway (29), the SACC83
cells were treated with a PI3K inhibitor, GDC-0941 (1 µM), to
inhibit PI3K activity. However, PI3K inhibitor treatment failed to
cause the expected decrease in WDR66 expression (Fig. 1C), suggesting that the inverse
association between PTEN and WDR66 expression levels may not depend
entirely on the function of PTEN in regulating the PI3K signaling
pathway. WDR66 belongs to the WD-repeat containing family of
proteins, which collectively functions in the formation of
protein-protein complexes in a variety of signaling pathways
(30). Thus, a co-IP experiment
between WDR66 and PTEN was performed, and the results suggested
that there may be an interaction between PTEN and WDR66 in SACC83
cells (Fig. 1D).
To further determine the effect of the PTEN-WDR66
interaction on SACC pathogenesis, double IF staining was performed
to examine the intracellular distribution of WDR66 in the control
and under PTEN-deficient conditions. As presented in Fig. 1E, WDR66 expression (green) was
inversely associated with PTEN expression (red).
WDR66 is essential for EMT and
regulates the expression of genes associated with CSCs in the
context of PTEN deficiency in SACC
To facilitate the functional studies of WDR66,
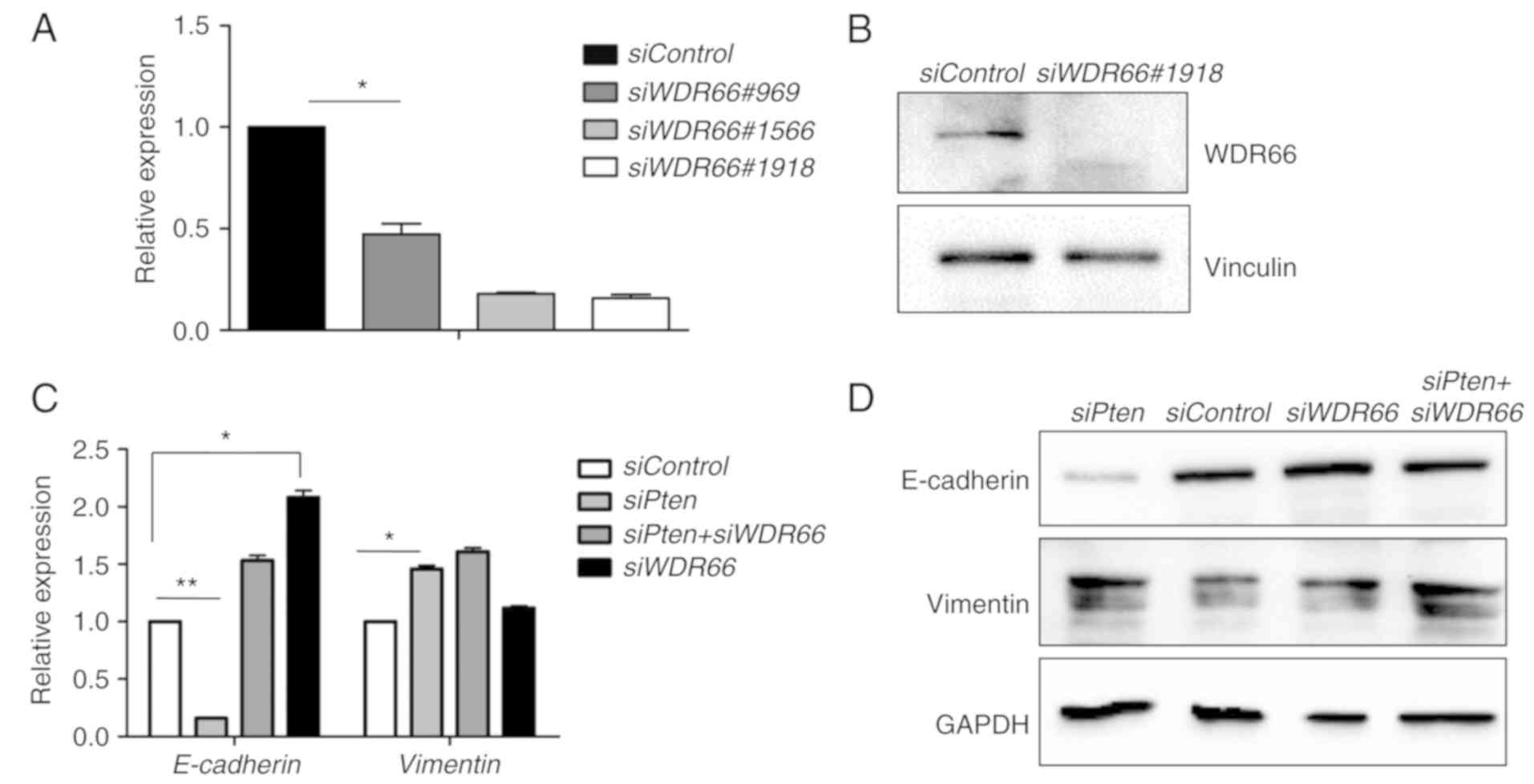
WDR66 mRNA was knocked down using three siRNAs, and siRNA
#1918 was selected on the basis of its optimal knockdown efficacy
as determined using RT-qPCR (Fig.
2A). Knockdown efficacy of siRNA #1918 was validated by western
blotting (Fig. 2B). siRNA #1918 was
used to determine the function of WDR66 in mediating the EMT
phenotype induced by PTEN deficiency. Downregulation of CDH1
and upregulation of VIM mRNAs were confirmed in the SACC83
cells with PTEN knockdown (Fig.
2C). However, simultaneous knockdown of WDR66 mRNA with
PTEN mRNA knockdown led to a complete reversal of the effect
of PTEN siRNA on decreasing the CDH1 expression level
(Fig. 2C). Similar observations
were made at the protein expression level on the basis of western
blot results for E-cadherin and vimentin (Fig. 2D). Collectively, these results
suggest that WDR66 may be essential for the maintenance of the EMT
phenotype induced by PTEN deficiency in SACCs.
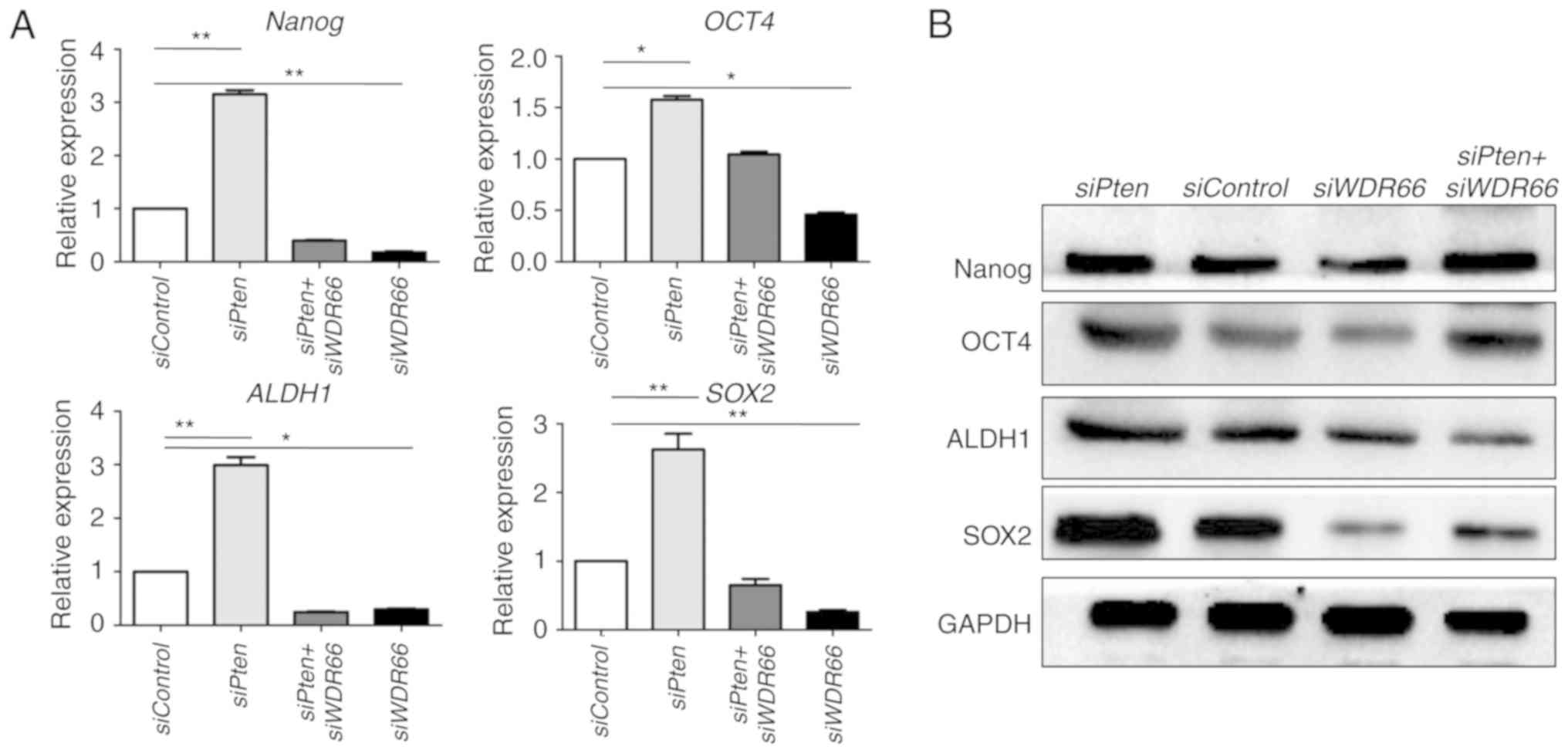
Since EMT has been suggested to confer CSC-like
properties on neoplastic cells (31), the function of WDR66 in regulating
the expression of genes associated with cancer cell stemness in the
context of PTEN deficiency was also investigated. Similar to the
gene changes in EMT, the embryonic stem cell genes NANOG and
OCT4, and the putative CSC genes ALDH1 and
SOX2 in SG cancer (32) were
all increased following PTEN knockdown in SACC cells. The
expression of NANOG and OCT4 genes was decreased
following WDR66 knockdown with or without PTEN
knockdown (Fig. 3A). Similar
observations were made at the protein expression level on the basis
of western blot results for NANOG and OCT4 (Fig. 3B). Collectively, these results
suggest that WDR66 may be required to maintain the expression of
genes associated with CSCs in the context of PTEN deficiency.
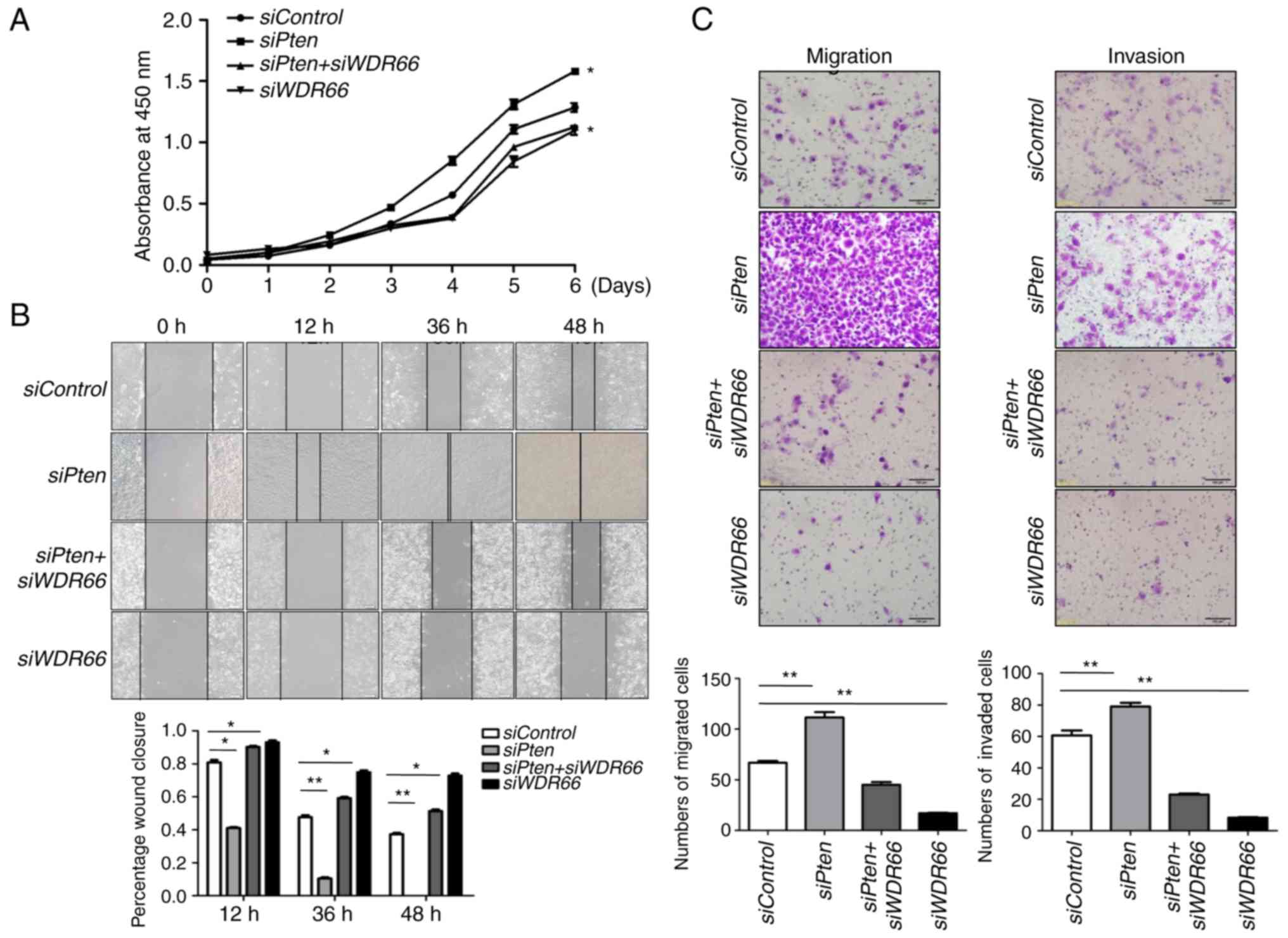
Knockdown of WDR66 decreases cell
proliferation, migration and invasion in the context of PTEN
deficiency
To determine the cellular effects of WDR66 in the
context of PTEN deficiency, cellular proliferation assays were
performed in SACC83 cells carrying WDR66 knockdown with or
without PTEN knockdown. As expected, knockdown of
PTEN promoted cellular proliferation rates, but this effect
was fully nullified with WDR66 knockdown (Fig. 4A). Similarly, knockdown of
PTEN accelerated wound closure, but simultaneous knockdown
of WDR66 led to the attenuation of this effect to the point that
there was no significant difference between the siPten + siWDR66
group and the siControl group (Fig.
4B). Finally, the migratory and invasive abilities that were
enhanced by PTEN knockdown were also decreased with
WDR66 knockdown (Fig.
4C).
WDR66 expression is inversely
associated with PTEN expression and is negatively associated with
overall survival of patients with SACC
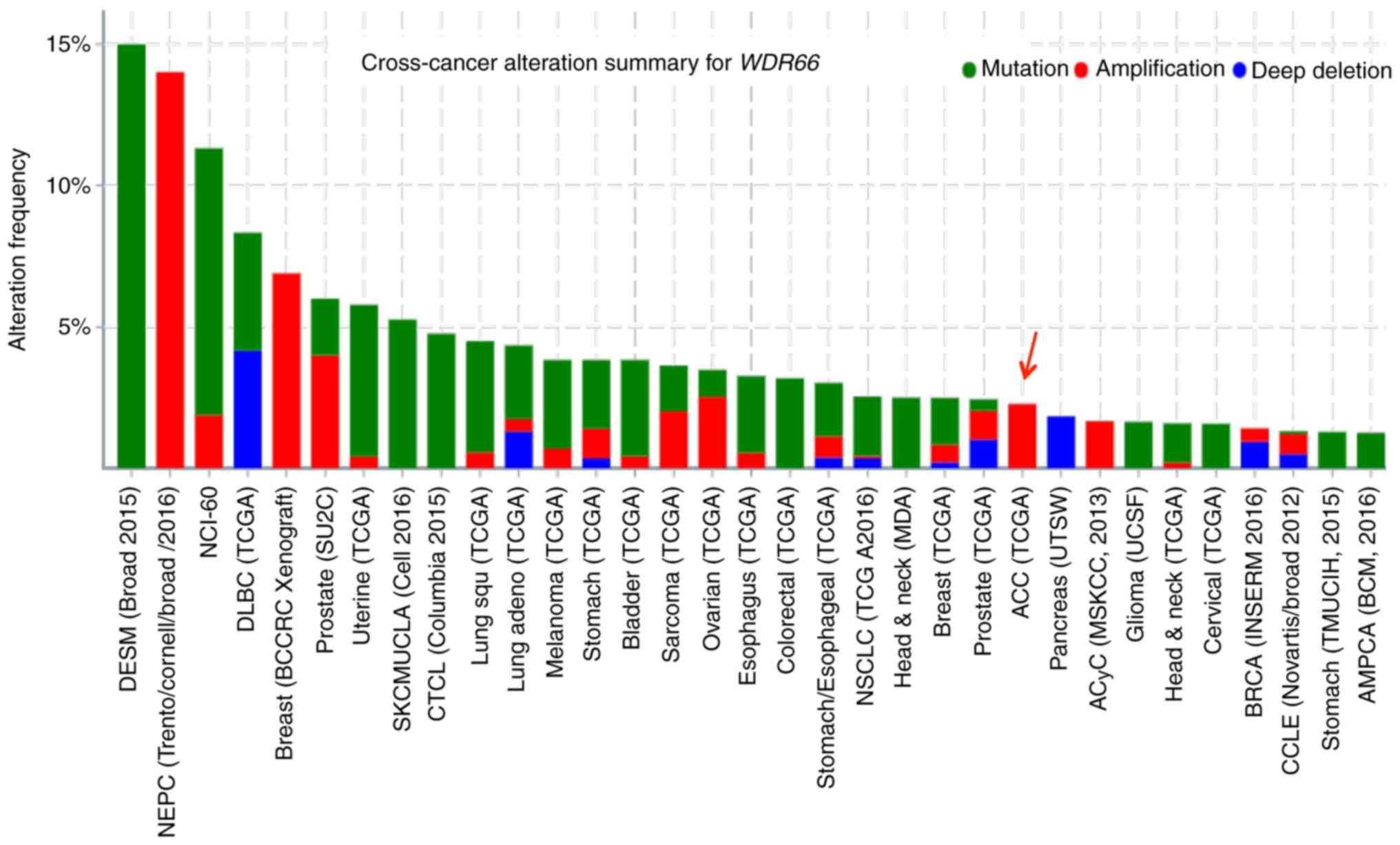
To search for genetic alterations of WDR66 in
human malignancies, a cross-cancer genomic analysis of WDR66
was performed using The Cancer Genome Atlas (TCGA) database.
Somatic point mutations and gene amplification were the most common
genetic alterations in WDR66 across various types of cancer.
Notably, amplification of WDR66 was the only genetic
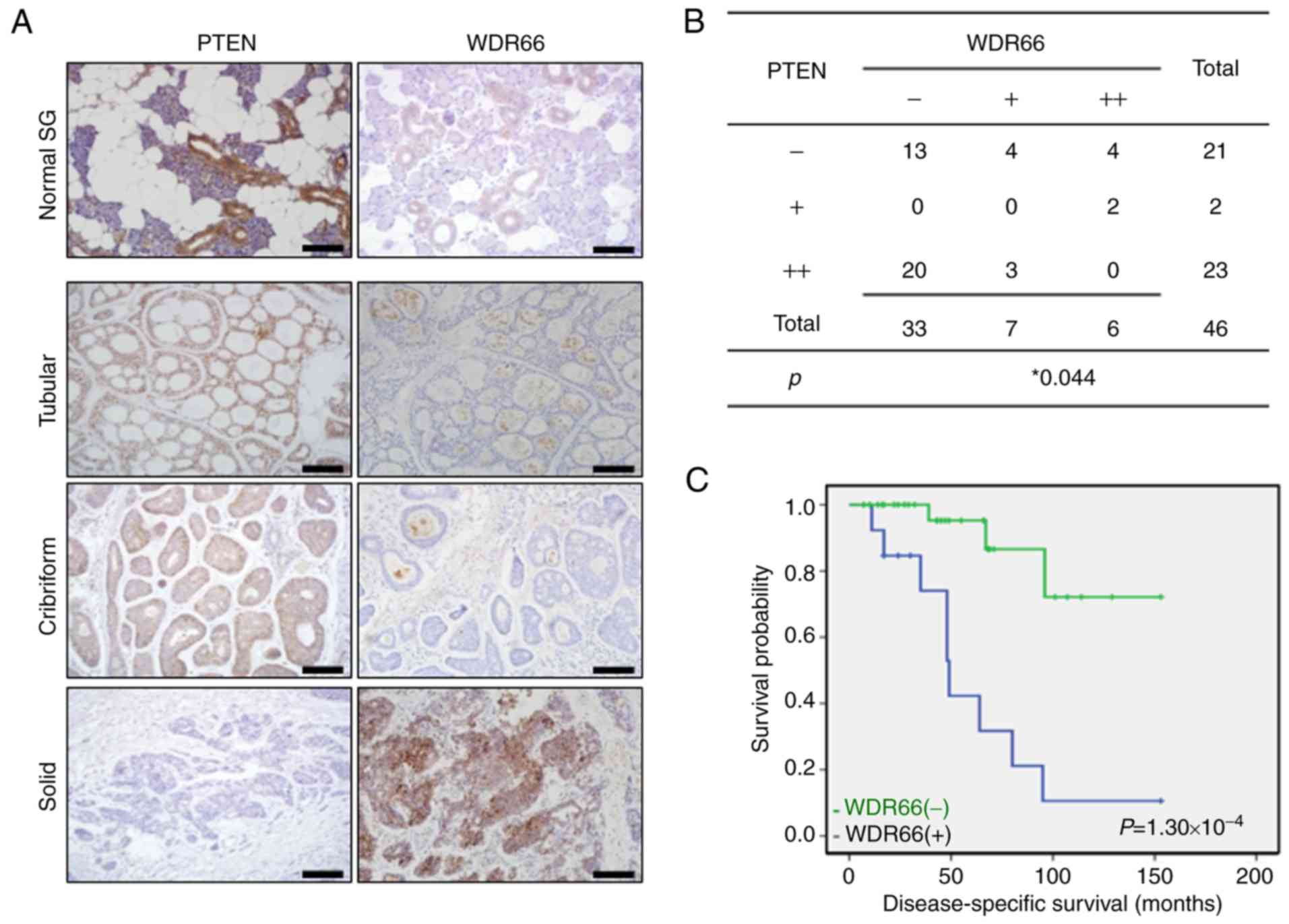
alteration noted in patients with SACC (Fig. 5). Next, WDR66 expression levels were
determined in patient samples and associated with PTEN expression
in SACCs via IHC. A total of 46 SACC and 20 normal SGs were
included. WDR66 was weakly expressed in normal SGs, and the
majority of the tubular and cribriform SACCs, but it was markedly
expressed in tumor cells of solid-pattern SACCs (Fig. 6A). This staining pattern of WDR66
was the opposite to the staining pattern of PTEN, where it was
markedly expressed in normal SGs, tubular and cribriform SACCs, but
was lost from the solid pattern SACCs (Fig. 6A). This inverse association of WDR
and PTEN expression is exemplified in their opposite staining
pattern in the same fields of tumor cells, as presented in Fig. 6A. The IHC staining results were
summarized in Fig. 6B, and revealed
a statistically significant inverse correlation between WDR66 and
PTEN expression in patient tissues (P=0.044). Finally, the
significance of WDR66 on prognosis of the 46 patients with SACC was
investigated. Kaplan-Meier analysis demonstrated that the patients
with positive WDR66 expression had significantly decreased overall
survival times compared with patients with negative WDR66
expression (70.1 months vs. 121.8 months, P=1.30×10−4;
log-rank test; Fig. 6C), which
revealed that WDR66 expression was negatively associated with the
overall survival periods of patients with SACC.
Discussion
We have demonstrated previously that the loss of
PTEN or decreased PTEN expression is common in human SACCs,
particularly in the most aggressive solid pattern form (24). To further understand the molecular
mechanism of SACC pathogenesis in the context of PTEN deficiency, a
gene expression profile array was performed in the SACC cells with
ectopic PTEN knockdown (24). Although the knockdown approach using
siRNA may not be able to completely eliminate PTEN expression, it
represents at least a significant portion of patients with SACC who
exhibited decreased PTEN expression. A complete knockout of PTEN
using the clustered regularly interspaced short palindromic repeats
system should provide additional information for patients with SACC
with complete loss of PTEN in future studies. In the present study,
the array results were further analyzed, specifically for genes
associated with the EMT process which was one of the enriched
signatures in SACC cells with PTEN knockdown. Individual
gene expression validation of EMT regulators and EMT-associated
transcription factors confirmed the microarray data analysis, and
one EMT-associated gene, WDR66, was selected to be the focus
of the present study due to the relative novelty of the function of
WDR66 in human cancer and interaction with PTEN.
WDR66 belongs to the WD-repeat containing family of
proteins which functions in the formation of protein-protein
complexes in a variety of signaling pathways (30), and affects a wide variety of
physiological and pathological conditions, including cancer.
Perhaps the best example of a tumor regulator within this family is
WDR7, which is an E3 ubiquitin ligase involved in human cancer
through multiple mechanisms (33,34).
Compared with WDR7, reports of oncogenic relevance of other WD
repeat domain-containing protein family members is relatively
limited. Overexpression of WDR5 has been identified to be
correlated with the aggressiveness of head and neck squamous cell
carcinoma (35), whereas WDR79 has
been suggested to promote the proliferation of lung cancer cells
through mediation of the p53 pathway (36). To date, the best-known function of
WDR66 has been in the determination of mean platelet volume
(37). In the oncology field, to
the best of our knowledge, the only previous study on WDR66 has
suggested its association with EMT in esophageal tumors (38).
SACC is characterized as a long indolent growth, but
it is a disease that is manifested with a disproportionally high
rate of hematogenous spread compared with the relatively low rate
of local node metastasis (9). The
EMT process has been well-established as the primary mechanism for
neoplastic cells to invade blood vessels, survive in the harsh
circulatory system and regenerate new tumors by the acquisition of
CSC properties (39,40). PTEN deficiency and WDR66
overexpression have been reported individually in colon cancer
(41) and esophageal cancer
(38), respectively. However, to
the best of our knowledge, there have not been any studies that
suggest the connection of PTEN and WDR66 in the EMT process within
a single tumor context. The results of the present study indicated
that PTEN loss significantly decreased E-cadherin expression at the
mRNA and protein levels. In contrast, knocking down WDR66
expression increased the CDH1 mRNA level, but no significant
increase of E-cadherin at the protein level was identified. It is
possible that the protein level changes may take longer in WDR66
compared with those of PTEN. Nonetheless, the results indicated an
association of EMT/CSC with the expression levels of PTEN and
WDR66. More importantly, the attenuation of
PTEN-knockdown-induced EMT/CSC phenotypes upon WDR66
knockdown highlighted the functional dependence on WDR66 status of
the PTEN-deficiency phenotype in SACCs. The present study was not
able to fully elucidate the molecular mechanisms underlying the
interaction between PTEN and WDR66 in the context of the EMT
process in SACCs. It may be useful to investigate the mechanism of
WDR66 in mediating platelet volume (37). Platelets have been revealed to
affect the maintenance of the EMT phenotype (42,43),
therefore improved understanding the function of WDR66 in platelet
volume may lead to an improved understanding of its function in
EMT.
The inverse association between PTEN and WDR66
expression has been convincingly documented in the IHC analysis of
patient tissues. A question is whether this association is
dependent on the enzyme activity of PTEN in the PI3K signaling
pathway. In contrast with the predicted decrease of WDR66
expression if the inverse correlation between PTEN and WDR66
depends on PI3K activity, it was identified that WDR66 expression
is paradoxically increased. The reason for this increase is
unclear, but the results of the present study suggested at least
that this inverse correlation may not depend on PI3K activity. One
of the primary functions of the WD repeat-containing protein family
is forming protein-protein complexes as a structural protein
(30). The physical interaction
between WDR66 and PTEN demonstrated by the co-IP experiment
suggested potential mechanisms associated with physical
interactions between WDR66 and PTEN molecules. One limitation of
the present study is that there are no data from SACC cells
overexpressing WDR66, which will be helpful to improve
understanding of the function of WDR66 in mediating PTEN
deficiency-associated SACC pathogenesis. This represents a
direction for future studies.
Despite the limitations, the present study revealed
several translational effects for patients with SACC. First, the
study identified WDR66 to be a key mediator of oncogenic effects
caused by PTEN deficiency, suggesting WDR66 to be a novel
therapeutic target in SACC tumors with PTEN deficiency. WDR66 gene
amplification was noted in SACCs via analysis of TCGA database.
Furthermore, the inverse association of WDR66 and PTEN expression
in patient tissue samples suggested that WDR66 can also be
increased in tumors with PTEN deficiency. Whether this negative
association between WDR66 and PTEN expression exists in other types
of cancer is a question to investigate further. However, the
mutational status of PTEN in the patients of the present study is
unknown, and patients with wild-type PTEN are unable to be selected
for survival comparison. Future study of PTEN mutation status in
the patient population used will provide additional information to
further address PTEN deficiency in SACC pathogenesis, although the
results of the present study have identified that the survival of
patients with SACC with PTEN loss or decreased expression is
significantly poorer compared with those with normal PTEN
expression level (24). Secondly,
the independence of the PI3K signaling pathway for this negative
association between WDR66 and PTEN suggested that in PTEN-deficient
SACC tumors, targeting the PI3K signaling pathway may not be a good
option, particularly in controlling tumor progression and
metastasis. Instead, WDR66 itself may appear as a novel target for
targeted therapy, particularly in PTEN-deficient human cancer,
which includes a certain proportion of SACCs in the present study,
and more broadly for expanding to other types of human cancer with
PTEN deficiency (44). Thus,
targeting WDR66 may be more effective in controlling tumor
progression and metastasis in patients with cancer with PTEN
deficiency.
Acknowledgements
Not applicable.
Funding
The present study was supported by the China
Postdoctoral Science Foundation (grant nos. 2017M621178 and
2015M580227), the National Institutes of Health (grant no.
R01DE021788) and Cancer League of Colorado.
Availability of data and materials
All data and materials used in the present study are
available from the corresponding authors upon request.
Authors' contributions
YC, HL, SLL and JX conceived and designed the study.
YC, HL, SLX, XZ, HB, QY and LG performed the experiments. JL, FJ
and MJW contributed to the study design, analysis and
interpretation of data. YC, HL, SLL and JX wrote the paper. SLL and
JX supervised the study. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
All patient samples were collected under the
approved guidelines provided by the Institutional Review Boards at
Dalian Medical University (Dalian, China). Written informed consent
was provided by all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Seethala RR: Salivary gland tumors:
Current concepts and controversies. Surg Pathol Clin. 10:155–176.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dunn LA, Ho AL, Laurie SA and Pfister DG:
Unmet needs for patients with salivary gland cancer. Oral Oncol.
60:142–145. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adelstein DJ, Koyfman SA, El-Naggar AK and
Hanna EY: Biology and management of salivary gland cancers. Semin
Radiat Oncol. 22:245–253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Keller G, Steinmann D, Quaas A, Grunwald
V, Janssen S and Hussein K: New concepts of personalized therapy in
salivary gland carcinomas. Oral Oncol. 68:103–113. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Seethala RR and Stenman G: Update from the
4th edition of the world health organization classification of head
and neck tumours: Tumors of the salivary gland. Head Neck Pathol.
11:55–67. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dillon PM, Chakraborty S, Moskaluk CA,
Joshi PJ and Thomas CY: Adenoid cystic carcinoma: A review of
recent advances, molecular targets, and clinical trials. Head Neck.
38:620–627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J, Shao C, Tan ML, Mu D, Ferris RL and
Ha PK: Molecular biology of adenoid cystic carcinoma. Head Neck.
34:1665–1677. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Moskaluk CA: Adenoid cystic carcinoma:
Clinical and molecular features. Head Neck Pathol. 7:17–22. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coca-Pelaz A, Rodrigo JP, Bradley PJ,
Vander Poorten V, Triantafyllou A, Hunt JL, Strojan P, Rinaldo A,
Haigentz M Jr, Takes RP, et al: Adenoid cystic carcinoma of the
head and neck-an update. Oral Oncol. 51:652–661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bell D and Hanna EY: Head and neck adenoid
cystic carcinoma: What is new in biological markers and treatment?
Curr Opin Otolaryngol Head Neck Surg. 21:124–129. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bell D and Hanna EY: Salivary gland
cancers: Biology and molecular targets for therapy. Curr Oncol Rep.
14:166–174. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ho AS, Kannan K, Roy DM, Morris LG, Ganly
I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al: The
mutational landscape of adenoid cystic carcinoma. Nat Genet.
45:791–798. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stephens PJ, Davies HR, Mitani Y, Van Loo
P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR,
Butler AP, et al: Whole exome sequencing of adenoid cystic
carcinoma. J Clin Invest. 123:2965–2968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rettig EM, Talbot CC Jr, Sausen M, Jones
S, Bishop JA, Wood LD, Tokheim C, Niknafs N, Karchin R, Fertig EJ,
et al: Whole-genome sequencing of salivary gland adenoid cystic
carcinoma. Cancer Prev Res. 9:265–274. 2016.
|
|
15
|
Drier Y, Cotton MJ, Williamson KE,
Gillespie SM, Ryan RJ, Kluk MJ, Carey CD, Rodig SJ, Sholl LM,
Afrogheh AH, et al: An oncogenic MYB feedback loop drives alternate
cell fates in adenoid cystic carcinoma. Nat Genet. 48:265–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yin LX and Ha PK: Genetic alterations in
salivary gland cancers. Cancer. 122:1822–1831. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mitani Y, Liu B, Rao PH, Borra VJ, Zafereo
M, Weber RS, Kies M, Lozano G, Futreal PA, Caulin C, et al: Novel
MYBL1 gene rearrangements with recurrent MYBL1-NFIB
fusions in salivary adenoid cystic carcinomas lacking t(6;9)
translocations. Clin Cancer Res. 22:725–733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferrarotto R, Heymach JV and Glisson BS:
MYB-fusions and other potential actionable targets in adenoid
cystic carcinoma. Curr Opin Oncol. 28:195–200. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grünewald I, Vollbrecht C, Meinrath J,
Meyer MF, Heukamp LC, Drebber U, Quaas A, Beutner D, Hüttenbrink
KB, Wardelmann E, et al: Targeted next generation sequencing of
parotid gland cancer uncovers genetic heterogeneity. Oncotarget.
6:18224–18237. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang K, Russell JS, McDermott JD, Elvin
JA, Khaira D, Johnson A, Jennings TA, Ali SM, Murray M, Marshall C,
et al: Profiling of 149 salivary duct carcinomas, carcinoma ex
pleomorphic adenomas, and adenocarcinomas, not otherwise specified
reveals actionable genomic alterations. Clin Cancer Res.
22:6061–6068. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ettl T, Schwarz-Furlan S, Haubner F,
Müller S, Zenk J, Gosau M, Reichert TE and Zeitler K: The
PI3K/AKT/mTOR signalling pathway is active in salivary gland cancer
and implies different functions and prognoses depending on cell
localisation. Oral Oncol. 48:822–830. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ettl T, Baader K, Stiegler C, Müller M,
Agaimy A, Zenk J, Kühnel T, Gosau M, Zeitler K, Schwarz S and
Brockhoff G: Loss of PTEN is associated with elevated EGFR and HER2
expression and worse prognosis in salivary gland cancer. Br J
Cancer. 106:719–726. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Griffith CC, Seethala RR, Luvison A,
Miller M and Chiosea SI: PIK3CA mutations and PTEN
loss in salivary duct carcinomas. Am J Surg Pathol. 37:1201–1207.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu H, Du L, Wang R, Wei C, Liu B, Zhu L,
Liu P, Liu Q, Li J, Lu SL, et al: High frequency of loss of PTEN
expression in human solid salivary adenoid cystic carcinoma and its
implication for targeted therapy. Oncotarget. 6:11477–11491.
2015.PubMed/NCBI
|
|
25
|
Cong W, Liu B, Liu S, Sun M, Liu H, Yang
Y, Wang R and Xiao J: Implications of the Wnt5a/CaMKII pathway in
retinoic acid-induced myogenic tongue abnormalities of developing
mice. Sci Rep. 4:60822014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He W, Li AG, Wang D, Han S, Zheng B,
Goumans MJ, Ten Dijke P and Wang XJ: Overexpression of Smad7
results in severe pathological alterations in multiple epithelial
tissues. EMBO J. 21:2580–2590. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li D and Roberts R: WD-repeat proteins:
Structure characteristics, biological function, and their
involvement in human diseases. Cell Mol Life Sci. 58:2085–2097.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adams A, Warner K and Nör JE: Salivary
gland cancer stem cells. Oral Oncol. 49:845–853. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takada M, Zhang W, Suzuki A, Kuroda TS, Yu
Z, Inuzuka H, Gao D, Wan L, Zhuang M, Hu L, et al: FBW7 loss
promotes chromosomal instability and tumorigenesis via cyclin
E1/CDK2-mediated phosphorylation of CENP-A. Cancer Res.
77:4881–4893. 2017.PubMed/NCBI
|
|
34
|
Kitagawa K and Kitagawa M: The SCF-type E3
ubiquitin ligases as cancer targets. Curr Cancer Drug Targets.
16:119–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu Y, Diao P, Li Z, Zhang W, Wang D, Wang
Y and Cheng J: Overexpression of WD repeat domain 5 associates with
aggressive clinicopathological features and unfavorable prognosis
in head neck squamous cell carcinoma. J Oral Pathol Med.
47:502–510. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun Y, Cao L, Sheng X, Chen J, Zhou Y,
Yang C, Deng T, Ma H, Feng P, Liu J, et al: WDR79 promotes the
proliferation of non-small cell lung cancer cells via USP7-mediated
regulation of the Mdm2-p53 pathway. Cell Death Dis. 8:e27432017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Meisinger C, Prokisch H, Gieger C, Soranzo
N, Mehta D, Rosskopf D, Lichtner P, Klopp N, Stephens J, Watkins
NA, et al: A genome-wide association study identifies three loci
associated with mean platelet volume. Am J Hum Genet. 84:66–71.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Q, Ma C and Kemmner W: Wdr66 is a
novel marker for risk stratification and involved in
epithelial-mesenchymal transition of esophageal squamous cell
carcinoma. BMC Cancer. 13:1372013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nieto MA: Epithelial plasticity: A common
theme in embryonic and cancer cells. Science. 342:12348502013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bowen KA, Doan HQ, Zhou BP, Wang Q, Zhou
Y, Rychahou PG and Evers BM: PTEN loss induces epithelial -
mesenchymal transition in human colon cancer cells. Anticancer Res.
29:4439–4449. 2009.PubMed/NCBI
|
|
42
|
Labelle M, Begum S and Hynes RO: Direct
signaling between platelets and cancer cells induces an
epithelial-mesenchymal-like transition and promotes metastasis.
Cancer Cell. 20:576–590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gay LJ and Felding-Habermann B:
Contribution of platelets to tumour metastasis. Nat Rev Cancer.
11:123–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hollander MC, Blumenthal GM and Dennis PA:
PTEN loss in the continuum of common cancers, rare syndromes and
mouse models. Nat Rev Cancer. 11:289–301. 2011. View Article : Google Scholar : PubMed/NCBI
|