Introduction
The majority of the current cancer immunotherapy
treatments involve the generation and activation of
antigen-specific and non-specific killer cells. However, successful
induction of tumor-specific immune responses is not always followed
by tumor rejection in patients. Indeed, several mechanisms have
been associated with the acquisition of tumor resistance to
cell-mediated cytotoxicity. Tumor cells can evade adaptive immunity
through the selection of variants that are resistant to specific
cytotoxic T lymphocyte (CTL) pressure. Indeed, the interaction
between immune and tumor cells can either eliminate the developing
tumor or generate a tumor cell repertoire that is able to survive
in immunocompetent hosts (1–3). In
this regard, Dunn et al reported that tumor specific T-cell
responses can select tumor-associated antigen-negative cells and
variants resistant to CTLs in vivo (4,5). We
previously reported that the reorganization of the actin
cytoskeleton may be used by tumor cells as a strategy to promote
their resistance to CTL-mediated lysis (6). Therefore, it is likely that tumor
variants resistant to T cells will emerge, most frequently in the
context of effective immunotherapies (7). Consequently, even if a strong and
sustained cytotoxic response is induced, there remain complex
issues, such as tumor evasion and selection of tumor-resistant
variants. Natural killer (NK) cells are also involved in the
control of tumor progression (8,9) and
several reports have indicated that solid tumor infiltration by NK
cells is a favorable prognostic marker (10–12).
NK cell-mediated cytotoxic activity can eliminate tumor cells that
have evaded CD8+ T cells through loss of antigen or MHC
class I molecules. Moreover, through their secretion of cytokines
or by mutual activation links established with dendritic cells
(DCs) (13–15), NK cells can positively modulate
adaptive immune responses against tumor cells and their
susceptibility to CD8+ T cells (16). However, it should be noted that
in vitro and in vivo studies demonstrated a poor or
defective activity of NK cells in cancer, as well as resistance of
tumor cells to NK cell-mediated lysis. Mechanistically, the
imbalance between activating and inhibitory signals, which may be
caused by low expression of ligands to activating receptors and
high expression of ligands to inhibitory receptors of NK cells on
tumor cells, was suggested as being one of the major mechanisms
underlying the poor anticancer activity of NK cells (17).
Although NK cells have been established as the main
effectors of antitumor immune response, their putative role on the
emergence of tumor cytotoxic resistant variants remains poorly
understood. Evidence of NK cell immunoediting was reported by
studies using NK-deficient models, and demonstrated how exposure to
NK cells causes modification of cancer immunogenicity to allow
survival and progression of the tumor clone in an immunocompetent
environment. In addition, Moretta et al reported that NK
cells play an important regulatory role by selectively editing DCs
during the course of the immune response, and that NK cell-mediated
killing of immature DCs results in selection of immunogenic DCs
during the initiation of anticancer immune responses (18). While several studies have revealed
the contribution of adaptive and innate immunity to cancer
immunoediting (1,5,19–25),
whether the innate immune system can suppress tumor formation
without adaptive immunity remains elusive. We previously used a
lung cancer model to study the emergence of tumor resistance
following specific cytotoxic T lymphocyte (CTL) selection pressure
(6). We also described various
mechanisms of resistance to NK cells that may differ between tumor
types, tumor aggressiveness and environmental contexts (26–28).
The focus of the present study was to investigate the consequences
of sustained exposure of melanoma cells to NK cell-mediated immune
stress, in order to determine whether sustained pressure on tumor
cells drives the selection of variants resistant to NK
cell-mediated lysis and acquisition of aggressive properties, and
to assess the role of the innate immunity in tumor editing.
Materials and methods
Cell lines
The T1 melanoma cell line was derived from a primary
lesion, as previously described (29). T1 cells and their derivatives were
grown in RPMI-1640/Glutamax™ (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 5% heat-inactivated fetal calf serum (FCS),
1 mM sodium pyruvate and 1% penicillin-streptomycin (all from
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere containing 5% CO2. Autologous LT2 CTL clone,
specific for the peptide Melan-A25-36, was isolated from
tumor-infiltrating lymphocytes as previously described (29), and grown in RPMI-1640/Glutamax™
supplemented with 8% AB human serum (Institut Jacques Boy), 1 mM
sodium pyruvate, 150 U/ml recombinant IL-2 and in the presence of
the irradiated autologous tumor cell line T1, lymphoblastoid LAZ
cells (B cells transformed by Epstein-Barr virus) and allogeneic
peripheral blood mononuclear cells (PBMCs). NK cells were isolated
from human healthy donor blood using a human NK Cell Isolation Kit
(Miltenyi Biotec), grown in RPMI-1640/Glutamax™ supplemented with
8% AB human serum, 1 mM sodium and 300 U/ml recombinant IL-2 in the
presence of irradiated lymphoblastoid LAZ cells and allogeneic
PBMCs. The purity of CD56+ CD3− NK cells was
>95%, as determined by flow cytometry.
Selection of NK cell-resistant
variants
The three T1 cell lines corresponded to three
independent batches of T1 reference cells. The three NK
cell-resistant T1 cell lines (T1R) were obtained following several
months of co-culture with NK cells. A total of 105 tumor
cells/well were plated in 6-well plates. On the following day, NK
cells were added in RPMI-1640 medium supplemented with 5%
heat-inactivated FCS, 1 mM sodium pyruvate and 1%
penicillin-streptomycin (Thermo Fisher Scientific, Inc.) and 300
U/ml of IL-2. Surviving tumor cells were amplified with regular
addition of NK cells in order to obtain a sustained selective
pressure.
Cell morphology and actin cytoskeleton
staining
Actin and nuclear staining were performed using
Alexa Fluor 488-coupled Phalloidin and DAPI
(4′,6-diamidino-2-phenylindole, dihydrochloride; Thermo Fisher
Scientific, Inc.), respectively. Cells were cultured overnight on
glass slides and fixed with 4% paraformaldehyde (Sigma-Aldrich;
Merck KGaA) for 30 min at room temperature. Cells were
permeabilized with 0.1% Triton X100 for 10 min at room temperature,
stained with Rhodamin-Phalloidin R415 and To-PRO3 iodide
(Invitrogen; Thermo Fisher Scientific, Inc.) for 45 min at 4°C,
mounted in Fluoromount-G (Southern Biotech) and analyzed with a
Zeiss laser scanning confocal microscope (LSM-510 Meta; Carl Zeiss
AG) using an objective of ×40 oil immersion lens. Images were later
analyzed by LSM Image Examiner software, version 4.2.0.121 (Carl
Zeiss AG).
Cytotoxicity experiments
NK cell and LT12 cytotoxic activity was measured by
a 4 h 51Cr release assay by using triplicate co-cultures
in U-bottomed 96-well plates. Various effector-to-target (E:T)
ratios were used, with 2,000 target cells per well. Following
co-culture, the supernatants were transferred to LumaPlate-96 wells
(PerkinElmer, Inc.), dried down, and 51Cr release was
measured on a Packard TopCount NXT. Data are expressed as the
percentage of specific 51Cr release from target cells,
calculated as (experimental release-spontaneous release)/(maximum
release-spontaneous release) ×100.
Confocal microscopy analysis of
immunological synapse assembly
Tumor cells and NK cells were co-cultured for 30 min
at a 1:2 ratio on poly-L-lysin slides (MatTek Corporation) at 37°C.
The cells were then fixed with 4% (w/v) paraformaldehyde/PBS
(Sigma-Aldrich; Merck KGaA) for 30 min at room temperature and
permeabilized with 0.1% (w/v) SDS solution in PBS for 10 min at
room temperature, followed by blocking with 10% FCS (v/v) solution
in PBS for 20 min at room temperature. The cells were stained at
4°C with anti-phosphotyrosine mAb (clone 4G10, cat. no. 05-321,
Upstate Biotechnology) diluted at 1:400 or mAb mouse anti-human
granzyme B (GzmB), clone GB11 (cat. no. MA1-80734, Caltag
Laboratories) diluted at 1:50, followed by a secondary mAb
conjugated to Alexa Fluor 488 (cat. no. A-11094, Thermo Fisher
Scientific, Inc.) diluted at 1:200 for 30 min at 4°C combined with
nuclear staining with TO-Pro 3 iodide (Invitrogen; Thermo Fisher
Scientific, Inc.). Fluoromount G (Southern Biotech) was added to
each slide and analysis was performed with a Zeiss LSM-510 Meta
Laser Scanning Confocal microscope. Images were analyzed using LSM
Image Examiner software, version 4.2.0.121 (Carl Zeiss AG).
Analysis of autophagosome
formation
T1 cells were transfected with green fluorescent
protein (GFP)-light chain 3 (LC3) encoding vector or its
corresponding empty vector using Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.) according to the manufacturer's recommendations.
The presence of autophagosomes was assessed at 24 h
post-transfection by monitoring the formation of dot-like
structures by confocal microscopy (LSM-510 Meta Laser Scanning
Confocal microscope; Carl Zeiss AG) using an objective of ×40 oil
immersion lens.
Western blot analysis of autophagy
markers
The expression of the autophagy markers LC3-I and
LC3-II (Cell Signaling Technology, Inc., cat. no. 2775), SQSTM1/p62
(Cell Signaling Technology, Inc., cat. no. 8025), ATG5 (Cell
Signaling Technology, Inc., cat. no. 2630) and Beclin-1 (Cell
Signaling Technology, Inc., cat. no. 3738) was assessed using total
cell extract. Proteins (30 µg) were resolved on 10% SDS-PAGE,
blotted on nitrocellulose membranes and then incubated with the
appropriate primary antibodies and secondary antibody,
Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (Jackson
ImmunoResearch, cat. no. 111-035-003).
siRNA targeting of autophagy
markers
Autophagy-defective cells were generated by
transfection with ATG5 siRNA. Briefly, cells were transfected by
electroporation of 50 nmol/l of siRNA targeting human ATG5 (Qiagen
APG5L-6 FlexiTube siRNA SI02655310). Luciferase siRNA was used as a
negative control. The silencing of ATG5 was assessed by western
blotting 48 h after transfection using appropriate Abs.
Microarray
RNA was extracted with TRIzol and purified on RNeasy
Micro Kit spin columns (Qiagen GmbH). The RNA amount, purity and
integrity were evaluated by NanoDrop and Bioanalyser (Agilent
Technologies, Inc.). For array hybridization, RNA samples (100 ng)
were amplified and stained with fluorophores using the Low RNA
Input Linear Amplification Labeling kit (Agilent Technologies,
Inc.). Cy3-stained cRNA were hybridized on Agilent Human Whole
Genome Oligo Microarray format 8×60K, design 028004 (Agilent
Technologies, Inc.), then washed and scanned. Data were extracted
by Feature Extraction software (v10.5.1.1; Agilent Technologies,
Inc.) and analysis was performed by the limma package (30) of the Bioconductor Project. An
intra-array normalization was performed, followed by a quantile
inter-array normalization. The median of all probes for a given
transcript was then recovered. Differential expression level
analysis was performed using the following criteria: Absolute fold
change >2 and corrected P-value [false discovery rate (FDR)]
<0.05. The microarray data and protocols are available at the
European Molecular Biology Laboratory European Bioinformatics
Institute database (https://www.ebi.ac.uk/arrayexpress/) under accession
no. E-MTAB-8777.
Anchorage-independent growth
A total of 500 and 1,000 cells were incubated in 1%
(w/v) methylcellulose (Methocell MC4000, Fluka) culture medium in
35-mm non-culture-treated Petri dishes. After 21 days of
incubation, cells able to grow in an anchorage-independent manner
gave rise to visible colonies that were then counted.
Tumor cell migration ability
A total of 10×106 cells/ml were plated in
the channels of 24-well BioFlux plates (Labtech) precoated with
fibronectin (20 µg/ml). After 48 h, the microfluidic device enables
to submit only half of the channel to a 5 dyn/cm2
trypsin laminar flow for removing cells only on half of the
channel. Then, the channel was washed by a 5 dyn/cm2
culture medium flow and cell migration was observed under a 2
dyn/cm2 passive culture medium flow by video microscopy
with a Zeiss AxioVert 200 fluorescence inverted video microscope
and analyzed with AxioVision software, version 4.8.2 (Carl Zeiss
AG).
Tumor cell adhesion on extracellular
matrix
Flat-bottomed 96-well plates were coated overnight
at 4°C with 100 µl of matrix composed of fibronectin (5 µg/ml),
collagen-I (5 or 200 µg/ml), osteopontin (1 µg/ml), thrombospondin
(1 µg/ml), laminin (1 µg/ml) and Matrigel (1:10 dilution). After
washing and blocking with 1% PBS-BSA solution for 1 h at 37°C
(Sigma-Aldrich; Merck KGaA), 10,000 EDTA-detached cells were added
in serum-free RPMI-1640 and plated for 2 h. PBS washing removed
unattached cells and a mixture of medium and MTT (10% v/v) was
added. After 4 h, formazan crystals were lysed with 10% SDS (w/v,
Bio-Rad Laboratories, Inc.) and 50% dimethyl formamide
(Sigma-Aldrich; Merck KGaA) solution (pH 4.7). The optical density
at 570 and 630 nm was measured to estimate the attached cell
number.
Flow cytometry analysis of main KIR
ligand expression
A total of 2×105 cells were washed in PBS
and incubated at 4°C for 20 min with the specified mAbs. Following
incubation and washing, the samples were analyzed on LSR Fortessa
or FACS Canto II (Becton, Dickinson and Company) using DIVA
software, version 6.1.3 (BD Biosciences). The following Abs were
used: PE-conjugated anti-CD112 mAb (cat. no. IM3452; Beckman
Coulter, Inc.); PE-conjugated anti-CD155 mAb (cat. no. 337610;
BioLegend, Inc.); PE-conjugated anti-MICA/B mAb (cat. no. 558352;
BD Biosciences); PE-conjugated anti-ULBP1 mAb (cat. no. IC1380P;
R&D Systems, Inc.); PE-conjugated anti-ULBP2 mAb (cat. no.
FAB1298P; R&D Systems, Inc.); PE-conjugated anti-ULBP3 mAb
(cat. no. FAB1517P; R&D Systems, Inc.); PE-conjugated
anti-HLA-E mAb (cat. no. 12-9953-42; eBioscience); PE-conjugated
anti-HLA-G mAb (cat. no. ab24384; Abcam); and FITC-conjugated
anti-HLA ABC (cat. no. IM1838U; Beckman Coulter, Inc.).
Flow cytometry conjugate
formation
The NK cells and tumor cells were stained with
CellTracker Green 5-chloromethylfluoresceindiacetate (CMFDA) and
Orange [5-(and-6)-(((4-chloromethyl)benzoyl)amino)
tetramethylrhodamine] (CMTMR) (Thermo Fisher Scientific, Inc.) for
30 min at 37°C. On the following day, the cells were co-cultured
for 45 min, fixed for at least 30 min in 4% (w/v)
paraformaldehyde/PBS (Sigma-Aldrich; Merck KGaA) at 4°C and
analyzed on FACScalibur (BD Biosciences) or AccuriC6 (BD
Biosciences) and the data were entered into the respective
CellQuest (version 4.0) or C6 Flow (version 227.4) software (both
from BD Biosciences). Double-stained events, corresponding to the
interaction between a tumor cell and at least one NK cell, were
compared to the whole stained tumor cell population.
CD107a externalization assay
NK cells were stained with CMFDA and then
co-cultured with tumor cells for 90 or 180 min in the presence of
APC-conjugated anti-CD107a antibody (clone H4A3, cat. no. 560664,
BD Pharmingen; BD Biosciences). The cells were then fixed in 4
(w/v) paraformaldehyde/PBS (Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature and analyzed with AccuriC6 flow cytometer (BD
Biosciences). Data were processed with C6 Flow software, version
227.4 (BD Biosciences).
Statistical analysis
Data are expressed as mean ± standard deviation.
P-values were determined by unpaired two-tailed Student's t-tests.
*P<0.05, **P<0.01 and ***P<0.001 were considered to
indicate statistically significant differences and error bars were
used to indicate standard deviation.
Results
NK cell pressure induced the selection
of tumor variants resistant to NK cell-mediated lysis and
displaying a differential susceptibility to other cell death
inducers
For this study, the T1 melanoma cell line and NK
cells isolated from a healthy donor were used. T1 cells were
continuously co-cultured with NK cells during several months or
left untreated during the same period to serve as control. Three
independent batches of T1 cells were co-cultured with healthy donor
NK cells in parallel. The sensitivity of control or NK-treated
cells to NK cell-mediated lysis was then assessed. The data
depicted in Fig. 1A demonstrated
that T1 cells isolated following sustained NK cell pressure (T1R)
exhibited a strong decrease in their susceptibility to NK
cell-mediated lysis compared with the parental cell line (T1). Of
note, NK cell-mediated lysis of both T1 and T1R cells was
completely inhibited by concanamycin A (a Ca2+ chelator
that inhibits cytotoxic granule exocytosis), indicating that their
killing by NK cells is mostly dependent on the perforin (PFN)/GzmB
pathway (data not shown). In parallel, we examined the putative
cross-resistance of T1R cells to additional cell death inducers,
including an autologous CTL clone (LT12) and dacarbazine (DTIC),
which are commonly used in melanoma chemotherapy. No significant
differences were observed between T1 and T1R cells regarding their
sensitivity to LT12-mediated lysis or to DTIC-induced cell death
(Fig. 1B and C). It should be noted
that the LT12 CTL clone also destroys T1 cells trough the PFN/GzmB
pathway (26), suggesting that T1R
resistance to NK cell-mediated lysis is not associated with a
resistance to GzmB-mediated cell death. However, a slight but
significant decrease in T1R cell susceptibility to tumor necrosis
factor (TNF)-α as compared to the parental T1 cell line was
observed (Fig. 1D), which was
associated with a decrease in TNF-R1 expression (Fig. 1E). Taken together, these results
indicated that NK cell pressure can select variants that are
resistant to NK cell-mediated lysis by a probable multiparametric
mechanism.
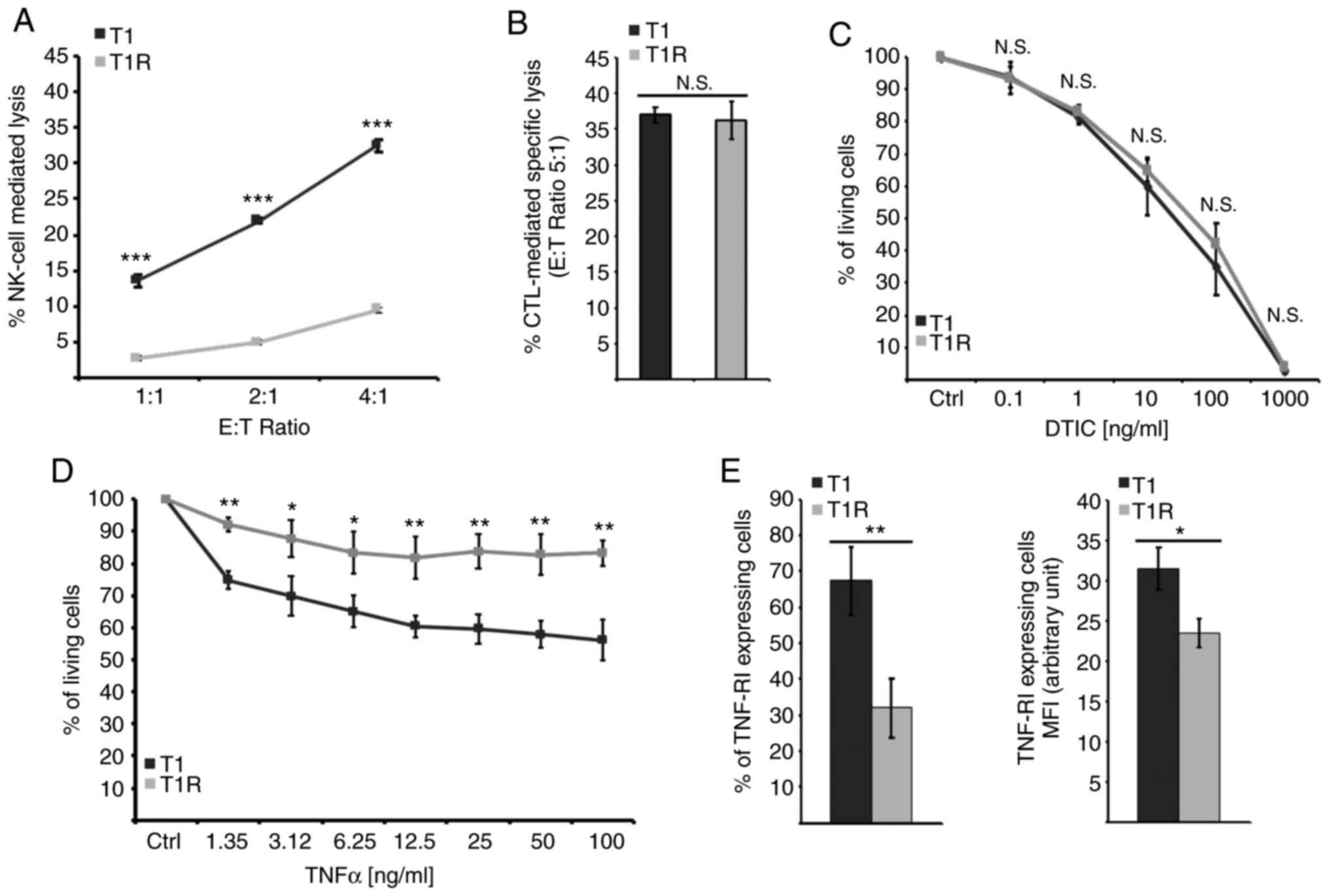 | Figure 1.The acquisition of resistance to NK
cell-mediated lysis induced by sustained NK cell pressure does not
confer resistance to autologous CTL-mediated specific lysis or to
DTIC, but does attenuate TNF response in association with a
decreased membrane expression of TNF-R1 receptor. (A)
Susceptibility to NK cell-mediated lysis for cocultured cells (T1R)
in comparison to reference cells (T1) was analyzed by a
conventional 4-h 51Cr release test. Data of a
representative experiment are presented as means of the three
independent batches of T1 and the three independent batches of T1R
cells, each performed in triplicate ± standard deviation. (B)
Susceptibility to autologous CTL-mediated specific lysis was
observed by a conventional 4-h 51Cr release test. Data
of a representative experiment are presented as means of the three
independent batches of T1 and the three independent batches of T1R
cells, each performed in triplicate ± standard deviation. (C)
Susceptibility to DTIC was evaluated by MTT assay following a 24-h
treatment by 0.1-1,000 µg/ml of DTIC. Data represent the means of
three experiments for the three independent batches of T1 and the
three independent batches of T1R cells, each performed in
triplicate±standard deviation. (D) MTT assay evaluation of
susceptibility to a 72-h TNF-α treatment at a concentration ranging
between 1.55 and 100 ng/ml. Data of a representative experiment are
presented as the means of the three independent batches of T1 and
the three independent batches of T1R cells, each performed in
duplicate ± standard deviation. (E) Percentage of positive cells by
flow cytometry for evaluation of TNF-R1 membrane expression and
TNF-R1-expressing cells MFI. Data of a representative experiment
are presented as the means of the three independent batches of T1
and the three independent batches of T1R cells ± standard
deviation. NK, natural killer; CTL, cytotoxic T lymphocyte; DTIC,
dacarbazine; TNF, tumor necrosis factor; MFI, mean fluorescence
intensity; E:T, effector:target; N.S., not significant. *P<0.05,
**P<0.01, ***P<0.001. |
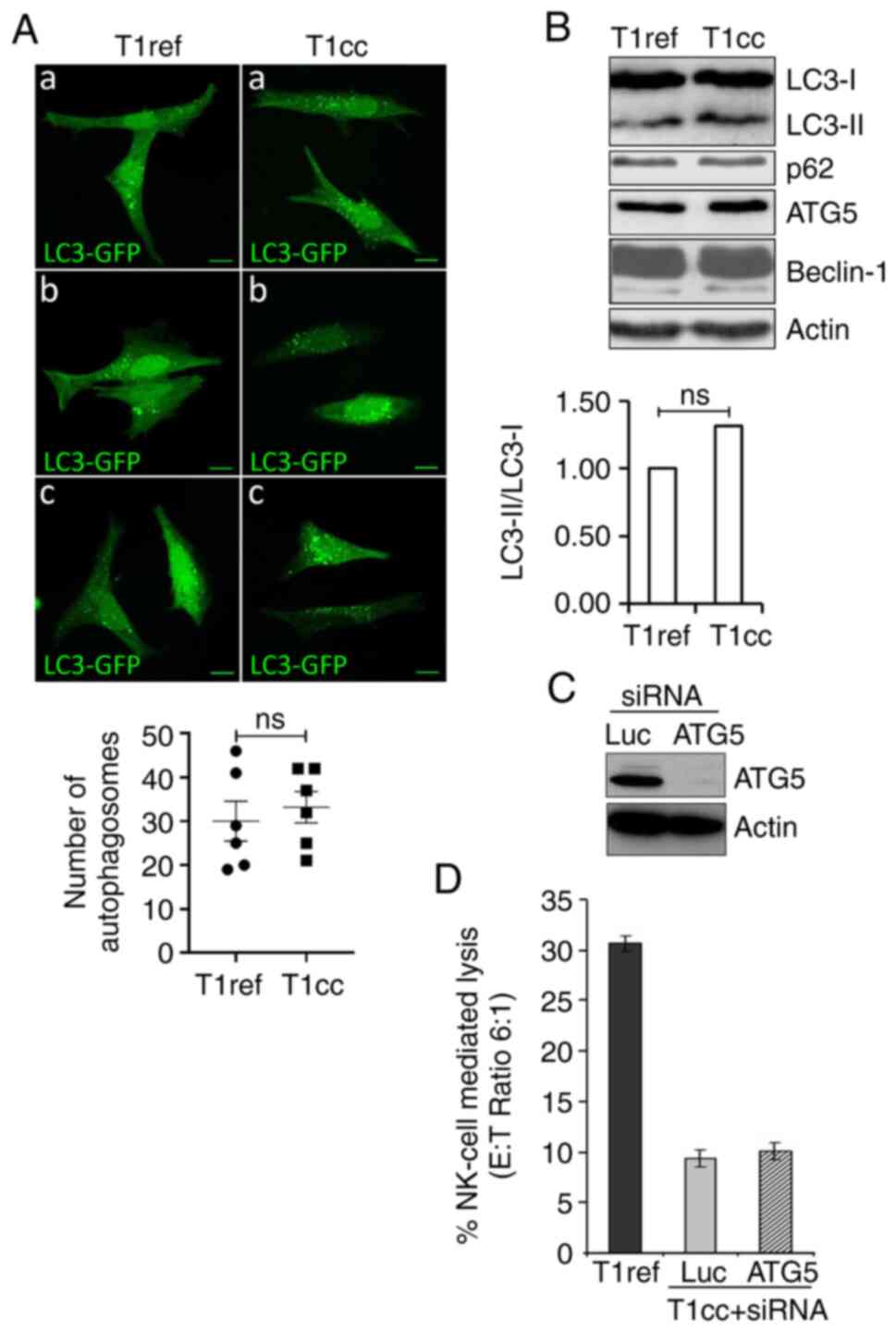
T1R cell resistance to NK
cell-mediated lysis is independent of autophagy induction
We have previously reported that autophagy can
protect tumor cells against NK cell- and CTL-mediated cytotoxicity
(27,28). Therefore, we sought to determine
whether the selection of resistant cells following NK cell pressure
involves activation of autophagy in these cells. The autophagy
level was first evaluated by following the distribution pattern of
the microtubule-associated protein-LC3 (referred to as LC3
hereafter) fused with a GFP tag (LC3-GFP). T1 and T1R cells were
transfected with a vector encoding the LC3-GFP fusion protein
before visualizing the autophagosomes. Confocal microscopy data
revealed a significant basal level of autophagy in T1 cells, with
punctate LC3-GFP staining, but no significant increase was observed
in T1R cells (Fig. 2A). This was
also supported by assessing the expression of the
phosphatidylethanolamine-conjugated form of LC3 (or LC3-II), which
is similar between T1 and T1R cells. Moreover, no difference in the
expression of other main autophagy-related proteins, such as p62,
ATG5 and Beclin-1 (involved in autophagosome formation and the
autophagy process) was observed between T1 and T1R cells (Fig. 2B). Finally, silencing of ATG5 using
siRNAs (Fig. 2C) had no impact on
T1R cell susceptibility to NK cell-mediated lysis (Fig. 2D). Taken together, these results
clearly indicated that, in our model, autophagy was not implicated
in T1R cell resistance to NK cell-mediated lysis.
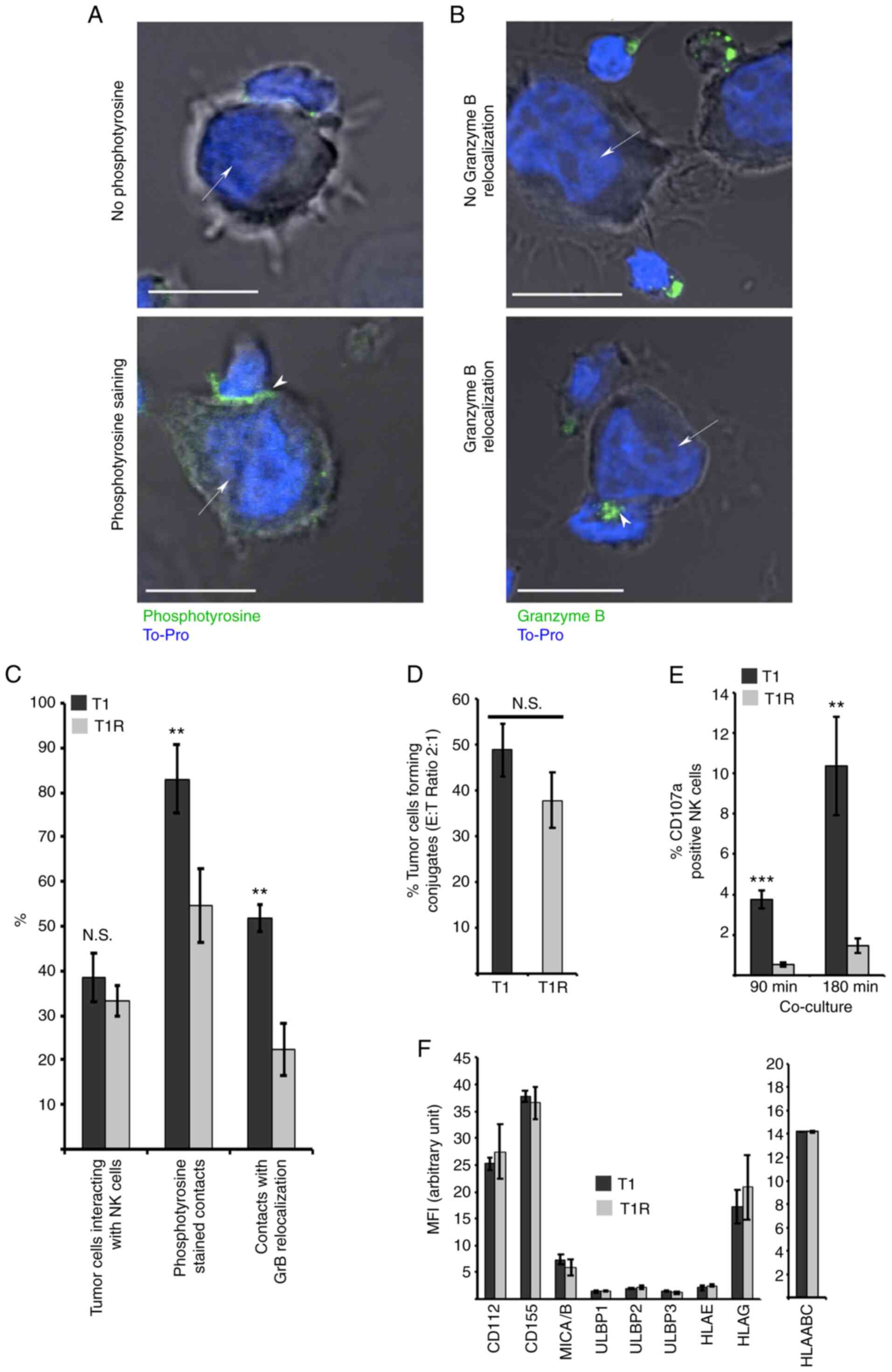
T1R cell resistance to NK
cell-mediated lysis is associated with an alteration of the
effector/target interaction
It was next investigated whether T1R cell resistance
to NK cell-mediated lysis was associated with alteration in the
target cell recognition process. For this purpose, the immune
synapse formation between NK and T1 or T1R cells was analyzed using
confocal microscopy. The percentage of tumor cells interacting with
NK cells, and the establishment of active immune synapses,
evaluated by both phosphotyrosine staining and GzmB relocalization
at the contact zone, was measured in at least 200 target cells from
each of the three independent batches of T1 and T1R cells. The data
revealed a mild decrease in the percentage of NK cells in contact
with T1R compared to T1 cells. More interestingly, a strong
decrease was observed in active immune synapse formation between
T1R and NK cells, compared with T1 control cells, as shown by the
decrease in phosphotyrosine staining and GzmB relocalization at the
immune synapse (Fig. 3A-C). To
corroborate those results, flow cytometry analysis was performed to
quantify the percentage of conjugate formation between NK and T1 or
T1R cells. The data depicted in Fig.
3D revealed a mild but non-statistically significant reduction
of contacts between T1R and NK cells as compared to T1 control
cells. In addition, using a degranulation assay based on CD107
externalization following target cell recognition leading to
cytotoxic granule exocytosis, it was demonstrated that T1R
triggering of CD107 externalization by NK cells was significantly
reduced in comparison to T1 parental cells (Fig. 3E). Finally, it was observed that
this alteration of the T1R cell recognition by NK cells occurred in
a KIR-, DNAM1- and NKG2D-independent manner, as the expression of
the ligands for these receptors (CD112, CD155, MICA/B, ULBP1-3,
HLA-E and HLA-G) was similar between the parental cells and their
resistant counterparts (Fig. 3F).
Taken together, these results indicated that T1R cell resistance to
NK cell-mediated lysis is, as least partly, dependent on defective
immune synapse signaling following effector/target conjugation, as
evidenced by the alteration of GzmB relocalization, phosphotyrosine
signaling and CD107 staining in NK cells interacting with T1R
cells.
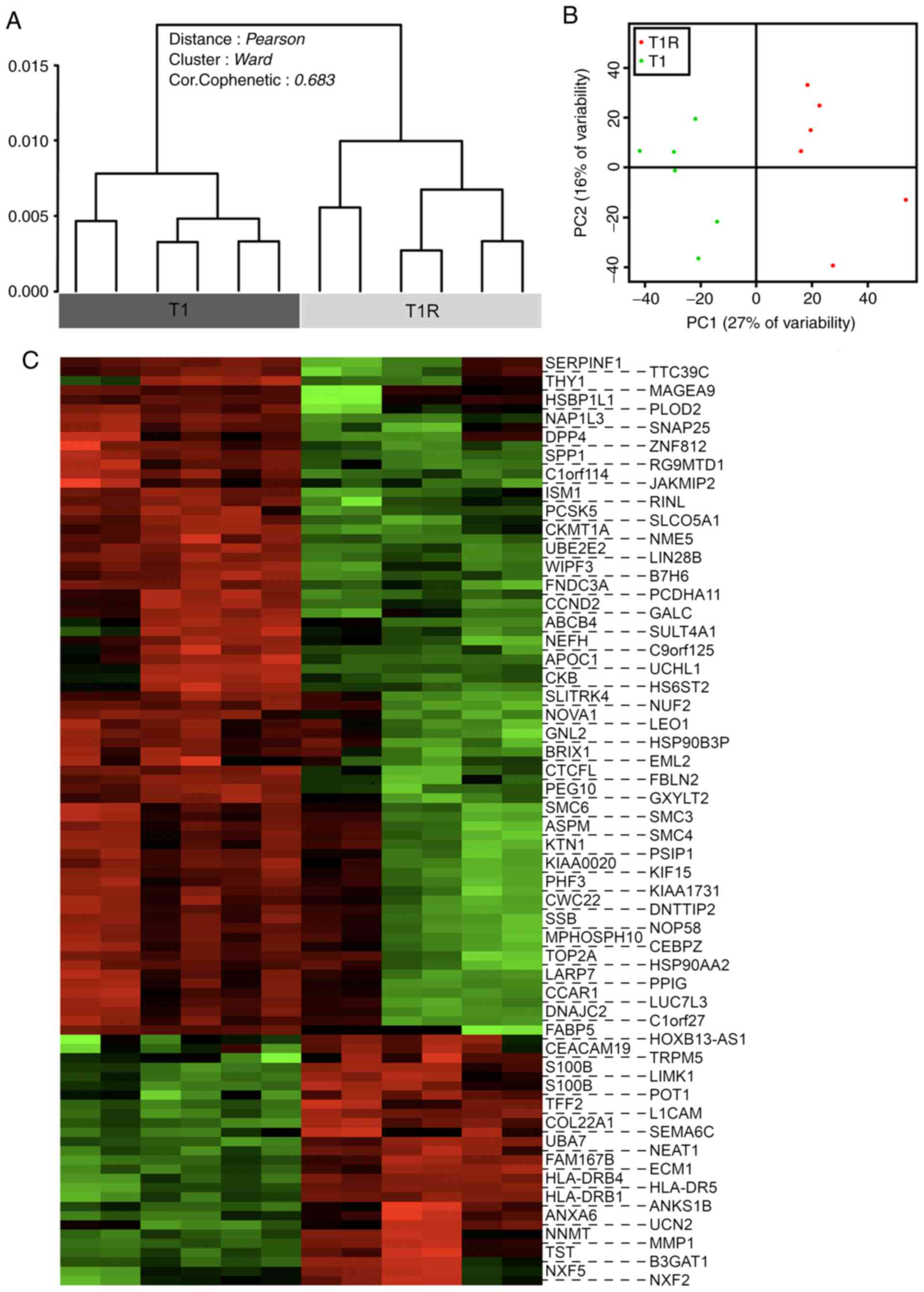
Impact of sustained NK cell pressure
on tumor cell transcriptional signature
A global transcriptional analysis comparing T1 and
T1R cells was next performed in order to evaluate the impact of NK
cell pressure on tumor cell behavior and to elucidate the putative
molecular basis of the NK cell-mediated lysis resistance
mechanisms. Transcriptional profiles of the three independent T1
and T1R series were compared by DNA microarray, performed in
duplicate. Dendrogram representation of the unsupervised analysis
of the whole data set revealed a significant discrimination between
the T1 and T1R cell populations (Fig.
4A). These data, confirmed by principal component analysis
(Fig. 4B), enabled us to define a
particular genomic signature of tumor cells subjected to sustained
NK cell pressure with robust statistical significance. A supervised
analysis was then performed to select differentially expressed
genes [absolute fold-change (FC) >2 and a corrected P-value
(FDR) <0.05]. The heatmap in Fig.
4C represents the 99 identified genes, including 26
overexpressed and 73 downregulated genes in T1R compared with T1
cells. As three independent biological replicates (all performed in
technical duplicates) were used for each condition, the impact of
variability in basal gene expression due to cell line heterogeneity
was reduced to a minimum. Therefore, the number of genes in this
signature is relatively limited. However, no main modifications of
functional gene groups or signaling pathways were significantly
detectable with DAVID or INGENUITY analysis tools (data not shown).
Similarly, no particular pathways were found to be significantly
enriched on Gene Set Enrichment Analysis (data not shown).
Nevertheless, among the overexpressed or
downregulated genes in T1R cells, several may be of interest,
either regarding the description of newly acquired characteristics
of migration and invasiveness in the T1R cell model, or regarding
their resistance to NK cell-mediated lysis. These overexpressed
genes include matrix metallopeptidase-1 (FC +7.561) involved in
extracellular matrix reorganization, in metastatic process and in
resistance to NK cell-mediated lysis (31). Trefoil factor 2 (FC +2.631) is
involved in cell migration and apoptosis regulation in
gastrointestinal mucosa. HLA-DRB5 (FC +2.381), -DRB4
(FC +2.287) and -DRB1 (FC +2.211) may be implicated in
resistance to NK cells, as previously described (32). More importantly, inhibition of
protection of telomeres protein 1 (POT1; FC +2.381) has been
demonstrated to increase apoptosis and to limit gastric cancer cell
proliferation (33). In our model,
POT1 overexpression, following NK cell pressure, may have
induced the emergence of cells more resistant to apoptosis.
Moreover, given that POT1 has been identified in a
high-scale screening of molecules affecting NK cell-mediated lysis
susceptibility (34), it may be a
potential candidate associated with resistance in our T1R model.
L1CAM (FC +2.321) encodes L1 cell adhesion molecule, which
is overexpressed in several types of cancer and contributes to
invasiveness, metastasis (35,36)
and apoptosis resistance (37).
S100B (FC +2.315) encodes S100 calcium-binding
protein B, which is involved in the regulation of various cellular
processes, such as cell cycle regulation or differentiation. Its
alteration has been implicated in melanoma cell proliferation and
metastatic progression (38).
ECM1 (FC +2.109) encodes extracellular matrix protein 1, the
overexpression of which has been shown to contribute to cancer cell
invasiveness (39). Finally,
CEACAM19 (FC +2.063), encoding carcinoembryonic
antigen-related cell adhesion molecule 19, may regulate NK
cell-mediated lysis, similar to CEACAM1 (40).
Regarding the downregulated genes in T1R cells,
ubiquitin carboxy-terminal hydrolase-L1 (UCHL1; FC −9.011)
was identified. UCHL1 encodes a deubiquitinating enzyme that
may play a proapoptotic role, and its downregulation may be
associated with an increase in cell survival (41). SERPINF1/PEDF (FC −3.313)
encodes pigment epithelium-derived factor, which is involved in
anti-angiogenic and anti-tumor activities. Therefore, its
downregulation in T1R cells may be associated with tumor-promoting
characteristics (42). Finally, our
data indicated downregulation of B7-H6 (FC −2,839), a
NKp30-activating ligand that may contribute to T1R cell resistance
to NK cell-mediated lysis. Thus, the present analysis revealed
putative contributors to T1R cell resistance to NK cell-mediated
lysis, as well as several genes involved in cell adhesion or
migration and invasiveness, strongly suggesting that NK cell
pressure may lead to the selection of more aggressive tumor
cells.
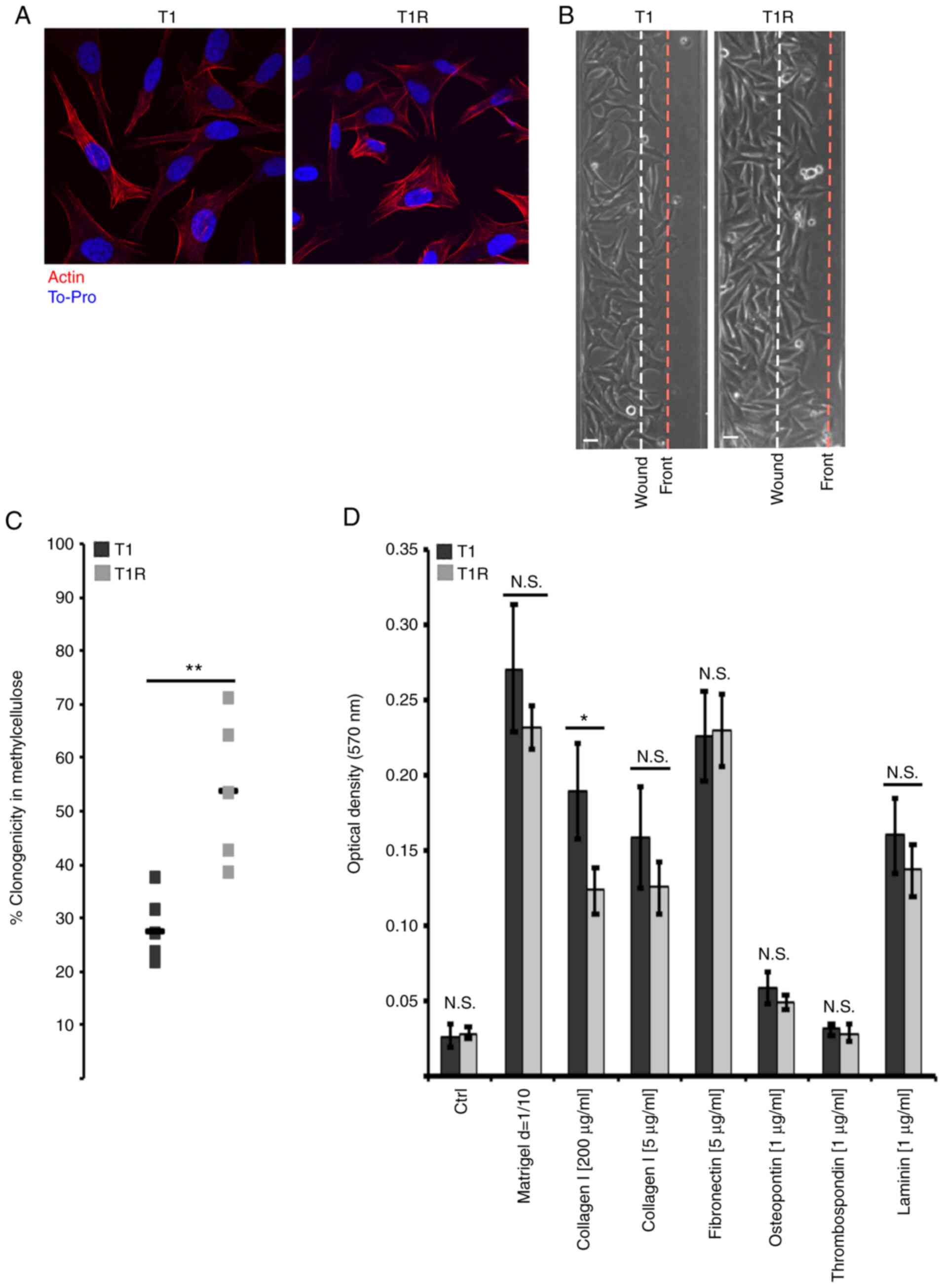
Effect of sustained NK cell pressure
on tumor cell phenotype
Tumor immunoediting is a phenomenon that may also
lead to tumor cell shaping through alteration of phenotypical
characteristics. As our transcriptomic analysis revealed putative
alteration of T1R cell adhesion, migration and invasiveness, their
migration ability was examined. First, no morphological changes
were observed by actin staining (Fig.
5A) that could contribute to migration or to an alteration of
target recognition and resistance to NK cell-mediated lysis
(6). As shown in Fig. 5B, T1R cells exhibited increased
migration ability compared with the T1 control cells, whereas the
proliferation rate was comparable between T1 and T1R cells (data
not shown). Anchorage-independent growth in methylcellulose was
also evaluated, and a significant increase in the clonogenic
ability of T1R cells was observed in comparison to T1 cells
(Fig. 5C). On the contrary,
adhesion assay on several matrices indicated a significant decrease
of T1R adhesion only to collagen at high concentration, whereas no
statistically significant differences were observed for other
proteins. Taken together, these results suggested that NK cell
pressure may lead to the selection of tumor cells exhibiting
phenotypical characteristics associated with a more aggressive
behavior.
Discussion
It has become clear that the host immune system is
involved in eliminating tumors, as well as in shaping the
immunogenic phenotypes of tumors that eventually form in
immunocompetent hosts, indicating that immunity plays a dual role
in the complex interactions between tumors and the host. Despite
some success with recent cancer immunotherapy approaches, the
majority of the patients do not respond to this type of treatment,
which is likely due to intrinsic tumor resistance that involves the
innate molecular qualities of the tumor inhibiting the antitumor
immune response. Several mechanisms have been proposed, including
the reduction in antigenic expression and the alteration of the
quality and number of immune effector cells in the tumor
microenvironment. The other major mechanism involves an acquired
resistance associated with several mechanisms by which tumor cells
develop resistance over the course of treatment, resulting in
cancer progression despite an initial response to immunotherapy.
This includes loss of T-cell function, lack of T-cell recognition
due to immunoediting, and the development of escape mutation
variant tumor cells.
Blurring the boundary between innate and adaptive
immune system, NK cells, a key component of innate immunity, are
recognized as potent anticancer mediators. Tumor cells may develop
several strategies to evade NK cell-mediated killing. In this
regard, the involvement of NK cells in immune editing has been
studied in relation to NKG2D and DNAM1 (21,43).
Guillerey and Smyth demonstrated the NK cell activity in the cancer
immune editing process, with particular emphasis on the elimination
and escape phases (44). NK cells
have been also shown to kill immature DCs due to their low amount
of surface HLA class I molecules (45) and, therefore, impact the quality of
adaptive immune response. These previous studies were undertaken in
an attempt to further unravel the involvement of NK cells in tumor
immunoediting and the emergence of tumor-resistant variants using a
melanoma model. It should be noted that tumor cells evading NK
cell-mediated lysis are well characterized, but few data are
available regarding the implication of NK cells in the selection of
such mechanisms.
The present study investigated the consequences of
sustained NK cell-mediated immune stress and demonstrated that NK
cells can contribute to immunoediting of the tumor, leading to
emergence of cytotoxic resistant variants by a selection process or
induction of tumor cell characteristics alteration. It was observed
that the established resistant variant T1R did not display
cross-resistance to autologous specific CTLs, suggesting that the
resistant cells may use different mechanisms to escape
cell-mediated cytotoxicity. In addition, no cross-resistance to
dacarbazin was observed, which emphasizes that NK cells do not
select tumor-resistant variants with an altered apoptotic signaling
pathway. A decrease was observed in the susceptibility to TNF-α
associated with a reduced TNF-R1 expression, which confirms the
putative broad effect of long-term sustained NK-cell selective
pressure, during which lysis pathways and the secreted molecules
may differ from 51Cr release standardized conditions. In
contrast to short-term 51Cr release, the death domain
receptor pathway may be used by NK cells in sustained co-culture
conditions, which may explain the decreased susceptibility to
TNF-α. Furthermore, it should be noted that, under 51Cr
release conditions, T1 and T1R NK cell-mediated lysis was
completely inhibited by concanamycin A (data not shown), indicating
that their lysis in such an assay is fully dependent on the
PFN/GzmB pathway.
Importantly, the data of the present study indicated
that the resistance of T1R cells to NK cell-mediated lysis was
associated with an alteration in immune synapse signaling. The
reduction of immune synapse signaling following effector/target
conjugation may be due to the loss in the expression of one of the
main KIR and NKG2D ligands (46),
as reported in the short 15 days without renewal of NK cell
population co-culture model described by Balsamo et al
(47). These investigators
demonstrated that melanoma cells co-cultured with NK cells could
induce, via IFN-γ, an increase of classical and non-classical MHC I
molecules and a decrease of NKG2D ligands. However, in our model,
no clear variation of these ligands, or MHC I and main KIR ligands,
was observed.
An interesting question raised by the present study
is whether the resistant variants pre-exist and/or adapt to NK
cell-mediated immune stress. It was hypothesized that both
selection and adaptation may be involved. However, further genetic,
transcriptomic and cell tracking analysis of the clones will be
required to address this question.
The transcriptomic analysis was performed using
three independent biological replicates for each condition, all
performed in technical duplicates. As evidenced by the relatively
low number of genes differentially expressed between the parental
T1 and T1R cells, this experimental design likely reduced much of
the basal variability in gene expression due to cell line
heterogeneity. Indeed, 99 genes were found to be differentially
expressed between the parental T1 and T1R cells, and no particular
pathways were found to be significantly enriched in subsequent
analyses (GSEA, DAVID or INGENUITY). Among these genes, reduced
expression of B7-H6 activator ligand of NKp30 was observed, which
may play a key role in the activation of NK cells by tumor cells
(46,48,49).
To the best of our knowledge, such an event in NK cell-mediated
selection has never been described to date, as the majority of
studies report NKG2D ligand reduction (47). Future studies should investigate
this possibility, as well as other putative mechanisms involved in
inhibiting immune synapse formation and signaling.
In the context of melanoma, NKG2D does not appear to
play a key role, and cytotoxic NK cell activity appears to be
preferentially triggered by DNAM1 and NCRs (50). T1R cell resistance may also involve
other molecules, and transcriptomic signature raises several
possibilities. For example, POT1 overexpression constitutes an
interesting candidate due to the fact that it has been identified
in a high-scale screening of molecules affecting NK cell- mediated
lysis susceptibility (34).
Importantly, the transcriptomic analysis also revealed the
acquisition of genes associated with pro-metastatic and
pro-invasive characteristics, which may reflect the aggressiveness
of tumor cells selected by NK cells. Measurements of
anchorage-independent growth ability as well as 2D migration
capacity tend to confirm this hypothesis.
Collectively, the results of the present study
demonstrated that NK cells are able to select tumor cells resistant
to their cytotoxic activity that exhibit enhanced tumor
aggressiveness characteristics. This should be considered in the
current immunotherapeutic strategies. Indeed, as regards melanoma,
the existence of numerous specific antigens has led to the
development of several immunotherapeutic strategies, but clinical
efficacy remains limited (51). As
melanoma cells often express low levels of MHC class I molecules
and a broad panel of NK receptor-activating ligands (52), NK cells represent a major
alternative strategy (50) and are
considered as key cytotoxic cells in adoptive antitumor
immunotherapies (53), but their
clinical benefits remain limited (52). Low level of tumor sites targeted by
adoptive transfer NK cells may explain the lack of efficiency. Some
targeting strategies may represent an interesting alternative
(54). In this context,
metalloproteinases have been reported to contribute to the ability
of NK cells to reach tumor sites (55,56).
Another obstacle in the efficiency of adoptive NK cell
immunotherapies is the immune escape mechanisms developed by tumor
cells. These processes may result from microenvironmental
modulations leading to immunosuppression disturbing NK cell
antitumor immune response through modification of activator and
inhibitor receptor expression levels (57–59).
In melanoma, indoleamine 2,3-dioxygenase and prostaglandin E2 have
been shown to target NKp30, NKp44 and NKG2D on NK cells, causing
loss of their activity (60).
Blockade of these pathways may increase the antitumor efficiency of
NK cells.
Tumor cell immune escape may also be due to
intrinsic resistance mechanisms, such as the loss of NK cell
activator ligands on tumor cells. Some treatments are being
developed to increase the expression of those ligands on tumor
cells in order to overcome this immune escape process (61–64).
All these data regarding NK cell homing and increased tumor cell
susceptibility through activator ligand re-expression or direct
stimulation of NK cells (65,66)
provide a rationale for the development of NK cell-based
immunotherapeutic strategies (67).
However, the sustained immune pressure of NK cells does not only
induce loss of expression of activating ligands, but may also
induce selection of tumor variants exhibiting aggressive
properties. This should be considered as a putative side effect of
such immunotherapies. We believe that the establishment of
combinatory strategies integrating NK cells together with other
cellular components and compounds, such as CD8+ T cells,
DCs, monoclonal antibodies or chemotherapeutics drugs, is essential
(65,68) for preventing tumor immunoediting and
the emergence of resistant aggressive variants.
The findings of the present study may provide
mechanistic insight into how NK cell pressure may lead to the
emergence of resistant variants. Future studies aimed at examining
whether such resistance occurs following NK-based cell therapy may
be key to the design of NK-based innovative cancer treatments.
Acknowledgements
The authors would like to thank Nathalie Droin
(Gustave Roussy, Genomics Platform) and Philippe Dessen (Gustave
Roussy, Bioinformatics Department) for their valuable help and
advice.
Funding
The present study was supported by grants from la
Ligue Contre le Cancer (EL2015.LNCC/SaC), Institut National Du
Cancer (INCa; Plan-Cancer), and ‘Action LIONS Vaincre le Cancer’
AUTOKIR project.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. The microarray data and protocols are available at the
European Molecular Biology Laboratory European Bioinformatics
Institute database (https://www.ebi.ac.uk/arrayexpress/) under accession
no. E-MTAB-8777.
Authors' contributions
Design of the study: TC and SC. Acquisition of data:
TC, BJ, GG, GM and DO. Data analysis and interpretation: TC, JT,
BJ, ST, GG, GM, CK, DO and SC. TC, JT, ST and SC drafted and
critically revised the article. All the authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Shankaran V, Ikeda H, Bruce AT, White JM,
Swanson PE, Old LJ and Schreiber RD: Pillars article: IFNγ and
lymphocytes prevent primary tumour development and shape tumour
immunogenicity. Nature. 2001. 410: 1107-1111. J Immunol.
201:827–831. 2018.PubMed/NCBI
|
|
2
|
Dunn GP, Old LJ and Schreiber RD: The
three Es of cancer immunoediting. Annu Rev Immunol. 22:329–360.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vesely MD, Kershaw MH, Schreiber RD and
Smyth MJ: Natural innate and adaptive immunity to cancer. Annu Rev
Immunol. 29:235–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dunn GP, Bruce AT, Ikeda H, Old LJ and
Schreiber RD: Cancer immunoediting: From immunosurveillance to
tumor escape. Nat Immunol. 3:991–998. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dunn GP, Old LJ and Schreiber RD: The
immunobiology of cancer immunosurveillance and immunoediting.
Immunity. 21:137–148. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abouzahr S, Bismuth G, Gaudin C, Caroll O,
Van Endert P, Jalil A, Dausset J, Vergnon I, Richon C, Kauffmann A,
et al: Identification of target actin content and polymerization
status as a mechanism of tumor resistance after cytolytic T
lymphocyte pressure. Proc Natl Acad Sci USA. 103:1428–1433. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Khong HT and Restifo NP: Natural selection
of tumor variants in the generation of ‘tumor escape’ phenotypes.
Nat Immunol. 3:999–1005. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trinchieri G: Biology of natural killer
cells. Adv Immunol. 47:187–376. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vivier E, Tomasello E, Baratin M, Walzer T
and Ugolini S: Functions of natural killer cells. Nature
Immunology. 9:503–510. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coca S, Perez-Piqueras J, Martinez D,
Colmenarejo A, Saez MA, Vallejo C, Martos JA and Moreno M: The
prognostic significance of intratumoral natural killer cells in
patients with colorectal carcinoma. Cancer. 79:2320–2328. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishigami S, Natsugoe S, Tokuda K, Nakajo
A, Che X, Iwashige H, Aridome K, Hokita S and Aikou T: Prognostic
value of intratumoral natural killer cells in gastric carcinoma.
Cancer. 88:577–583. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Villegas FR, Coca S, Villarrubia VG,
Jiménez R, Chillón MJ, Jareño J, Zuil M and Callol L: Prognostic
significance of tumor infiltrating natural killer cells subset CD57
in patients with squamous cell lung cancer. Lung Cancer. 35:23–28.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martín-Fontecha A, Thomsen LL, Brett S,
Gerard C, Lipp M, Lanzavecchia A and Sallusto F: Induced
recruitment of NK cells to lymph nodes provides IFN-γ for T(H)1
priming. Nat Immunol. 5:1260–1265. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morandi B, Mortara L, Chiossone L, Accolla
RS, Mingari MC, Moretta L, Moretta A and Ferlazzo G: Dendritic cell
editing by activated natural killer cells results in a more
protective cancer-specific immune response. PLoS One. 7:e391702012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moretta L, Ferlazzo G, Bottino C, Vitale
M, Pende D, Mingari MC and Moretta A: Effector and regulatory
events during natural killer-dendritic cell interactions. Immunol
Rev. 214:219–228. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kijima M, Saio M, Oyang G-F, Suwa T,
Miyauchi R, Kojima Y, Imai H, Nakagawa J, Nonaka K, Umemura N, et
al: Natural killer cells play a role in MHC class I in vivo
induction in tumor cells that are MHC negative in vitro. Int
J Oncol. 26:679–684. 2005.PubMed/NCBI
|
|
17
|
Farag SS, Fehniger TA, Ruggeri L, Velardi
A and Caligiuri MA: Natural killer cell receptors: New biology and
insights into the graft-versus-leukemia effect. Blood.
100:1935–1947. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ferlazzo G and Moretta L: Dendritic cell
editing by natural killer cells. Crit Rev Oncog. 19:67–75. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dunn GP, Bruce AT, Sheehan KCF, Shankaran
V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM
and Schreiber RD: A critical function for type I interferons in
cancer immunoediting. Nat Immunol. 6:722–729. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smyth MJ, Dunn GP and Schreiber RD: Cancer
immunosurveillance and immunoediting: The roles of immunity in
suppressing tumor development and shaping tumor immunogenicity. Adv
Immunol. 90:1–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smyth MJ, Swann J, Cretney E, Zerafa N,
Yokoyama WM and Hayakawa Y: NKG2D function protects the host from
tumor initiation. J Exp Med. 202:583–588. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Street SE, Hayakawa Y, Zhan Y, Lew AM,
MacGregor D, Jamieson AM, Diefenbach A, Yagita H, Godfrey DI and
Smyth MJ: Innate immune surveillance of spontaneous B cell
lymphomas by natural killer cells and gammadelta T cells. J Exp
Med. 199:879–884. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crowe NY, Smyth MJ and Godfrey DI: A
critical role for natural killer T cells in immunosurveillance of
methylcholanthrene-induced sarcomas. J Exp Med. 196:119–127. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takeda K, Smyth MJ, Cretney E, Hayakawa Y,
Kayagaki N, Yagita H and Okumura K: Critical role for tumor
necrosis factor-related apoptosis-inducing ligand in immune
surveillance against tumor development. J Exp Med. 195:161–169.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Smyth MJ, Crowe NY and Godfrey DI: NK
cells and NKT cells collaborate in host protection from
methylcholanthrene-induced fibrosarcoma. Int Immunol. 13:459–463.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ben Safta T, Ziani L, Favre L, Lamendour
L, Gros G, Mami-Chouaib F, Martinvalet D, Chouaib S and Thiery J:
Granzyme B-activated p53 interacts with Bcl-2 to promote cytotoxic
lymphocyte-mediated apoptosis. J Immunol. 194:418–428. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Noman MZ, Janji B, Kaminska B, Van Moer K,
Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib
F and Chouaib S: Blocking hypoxia-induced autophagy in tumors
restores cytotoxic T-cell activity and promotes regression. Cancer
Res. 71:5976–5986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Janji B, Viry E, Moussay E, Paggetti J,
Arakelian T, Mgrditchian T, Messai Y, Noman MZ, Van Moer K, Hasmim
M, et al: The multifaceted role of autophagy in tumor evasion from
immune surveillance. Oncotarget. 7:17591–17607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dufour E, Carcelain G, Gaudin C, Flament
C, Avril MF and Faure F: Diversity of the cytotoxic
melanoma-specific immune response: Some CTL clones recognize
autologous fresh tumor cells and not tumor cell lines. J Immunol.
158:3787–3795. 1997.PubMed/NCBI
|
|
30
|
Smyth GK: Linear models and empirical
bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article32004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Le Maux Chansac B, Missé D, Richon C,
Vergnon I, Kubin M, Soria JC, Moretta A, Chouaib S and Mami-Chouaib
F: Potentiation of NK cell-mediated cytotoxicity in human lung
adenocarcinoma: Role of NKG2D-dependent pathway. Int Immunol.
20:801–810. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Weichold FF, Jiang YZ, Dunn DE, Bloom M,
Malkovska V, Hensel NF and Barrett AJ: Regulation of a
graft-versus-leukemia effect by major histocompatibility complex
class II molecules on leukemia cells: HLA-DR1 expression renders
K562 cell tumors resistant to adoptively transferred lymphocytes in
severe combined immunodeficiency mice/nonobese diabetic mice.
Blood. 90:4553–4558. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wan SM, Tie J, Zhang YF, Guo J, Yang LQ,
Wang J, Xia SH, Yang SM, Wang RQ and Fang DC: Silencing of the
hPOT1 gene by RNA inference promotes apoptosis and inhibits
proliferation and aggressive phenotype of gastric cancer cells,
likely through up-regulating PinX1 expression. J Clin Pathol.
64:1051–1057. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bellucci R, Nguyen HN, Martin A, Heinrichs
S, Schinzel AC, Hahn WC and Ritz J: Tyrosine kinase pathways
modulate tumor susceptibility to natural killer cells. J Clin
Invest. 122:2369–2383. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hai J, Zhu CQ, Bandarchi B, Wang YH, Navab
R, Shepherd FA, Jurisica I and Tsao MS: L1 cell adhesion molecule
promotes tumorigenicity and metastatic potential in non-small cell
lung cancer. Clin Cancer Res. 18:1914–1924. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Meier F, Busch S, Gast D, Göppert A,
Altevogt P, Maczey E, Riedle S, Garbe C and Schittek B: The
adhesion molecule L1 (CD171) promotes melanoma progression. Int J
Cancer. 119:549–555. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stoeck A, Gast D, Sanderson MP, Issa Y,
Gutwein P and Altevogt P: L1-CAM in a membrane-bound or soluble
form augments protection from apoptosis in ovarian carcinoma cells.
Gynecol Oncol. 104:461–469. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Peric B, Zagar I, Novakovic S, Zgajnar J
and Hocevar M: Role of serum S100B and PET-CT in follow-up of
patients with cutaneous melanoma. BMC Cancer. 11:3282011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiong GP, Zhang JX, Gu SP, Wu YB and Liu
JF: Overexpression of ECM1 contributes to migration and invasion in
cholangiocarcinoma cell. Neoplasma. 59:409–415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stern N, Markel G, Arnon TI, Gruda R, Wong
H, Gray-Owen SD and Mandelboim O: Carcinoembryonic antigen (CEA)
inhibits NK killing via interaction with CEA-related cell adhesion
molecule 1. J Immunol. 174:6692–6701. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang WJ, Li QQ, Xu JD, Cao XX, Li HX, Tang
F, Chen Q, Yang JM, Xu ZD and Liu XP: Over-expression of ubiquitin
carboxy terminal hydrolase-L1 induces apoptosis in breast cancer
cells. Int J Oncol. 33:1037–1045. 2008.PubMed/NCBI
|
|
42
|
Wu QJ, Gong CY, Luo ST, Zhang DM, Zhang S,
Shi HS, Lu L, Yan HX, He SS, Li DD, et al: AAV-mediated human PEDF
inhibits tumor growth and metastasis in murine colorectal
peritoneal carcinomatosis model. BMC Cancer. 12:1292012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Iguchi-Manaka A, Kai H, Yamashita Y,
Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya
K and Shibuya A: Accelerated tumor growth in mice deficient in
DNAM-1 receptor. J Exp Med. 205:2959–2964. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guillerey C and Smyth MJ: NK cells and
cancer immunoediting. Curr Top Microbiol Immunol. 395:115–145.
2016.PubMed/NCBI
|
|
45
|
Ferlazzo G, Tsang ML, Moretta L, Melioli
G, Steinman RM and Münz C: Human dendritic cells activate resting
natural killer (NK) cells and are recognized via the NKp30 receptor
by activated NK cells. J Exp Med. 195:343–351. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Brandt CS, Baratin M, Yi EC, Kennedy J,
Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, et
al: The B7 family member B7-H6 is a tumor cell ligand for the
activating natural killer cell receptor NKp30 in humans. J Exp Med.
206:1495–1503. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Balsamo M, Vermi W, Parodi M, Pietra G,
Manzini C, Queirolo P, Lonardi S, Augugliaro R, Moretta A,
Facchetti F, et al: Melanoma cells become resistant to
NK-cell-mediated killing when exposed to NK-cell numbers compatible
with NK-cell infiltration in the tumor: Innate immunity. Eur J
Immunol. 42:1833–1842. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fiegler N, Textor S, Arnold A, Rölle A,
Oehme I, Breuhahn K, Moldenhauer G, Witzens-Harig M and Cerwenka A:
Downregulation of the activating NKp30 ligand B7-H6 by HDAC
inhibitors impairs tumor cell recognition by NK cells. Blood.
122:684–693. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kaifu T, Escalière B, Gastinel LN, Vivier
E and Baratin M: B7-H6/NKp30 interaction: A mechanism of alerting
NK cells against tumors. Cell Mol Life Sci. 68:3531–3539. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lakshmikanth T, Burke S, Ali TH, Kimpfler
S, Ursini F, Ruggeri L, Capanni M, Umansky V, Paschen A, Sucker A,
et al: NCRs and DNAM-1 mediate NK cell recognition and lysis of
human and mouse melanoma cell lines in vitro and in vivo. J Clin
Invest. 119:1251–1263. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fang L, Lonsdorf AS and Hwang ST:
Immunotherapy for advanced melanoma. J Invest Dermatol.
128:2596–2605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sutlu T and Alici E: Natural killer
cell-based immunotherapy in cancer: Current insights and future
prospects. J Intern Med. 266:154–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ljunggren HG and Malmberg KJ: Prospects
for the use of NK cells in immunotherapy of human cancer. Nat Rev
Immunol. 7:329–339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pachynski RK, Zabel BA, Kohrt HE, Tejeda
NM, Monnier J, Swanson CD, Holzer AK, Gentles AJ, Sperinde GV,
Edalati A, et al: The chemoattractant chemerin suppresses melanoma
by recruiting natural killer cell antitumor defenses. J Exp Med.
209:1427–1435. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Albertsson PA, Basse PH, Hokland M,
Goldfarb RH, Nagelkerke JF, Nannmark U and Kuppen PJK: NK cells and
the tumour microenvironment: Implications for NK-cell function and
anti-tumour activity. Trends Immunol. 24:603–609. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim MH, Kitson RP, Albertsson P, Nannmark
U, Basse PH, Kuppen PJ, Hokland ME and Goldfarb RH: Secreted and
membrane-associated matrix metalloproteinases of IL-2-activated NK
cells and their inhibitors. J Immunol. 164:5883–5889. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Balsamo M, Scordamaglia F, Pietra G,
Manzini C, Cantoni C, Boitano M, Queirolo P, Vermi W, Facchetti F,
Moretta A, et al: Melanoma-associated fibroblasts modulate NK cell
phenotype and antitumor cytotoxicity. Proc Natl Acad Sci USA.
106:20847–20852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Drake CG, Jaffee E and Pardoll DM:
Mechanisms of immune evasion by tumors. Adv Immunol. 90:51–81.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rabinovich GA, Gabrilovich D and Sotomayor
EM: Immunosuppressive strategies that are mediated by tumor cells.
Annu Rev Immunol. 25:267–296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pietra G, Manzini C, Rivara S, Vitale M,
Cantoni C, Petretto A, Balsamo M, Conte R, Benelli R, Minghelli S,
et al: Melanoma cells inhibit natural killer cell function by
modulating the expression of activating receptors and cytolytic
activity. Cancer Res. 72:1407–1415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai
Z, Mann HH, Strong RK, Groh V and Spies T:
Disulphide-isomerase-enabled shedding of tumour-associated NKG2D
ligands. Nature. 447:482–486. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Soriani A, Zingoni A, Cerboni C, Iannitto
ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C,
Petrucci MT, Guarini A, et al: ATM-ATR-dependent up-regulation of
DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic
agents results in enhanced NK-cell susceptibility and is associated
with a senescent phenotype. Blood. 113:3503–3511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Valés-Gómez M, Chisholm SE, Cassady-Cain
RL, Roda-Navarro P and Reyburn HT: Selective induction of
expression of a ligand for the NKG2D receptor by proteasome
inhibitors. Cancer Res. 68:1546–1554. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Waldhauer I, Goehlsdorf D, Gieseke F,
Weinschenk T, Wittenbrink M, Ludwig A, Stevanovic S, Rammensee H-G
and Steinle A: Tumor-associated MICA is shed by ADAM proteases.
Cancer Res. 68:6368–6376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Burke S, Lakshmikanth T, Colucci F and
Carbone E: New views on natural killer cell-based immunotherapy for
melanoma treatment. Trends Immunol. 31:339–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Veuillen C, Aurran-Schleinitz T,
Castellano R, Rey J, Mallet F, Orlanducci F, Pouyet L, Just-Landi
S, Coso D, Ivanov V, et al: Primary B-CLL resistance to NK cell
cytotoxicity can be overcome in vitro and in vivo by priming NK
cells and monoclonal antibody therapy. J Clin Immunol. 32:632–646.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chouaib S, Pittari G, Nanbakhsh A, El
Ayoubi H, Amsellem S, Bourhis JH and Spanholtz J: Improving the
outcome of leukemia by natural killer cell-based immunotherapeutic
strategies. Front Immunol. 5:952014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Terme M, Ullrich E, Delahaye NF, Chaput N
and Zitvogel L: Natural killer cell-directed therapies: Moving from
unexpected results to successful strategies. Nat Immunol.
9:486–494. 2008. View
Article : Google Scholar : PubMed/NCBI
|