Introduction
Malignant melanoma represents the most aggressive
and deadliest form of skin cancer. It is responsible for 1.7% of
newly diagnosed primary malignant cancers and leads to about 0.7%
of all cancer-related deaths worldwide each year (1). Almost 50% of metastatic melanoma
patients harbor a BRAF V600 mutation, among which the most common
is a single nucleotide mutation resulting in the substitution of
glutamic acid for valine (BRAF V600E) (2,3). BRAF
V600E leads to a constitutive activation of the mitogen-activated
protein kinase (MAPK)/extracellular signal-regulated kinase (ERK)
pathway, as well as to insensitivity to negative feedback
mechanisms (4). In recent years, the
use of molecular inhibitors targeting mutant BRAF and its
downstream effector MEK became a valid anti-melanoma therapeutic
strategy. The first approved selective inhibitor of mutant BRAF was
vemurafenib (PLX4032), which demonstrated highly beneficial early
therapeutic effects (5).
Unfortunately, a complete response is rarely observed due to either
acquired or intrinsic resistance to the therapy (6,7). The
combinational treatment with BRAF and MEK inhibitors resulted in an
improved activity, but acquired resistance to the BRAF inhibitor
remains the major obstacle to an effective cure of metastatic
melanoma (8–10). Increasing evidence suggests that in
addition to acquired mutations that confer drug resistance,
phenotype plasticity is a key mechanism that allows for adaptation
of melanoma cells to the drug and for drug resistance (11–14). Drug
exposure - along with other unfavorable environmental conditions,
such as hypoxia and nutrient starvation - leads to an early innate
cell response in melanoma cells, resulting in multidrug resistance
in so-called induced drug-tolerant cells (IDTCs) (15). The phenotype of IDTCs is reversible
upon drug withdrawal. However, continuous exposure to the treatment
eventually leads to transition of IDTCs into permanently resistant
tumor cells.
The lack of optimal treatment options for patients
with metastatic melanoma urged the cancer research community to
look for novel strategies aiming at subverting eventual drug
resistance and enhancing therapeutic efficacy. Targeting specific
molecules, involved in multiple signaling pathways in melanoma
cells and in several cellular functions important for cancer
spreading, has the potential to improve clinical outcomes.
One such candidate target molecule is chondroitin
sulfate proteoglycan 4 (CSPG4) (16,17).
Structurally, CSPG4 is a single-pass type I transmembrane protein
expressed as an either N-linked glycoprotein of approximately 280
kDa, or as an approximately 450 kDa chondroitin sulfate
proteoglycan (18,19). The core protein of CSPG4 contains
three major structural domains: A large, 2221-amino acid
extracellular domain, a 25-amino acid hydrophobic transmembrane
region, and a short 75-amino acid cytoplasmic tail (20). The extracellular region of CSPG4
contains the sequences for the modification with chondroitin
sulfate (CS) chains that may confer different attributes of the
cell (21–23). In the cytoplasmic domain of CSPG4,
there are sites critical for CSPG4 function: The threonine
phosphoacceptor sites phosphorylated by PKCα and ERK1/2, the PDZ
domain binding motif that is the site for attachment of scaffold
proteins, such as MUPP1, syntenin and GRIP1, and a proline-rich
region (PRR) comprising a non-canonical SH3 protein interaction
domain (16).
CSPG4 does not exhibit any known intrinsic catalytic
activity (20). It functions as a
scaffold protein, activating two major signaling pathways
associated with oncogenic transformation, in particular the
integrin-regulated FAK pathway and RTK signaling through the MAPK
cascade, with downstream ERK1/2 (20). Activation of these pathways by CSPG4
leads to the regulation of a number of cellular functions that
drive cytoskeletal reorganization, survival and chemoresistance,
invasion, migration and proliferation, as well as epithelial to
mesenchymal transition (EMT) in the radial growth phase of human
melanomas (24,25).
Although CSPG4 received much attention as a
potential therapeutic agent in recent years (16,17,26–31),
there is little information concerning the mechanisms that regulate
the expression of this proteoglycan (32). To the best of our knowledge, no study
has investigated whether CSPG4 expression is altered by inhibiting
signaling through the MAPK pathway.
In the present study, we demonstrated that a panel
of induced-drug tolerant and drug-resistant melanoma cells exhibit
lower levels of CSPG4 than the parental cells. Furthermore,
exposure of the cells to the BRAF inhibitor PLX4032 resulted in
markedly reduced levels of the CSPG4 protein in cell lysates, as
well as decreased levels of its mRNA, with no increase in protein
shedding into the culture supernatant. In addition, patient-derived
tumor samples pre- and post-treatment with kinase inhibitors showed
decreased numbers of CSPG4-positive cells after therapy. Our
results indicate that BRAF and MEK inhibition can be regulatory
factors of CSPG4 expression.
Materials and methods
Cell lines
The human melanoma cell line M14 was described
previously (29) and the human
melanoma cell lines WM9, WM35, WM164, WM1366, CJM, D24 and 451Lu
were provided by Helmut Schaider's laboratory (University of
Queensland, Australia). Cells were maintained in RPMI-1640 medium
with 2 mM L-glutamine and 25 mM HEPES (Lonza Group, Ltd.),
supplemented with either 10% FBS (M14, CJM and D24) or 5% FBS (WM9,
WM35, WM164, WM1366 and 451Lu) and 1% penicillin-streptomycin
(Gibco, Thermo Fischer Scientific, Inc.). Cells were cultured in a
humidified atmosphere containing 5% CO2 and 95% ambient
air at 37°C. The cell lines WM9, WM35, WM164, 451Lu and M14 were
BRAF V600-mutated, CJM and WM1366 were NRAS Q61L-mutated, and D24
carried neither BRAF nor NRAS mutations. Prior to the experiments,
all cell lines were tested negative for mycoplasma.
Antibodies
The mouse anti-CSPG4 monoclonal antibody clone
9.2.27 (#554275) was purchased from BD Biosciences. The donkey
anti-mouse Alexa Fluor 488® (#A-21202) and goat
anti-mouse Alexa Fluor 568® (#A-11004) secondary IgG
antibodies were obtained from Life Technologies Corporation.
Anti-CD271 (LNGFR) APC-conjugated human monoclonal antibody
(#130-091-884) was purchased from MACS Miltenyi Biotec. The APC
mouse IgG1, κ isotype control (FC) antibody (#400122) was obtained
from BioLegend, Inc. The mouse monoclonal antibodies anti-Ki67
(sc-23900), as well as mAbs against CSPG4: Anti-NG2 clone G-9
(sc-166251) and anti-NG2 clone LHM 2 (sc-53389), were purchased
from Santa Cruz Biotechnology, Inc. The primary anti-human CSPG4
antibody clone 132.38 (#ab50009) was purchased from Abcam. Control
mouse IgG (#I8765) was obtained from Sigma-Aldrich (Merck KGaA).
The mouse monoclonal antibodies anti-p44/42 MAPK (Erk1/2) clone
L34F12 (#4696), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204)
clone E10 (#9106), anti-Akt (pan) clone 40D4 (#2920),
anti-phospho-Akt (Ser473) clone 587F11 (#4051) and anti-β-actin
clone 8H10D10 (#3700) were purchased from Cell Signaling
Technology, Inc. Anti-mouse IgG, HRP-linked secondary antibody
(#7076) was obtained from Cell Signaling Technology, Inc.
Inhibitors and cell treatment
Inhibitors of mutant BRAF V600, vemurafenib
(PLX4032) and dabrafenib (GSK2118436) and the MEK1/2 inhibitor
trametinib (GSK1120212) were purchased from Selleckchem. Induced
drug tolerant cells (IDTCs) were generated by exposure to PLX4032
(250 and 1,000 nM) for a minimum of 7 days. Drug resistant cells
were obtained by exposure to PLX4032 (250 and 1,000 nM) for a
minimum of 30 days. To confirm multidrug resistance of the cells,
either additional MEK1/2 inhibitor GSK1120212 was applied (5 nM) or
PLX4032 concentration was increased up to 10 µM. The media
containing fresh drugs were replenished every third day for the
period of the experiments.
In order to determine suboptimal doses of PLX4032
for each cell line, a CytoSelect™ MTT Cell Proliferation Assay
(Cell Biolabs, Inc.) was performed according to the manufacturer's
instructions. Briefly, cells were seeded in triplicates at a
density of 6,000 per well in 96-well plates and subjected to the
following concentrations of PLX4032: 0, 0.1, 1, 10, 100, 250, 500,
1,000, 5,000 and 10,000 nM for 72 h. Cells were then incubated with
MTT reagent and solubilized. Absorbance was measured at 540 nm
using a Spark® multimode microplate reader (Tecan Group
Ltd.). Data are presented as percent of inhibition of
PLX4032-exposed cells compared to the untreated cells.
Flow cytometry
Parental, induced-drug tolerant and drug resistant
melanoma cells were harvested by scraping, washed with 1X PBS and
dispensed into 5 ml polystyrene round-bottom FACS tubes. Cells were
then incubated with Fixable Viability Dye eFluor® 506
(Affymetrix, eBioscience) according to the manufacturer's protocol.
Next, the cells were washed with FACS buffer [0.5% BSA and 0.05%
sodium azide (NaN3) in 1X PBS] and incubated with
anti-CSPG4 antibody 9.2.27 (1:1,000) for 10 min at 4°C, washed with
FACS buffer and incubated with donkey anti-mouse secondary IgG
antibodies Alexa Fluor 488® (1:500) for 15 min at 4°C,
protected from light. As an IgG control, cells were incubated with
Alexa Fluor 488® secondary antibody only. For additional
CD271 staining, the cells were incubated with anti-CD271-APC
antibody (1:100) for 15 min at 4°C, protected from light. As a
control, cells were incubated with the APC mouse IgG1, κ isotype
antibody (1:100). Cells were washed and resuspended in FACS buffer.
The samples were analyzed with a FACS Canto flow cytometer (BD
Biosciences). FlowJo software version 10.6.1 (TreeStar Inc.) was
used for analysis of the results.
Immunofluorescence
Melanoma cells were plated into 24-well cultivation
plates with glass coverslips and were allowed to attach overnight.
Cells were exposed to 1,000 nM PLX4032 for up to 7 days. At
specific time points, indicated in the results, cells were washed
with 1X PBS and fixed with 4% paraformaldehyde solution in 1X PBS
for 30 min at room temperature (RT). The slides were washed with 1X
PBS and the cells were blocked and permeabilized in blocking buffer
(1X PBS containing 5% goat serum and 0.5% saponine) for 1 h at RT.
Cells were incubated for 1 h at RT with anti-CSPG4 antibody 9.2.27,
diluted 1:1,000 in blocking buffer, washed three times with 1X PBS
and incubated for 1 h at RT with goat anti-mouse secondary IgG
antibodies Alexa Fluor 568®, diluted 1:2,000 in blocking
buffer. Finally, nuclei of the cells were counterstained with DAPI
(1:10,000), slides were washed three times with 1X PBS and mounted
on a glass slide using Fluoromount-G (Thermo Fisher Scientific,
Inc.). A wide-field fluorescence microscope (Imager Z1/Zeiss) in
combination with TissueFAXS software version 4.2.6245.1019
(TissueGnostic GmbH) was used to examine the slides.
RNA extraction and quantitative
reverse transcription-PCR
Total RNA from non-treated and drug-exposed melanoma
cells was isolated using an RNeasy Mini Kit (Qiagen) according to
the manufacturer's protocol. cDNA was synthetized from 1 µg of
total RNA using a High-Capacity cDNA Reverse Transcription kit
(Applied Biosystems, Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. The following primers were used (5′-3′):
CSPG4_F, CCTCCTGCTGCAGCTCTACT and CSPG4_R, CTGAGGAGGCGTTCAGAAAC;
GAPDH_F, ACGGATTTGGTCGTATTGGG and GAPDH_R, TGATTTTGGAGGGATCTCGC;
hUBC_F, ATTTGGGTCGCAGTTCTTG and hUBC_R, TGCCTTGACATTCTCGATGGT.
RT-qPCR analysis of CSPG4 and GAPDH or hUBC as
an internal standards were performed on a 7900HT Fast-Real Time PCR
System using the Power SYBR® Green PCR Master Mix
according to manufacturer's instructions (Applied Biosystems,
Thermo Fisher Scientific, Inc.). The thermocycling conditions were
as follows: Denaturation at 95°C for 10 min, followed by 40 cycles
of 95°C for 15 sec and 60°C for 1 min, and the melting curve stage
at 95°C for 15 sec, 60°C for 15 sec, and 95°C for 15 sec. The
results were analyzed using the Sequence Detection Systems (SDS)
software version 2.4 (Applied Biosystems, Thermo Fisher Scientific,
Inc.) and relative gene expression levels were calculated as ΔCT.
CSPG4 expression in melanoma cell lines was calculated as
100/ΔCT relative to GAPDH. Fold change expression of
CSPG4 after treatment was calculated using the
2−∆∆Cq method (33).
Western blot analysis
Non-treated and drug-exposed melanoma cells were
harvested by scraping and cell pellets were lysed in 1X RIPA buffer
(Sigma-Aldrich, Merck KGaA) with 1X Protease/Phosphatase Inhibitor
Cocktail (Cell Signaling Technology, Inc.). Lysates were incubated
with Chondroitinase ABC (Sigma-Aldrich, Merck KGaA) at the working
concentration 1 U/ml for 30 min at 37°C. Protein concentration in
cell lysates was measured by Pierce™ BCA Protein Assay Kit (Thermo
Fisher Scientific, Inc.) and equal amounts of proteins were
separated by SDS-PAGE (8% polyacrylamide gel) under reducing
conditions and transferred onto polyvinylidene fluoride (PVDF)
membranes (GE Healthcare Life Sciences, Thermo Fisher
Scientific).
Equal volumes of supernatants of non-treated and
drug-exposed melanoma cells were collected and concentrated eight
times (from 400 to 50 µl) using a Vacuum Concentrator Centrifuge
UNIVAPO 150 ECH (UniEquip GmbH). Next, 10 µl of concentrated
supernatants were centrifuged at 14,000 × g for 30 min at 4°C to
remove remaining aggregates. Five microliters of resulting
supernatants were carefully collected and mixed 1:1 with
ddH2O and with 4X reducing sample buffer. Samples were
then separated by SDS-PAGE (6% polyacrylamide gel) under reducing
conditions and transferred onto polyvinylidene fluoride (PVDF)
membranes (GE Healthcare Life Sciences, Thermo Fisher Scientific,
Inc.).
Membranes were blocked in 5% milk TBS-T for 1 h at
RT and incubated with primary antibodies overnight at 4°C. The
following dilutions of primary antibodies in 2% milk TBS-T were
used: Anti-Ki67 (1:500), anti-NG2 clone G-9 (1:1,000), anti-NG2
clone LHM 2 (1:800), anti-Erk1/2 (1:2,000), anti-phospho-Erk1/2
(1:2,000), anti-Akt (1:3,000), anti-phospho-Akt (1:1,000) and
anti-β-actin (1:1,000). Corresponding peroxidase-conjugated
secondary mAbs were used (1:5,000). Blots were developed using the
Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific,
Inc.) and bands were visualized using the ChemiDoc Imaging System
(Bio-Rad Laboratories, Inc.). The densitometric analysis of the
intensity of the bands was performed using the ImageJ software
(National Institutes of Health).
Immunohistochemistry
Formalin-fixed paraffin-embedded matched tumor
samples from five patients before and after progression during a
therapy with BRAF/MEK inhibitors from the archives of the
Department of Dermatology and the Department of Pathology at the
University Hospital St. Poelten, Karl Landsteiner University of
Health Sciences were processed. The collection and storage of
samples were performed according to local ethical guidelines. The
study was conducted in accordance with the Declaration of Helsinki
and was approved by the Ethics Committee of the Karl Landsteiner
University (EC number: 1011/2019). The tissue was deparaffinized
and stained using the BenchMark XT automated immune-staining
platform (Ventana Medical System) and the ultraView Universal DAB
detection system (Ventana Medical System). Antigen retrieval was
performed before using prediluted, commercially available
anti-CSPG4 antibodies (Abcam clone 132.38, dilution 1:500). Scoring
of tissue slides was performed independently by 2 investigators.
The percentage of positive cells and the intensity of staining were
graded from 0 to 3+: 0, no staining; 1+, weak positive staining;
2+, moderate positive staining; 3+, strong positive staining.
H&E staining was performed on 5-µm paraffin sections. The
stained sections were examined using an Olympus BX53 microscope and
photographed with an Olympus DP73 camera (Olympus Electronics).
Statistical analysis
Statistical analyses of CSPG4 expression in parental
and PLX4032-exposed melanoma cells in western blotting and RT-qPCR
as well as of ratios of pERK/ERK and pAKT/AKT in parental and
PLX4032-exposed melanoma cells was carried out using the one-way
ANOVA with Tukey's multiple comparison test. P-values <0.01 were
regarded as significant (***P<0.0001, **P<0.001, *P<0.01).
The Spearman's correlation test was used to determine the
association between cell-based CSPG4 expression levels and CSPG4
ectodomain shedding levels. The results of the densitometric
analyses of specific western blots were used for the calculation.
P-value <0.05 was considered statistically significant. All
statistical analyses were performed using GraphPad Prism software
version 4.03 (GraphPad Software, Inc.).
Results
Expression of CSPG4 in human melanoma
cell lines
The protein and gene expression of CSPG4 was
evaluated in a panel of melanoma cell lines: BRAF V600E-mutated
(WM9, WM35, 451Lu, WM164 and M14), NRAS Q61L-mutated (CJM, WM1366)
or carrying neither a BRAF nor an NRAS mutation (D24). CSPG4
protein expression on the cell surface was assessed by flow
cytometry using the CSPG4-specific 9.2.27 mAb. Six out of eight
cell lines tested showed a high percentage (>98%) of
CSPG4-positive cells (WM9, WM35, 451Lu, WM164, D24 and CJM)
(Fig. 1A). In the WM1366 cell line, a
lower percentage of cells (~84%) was recognized by the anti-CSPG4
antibody. In contrast, CSPG4 was not detected on the surface of M14
cells (Fig. 1A). Western blot
analysis (Fig. 1B) confirmed these
results, by demonstrating expression of CSPG4 in all tested cell
lines, except in M14. In WM9, WM35, 451Lu, WM164 and CJM cells, two
major forms of CSPG4 were detected: A single, non-chondroitin
sulfated glycoprotein of approximately 280 kDa (CSPG4-280), and a
450 kDa chondroitin sulfated proteoglycan (CSPG4-450). The D24 cell
line demonstrated expression of an additional, lighter band of
around 250 kDa, along with the 280 and 450 kDa forms of CSPG4. In
WM1366 cells, CSPG4 was detected as a single protein of 250 kDa,
which was not influenced by chondroitinase ABC treatment.
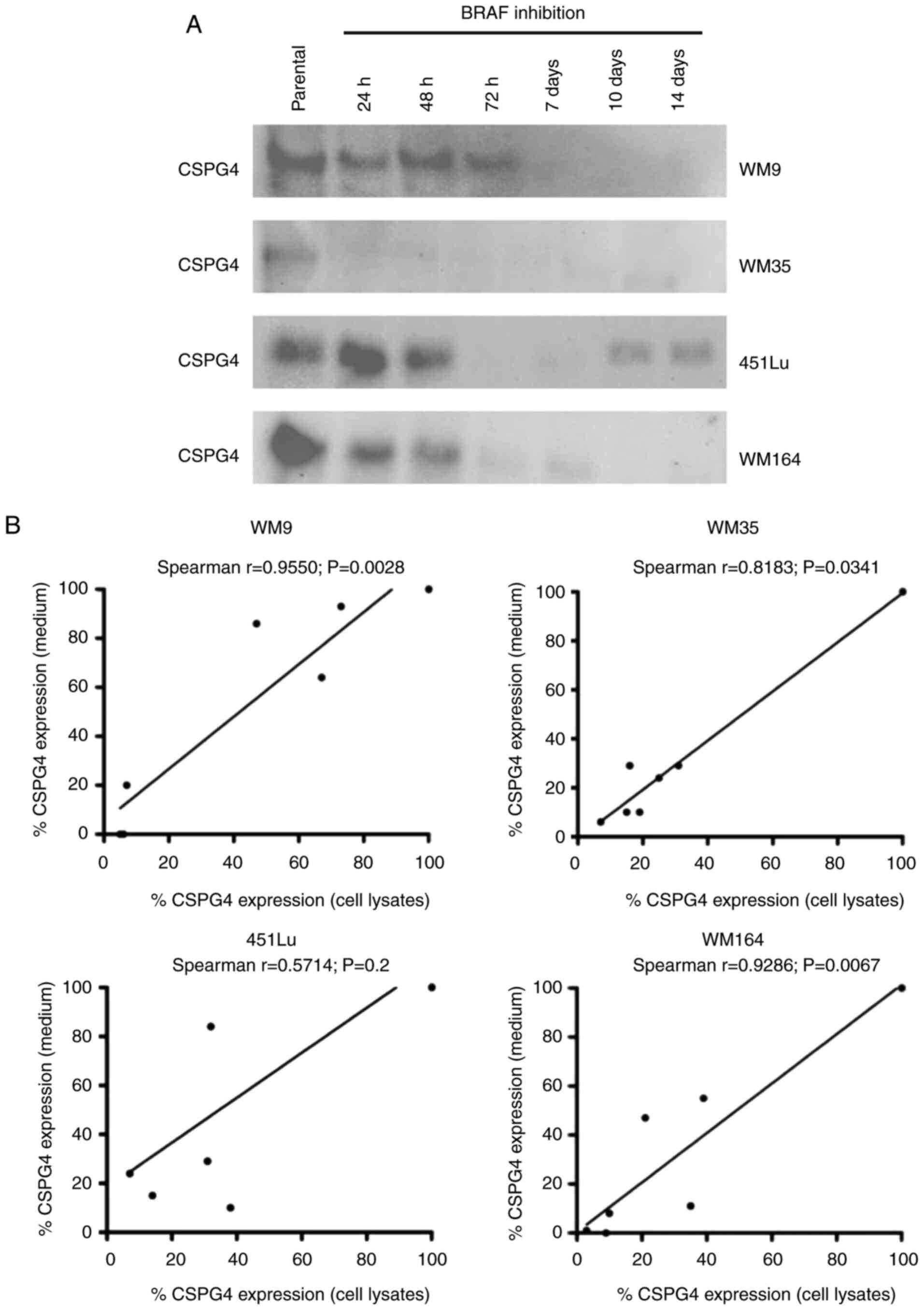
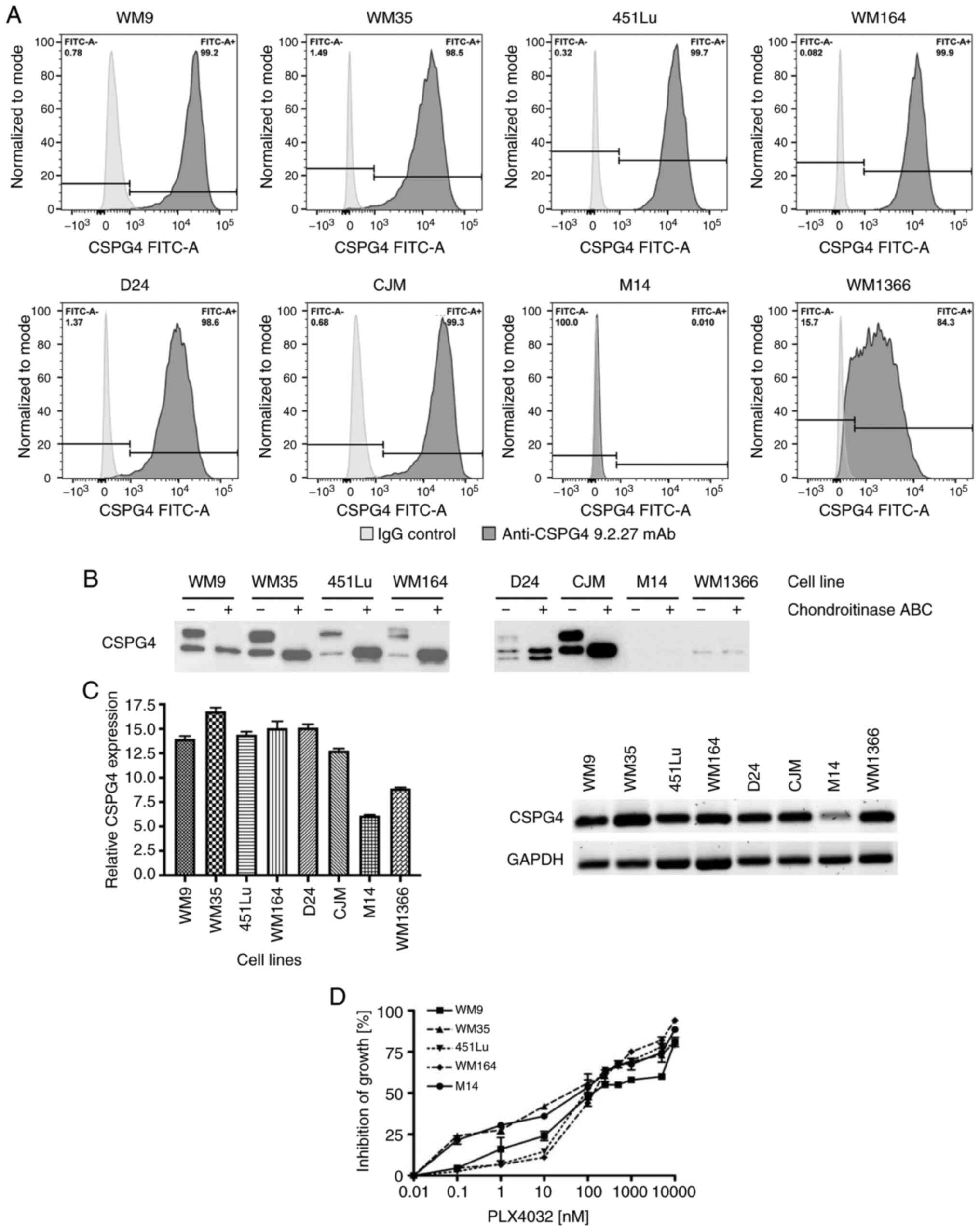 | Figure 1.CSPG4 expression in melanoma cell
lines. (A) WM9, WM35, 451Lu, WM164, D24, CJM, M14 and WM1366
melanoma cells were subjected to flow cytometry to determine the
percentage of CSPG4-positive cells using the anti-CSPG4 9.2.27 mAb.
Donkey anti-mouse secondary antibody, Alexa Fluor 488®,
was used as an IgG control. Data are representative of triplicates.
(B) CSPG4 expression in WM9, WM35, 451Lu, WM164, D24, CJM, M14 and
WM1366 cell lysates was evaluated by western blotting using the
anti-CSPG4 mAb (clone LHM 2). Samples were untreated or treated
with Chondroitinase ABC. In WM9, WM35, 451Lu, WM164 and CJM, CSPG4
was detected as a 280 kDa lower band and a 450 kDa upper band, in
D24 as 250, 280 and 450 kDa bands, in WM1366 as 250 kDa band. (C)
CSPG4 mRNA levels, normalized to the internal control
(GAPDH), were analyzed by qPCR using specific primers. Bars
represent mean ± SD from triplicates. Agarose gel electrophoresis
confirmed the specific qPCR products. (D) Dose-response effect of
PLX4032 on the growth of melanoma cell lines. The results are
presented as percent inhibition of cell growth, compared to
untreated cells. Bars represent mean ± SD from duplicates. PLX4032
at 0.01 nM corresponds to 0 nM PLX4032 (untreated cells), due to
Log 10 scale representation of the x-axis. CSPG4, chondroitin
sulfate proteoglycan 4. |
In addition, CSPG4 mRNA levels were assessed
in melanoma cells by qPCR. The presence of CSPG4 transcripts
was confirmed in all cell lines exhibiting the expression of the
proteoglycan (WM9, WM35, 451Lu, WM164, D24, CJM and WM1366)
(Fig. 1C). Only very low amounts of
CSPG4 mRNA were detected in the M14 cell line (Fig. 1C).
Differences in CSPG4 protein
expression in parental, induced drug-tolerant and drug-resistant
melanoma cells
Cell lines harboring the BRAF V600 mutation (WM9,
WM35, 451Lu and WM164) with a high CSPG4 expression were used for
the following experiments. The CSPG4-negative cell line M14 served
as a control. To determine the most appropriate doses of PLX4032 on
the different melanoma cell lines, dose-titration experiments were
performed. Treatment of WM9, WM35, 451Lu, WM164 and M14 cell lines
with increasing concentrations of PLX4032 resulted in a
dose-dependent inhibition of all cell lines. The growth of all five
cell lines exposed to PLX4032 in the concentration range of
250–1,000 nM was inhibited in 50–70% (Fig. 1D) and therefore, the concentrations of
250 and 1,000 nM PLX4032 were used for the study.
First, we generated induced drug-tolerant cells
(IDTCs) and drug-resistant cells by exposing the parental cells to
the BRAF inhibitor PLX4032. IDTCs demonstrated characteristic
morphological changes, such as elongated shape and dendrite-like
structures, as well as elevated CD271 expression (15). CD271 was used to verify the IDTC
status in all experiments. Continuous exposure of IDTCs to PLX4032
resulted in the transformation into permanent drug-resistant cells
that again proliferated (Fig. 2A). We
next evaluated the expression of CSPG4 in different stages of BRAF
inhibition by flow cytometry. Interestingly, the geometric mean
intensity of the CSPG4 signal was significantly lower in IDTCs and
drug-resistant cells, as compared to the parental cells for all
analyzed cell lines (Fig. 2B). In
451Lu and WM164 resistant cells, the expression of CSPG4 was
significantly higher than in IDTCs, but did not reach the level of
the corresponding parental cells (Fig.
2B). WM9 and WM35 drug-resistant cells demonstrated a slightly
but still significantly higher CSPG4 expression than IDTCs
(Fig. 2B). M14 underwent the same
morphological changes after exposure to PLX4032 as observed for the
other cell lines. Expression of CSPG4 by M14 cells was not observed
at any stage (Fig. 2A and B).
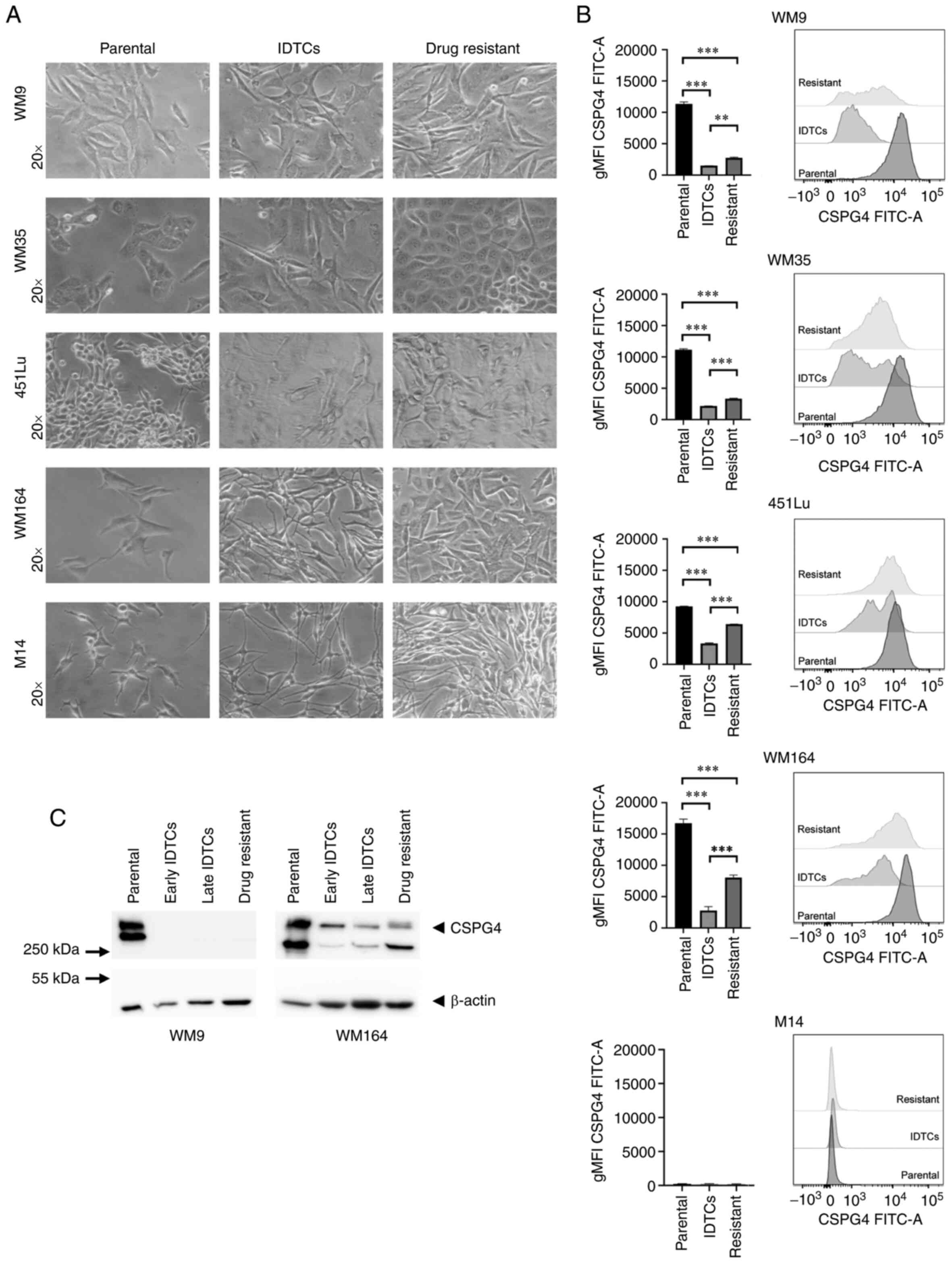 | Figure 2.Differences in CSPG4 expression in
parental, induced drug-tolerant and drug-resistant melanoma cells.
(A) WM9, WM35, 451Lu, WM164 and M14 cells were exposed to PLX4032
in order to generate induced drug-tolerant cells (IDTCs) and
drug-resistant cells. The parental cells and corresponding IDTCs
and resistant cells were examined by bright-field microscopy and
representative photos were taken. (B) The expression of CSPG4 in
parental, IDTCs and drug-resistant WM9, WM35, 451Lu, WM164 and M14
cells was evaluated by flow cytometry. Results are presented as
geometric mean of fluorescence intensity (gMFI) (left panel) and
representative histograms (right panel). Bars represent mean ± SD
from triplicates. Statistical analysis was carried out using the
one-way ANOVA with Tukey's multiple comparisons test.
***P<0.0001, **P<0.001. (C) Western blot analysis of CSPG4
(280 and 450 kDa) expression in WM9 and WM164 parental, early and
late IDTCs as well as drug-resistant cells. β-actin (45 kDa) was
used as a loading control. CSPG4, chondroitin sulfate proteoglycan
4; IDTCs, induced-drug tolerant cells. |
These results obtained by flow cytometry were
confirmed by western blotting. CSPG4 was not detected in IDTCs and
drug-resistant WM9 cell lysates (Fig.
2C). In WM164 early and late IDTCs, the amounts of CSPG4-280
and CSPG4-450 were decreased, as compared to the parental cells.
Interestingly, in drug-resistant cells the expression of CSPG4 was
increased, mainly the 280 kDa component not decorated with
chondroitin sulfate (Fig. 2C).
Overall, the expression of this proteoglycan decreased upon BRAF
inhibition in IDTCs and increased again in the corresponding
resistant cell lines, but the exact time points of these events
were cell line-specific.
CSPG4 protein expression over time
upon BRAF inhibition
To narrow down the time frame when changes in CSPG4
expression upon BRAF inhibition occur, we performed time course
experiments. We exposed four cell lines (WM9, WM35, 451Lu and
WM164) to PLX4032 for 14 days. At specific time points, samples
were collected and CSPG4 expression was analyzed by western
blotting. On days 7, 10 and 14 in the WM9, WM35 and WM164 cell
lines and on days 7 and 10 in the 451Lu cell line, cells presented
an IDTC phenotype, as shown in Fig.
2A. Interestingly, each cell line exhibited certain dynamics of
changes in CSPG4 expression (Fig.
3A). In WM9 cells, the CSPG4-280 and CSPG4-450 amounts were
lower after 72 h of BRAF inhibition and from day 7 on they were
detectable only in minute amounts. In the WM35 cell line, the
expression of CSPG4-280 and CSPG4-450 decreased as soon as 24 h
after drug exposure. Interestingly, the 280 kDa core protein was
detected at each time point, while the chondroitin sulfate modified
component decreased gradually over time. The amounts of CSPG4 in
451Lu cells were found to be lower already after 24 h. CSPG4-280
was detected until 72 h and continued to decrease until day 10,
while CSPG4-450 was downregulated at each time point. On day 14,
the 280 kDa core protein was detected again. The expression of the
proteoglycan in WM164 cells was markedly lower after 24 h of BRAF
inhibition and continued to decrease over time (Fig. 3A). Both CSPG4-280 and CSPG4-450 were
detected only until the 72 h time point. From day 7 on, only
CSPG4-450 was detectable, and its amounts decreased over time.
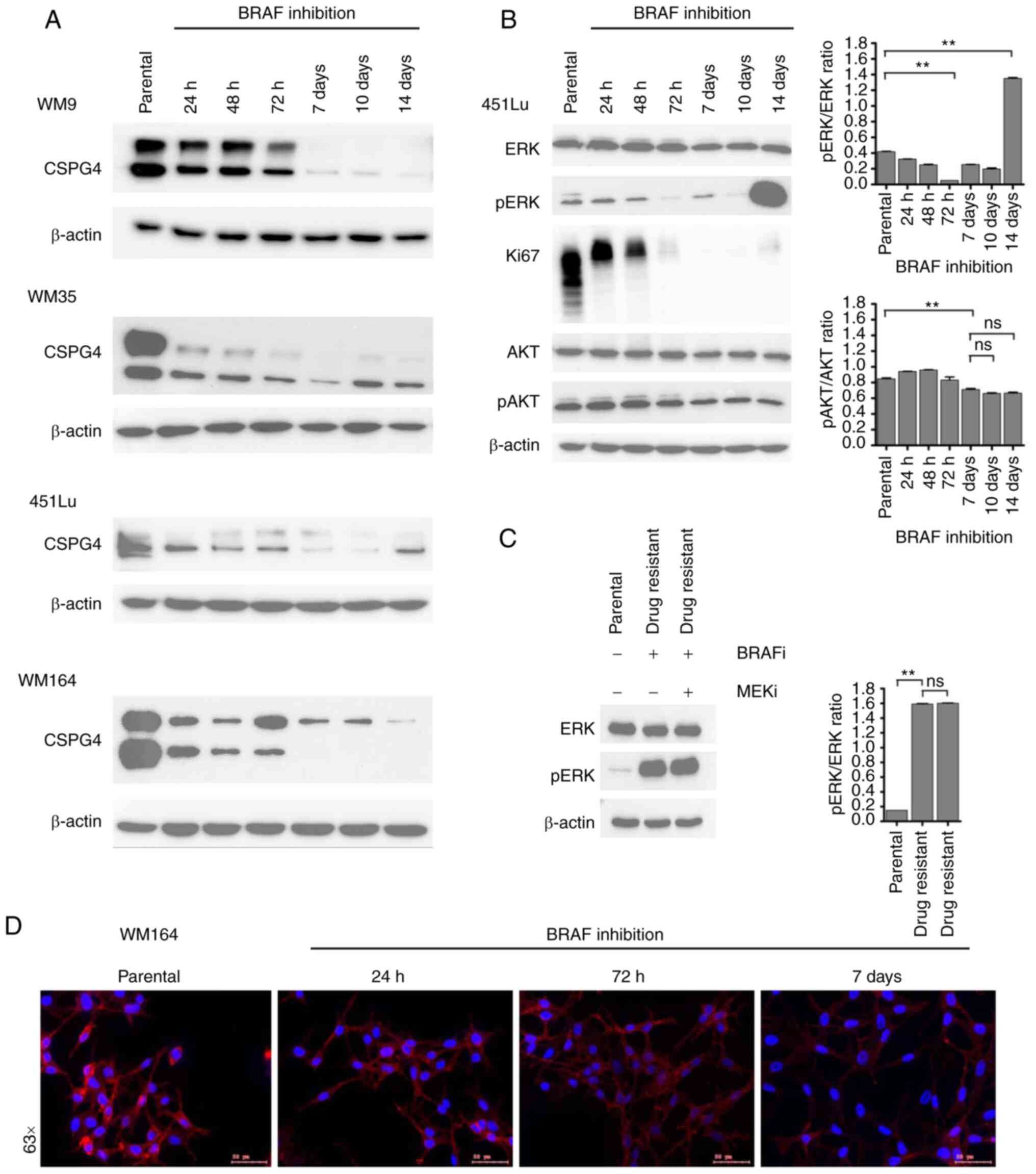 | Figure 3.Changes in CSPG4 expression over time
upon BRAF inhibition. (A) WM9, WM35, 451Lu and WM164 cells were
exposed to BRAF inhibitor (250 nM PLX4032) for 14 days. Samples
were collected at indicated time points and CSPG4 (280 and 450 kDa)
expression was analyzed in cell lysates by western blotting.
β-actin (45 kDa) was used as a loading control. (B) Protein lysates
of 451Lu parental and PLX4032-exposed cells were subjected to
western blotting for expression levels of Ki67 (345/395 kDa),
p44/42 MAPK (Erk1/2; 42/44 kDa), phospho-p44/42 MAPK (Erk1/2)
(Thr202/Tyr204; 42/44 kDa), total AKT (60 kDa), phospho-AKT
(Ser473; 60 kDa). β-actin (45 kDa) was used as a loading control.
Histograms represent pERK and pAKT expression normalized to ERK and
AKT, respectively (right panels). Statistical analysis was carried
out using one-way ANOVA with Tukey's multiple comparisons test.
**P<0.001; ns, not significant. (C) Lysates of 451Lu parental
and PLX-resistant cells, exposed to BRAF and/or MEK inhibitor (250
nM PLX4032 and/or 5 nM GSK1120212, respectively) were subjected to
western blotting to detect p44/42 MAPK (Erk1/2; 42/44 kDa) and
phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204; 42/44 kDa). β-actin
(45 kDa) was used as a loading control. Histogram represents pERK
expression normalized to ERK (right panel). Statistical analysis
was carried out using one-way ANOVA with Tukey's multiple
comparisons test. **P<0.001; ns, not significant. (D)
Immunofluorescent labelling of CSPG4 in WM164 parental and 1,000 nM
PLX4032-exposed cells was performed using anti-CSPG4 9.2.27 mAb and
goat anti-mouse IgG secondary antibody, Alexa Fluor
568®. CSPG4, chondroitin sulfate proteoglycan 4. |
In addition to western blot analysis,
immunofluorescence labelling of CSPG4 in WM164 parental and
drug-exposed cells was performed (Fig.
3D). The cells showed a homogenous pattern of CSPG4 staining,
with some punctate staining on the cell membrane, mainly in
untreated cells. The immunofluorescence analysis confirmed lower
CSPG4 levels in cells exposed to PLX4032, as compared to parental
cells (Fig. 3D).
CSPG4 is known to be involved in the activation of
two major cell signaling cascades, namely integrin/focal adhesion
kinase (FAK) signaling and MAPK pathway. Therefore, we investigated
whether - besides the fluctuations in CSPG4 expression - there were
also changes in the activation of the pathway components, ERK and
AKT. For this experiment, we chose 451Lu cells, as we previously
observed a cycle of CSPG4 downregulation within 10 days of drug
exposure and subsequent upregulation (after 14 days) (Fig. 3A). We evaluated the activity of ERK
and AKT, as well as the expression of Ki67 in 451Lu cell lysates at
specific time points of PLX4032 exposure by immunoblotting
(Fig. 3B). The inhibition of cell
proliferation was observed after 72 h and lasted until day 14, when
a faint band of Ki67 was again detected, suggesting that the cells
started to proliferate at this time point. Significant
downregulation of ERK signaling was achieved after 72 h and on day
14 a reactivation of signaling was observed. After exposure of
451Lu cells to PLX4032 for 14 days, we observed that
phosphorylation of ERK was still significantly high (Fig. 3C). The addition of the MEK inhibitor
did not lead to downregulation of ERK signaling, suggesting the
development of multidrug-resistance in the cells. Interestingly,
after 7 days of treatment, significantly reduced AKT (Ser473)
phosphorylation was detected and the similar phosphorylation level
remained until day 14 (Fig. 3B).
Decreased levels of CSPG4 in WM9,
WM35, 451Lu and WM164 melanoma cells after drug exposure are not
due to ectodomain shedding
The extracellular domain of CSPG4 contains a number
of putative proteolytic cleavage sites (34). Hence, in the next step, we
investigated whether the observed low levels of CSPG4 in
PLX4032-exposed cells were the result of protein shedding. Four
cell lines (WM9, WM35, 451Lu and WM164) were exposed to PLX4032 for
14 days. Medium from cultured cells was collected at specific time
points and analyzed for CSPG4 expression by western blotting
(Fig. 4A). The analysis detected
CSPG4 component of approximately 130 kDa. The highest amounts of
shed protein were observed in all parental cells tested and
decreased over time upon BRAF inhibition. The Spearman correlation
test demonstrated a significant correlation between cell-based
CSPG4 expression levels and CSPG4 ectodomain shedding levels for
WM9 (Spearman r=0.9550), WM35 (Spearman r=0.8183) and WM164
(Spearman r=0.9286) cell lines (Fig.
4B). This indicates that increased synthesis of CSPG4 in these
cell lines was associated with increased shedding of the CSPG4
ectodomain, suggesting that the inhibition of expression may occur
on the genomic level.
Effect of BRAF inhibition on the CSPG4
gene level
To investigate the gene expression of CSPG4
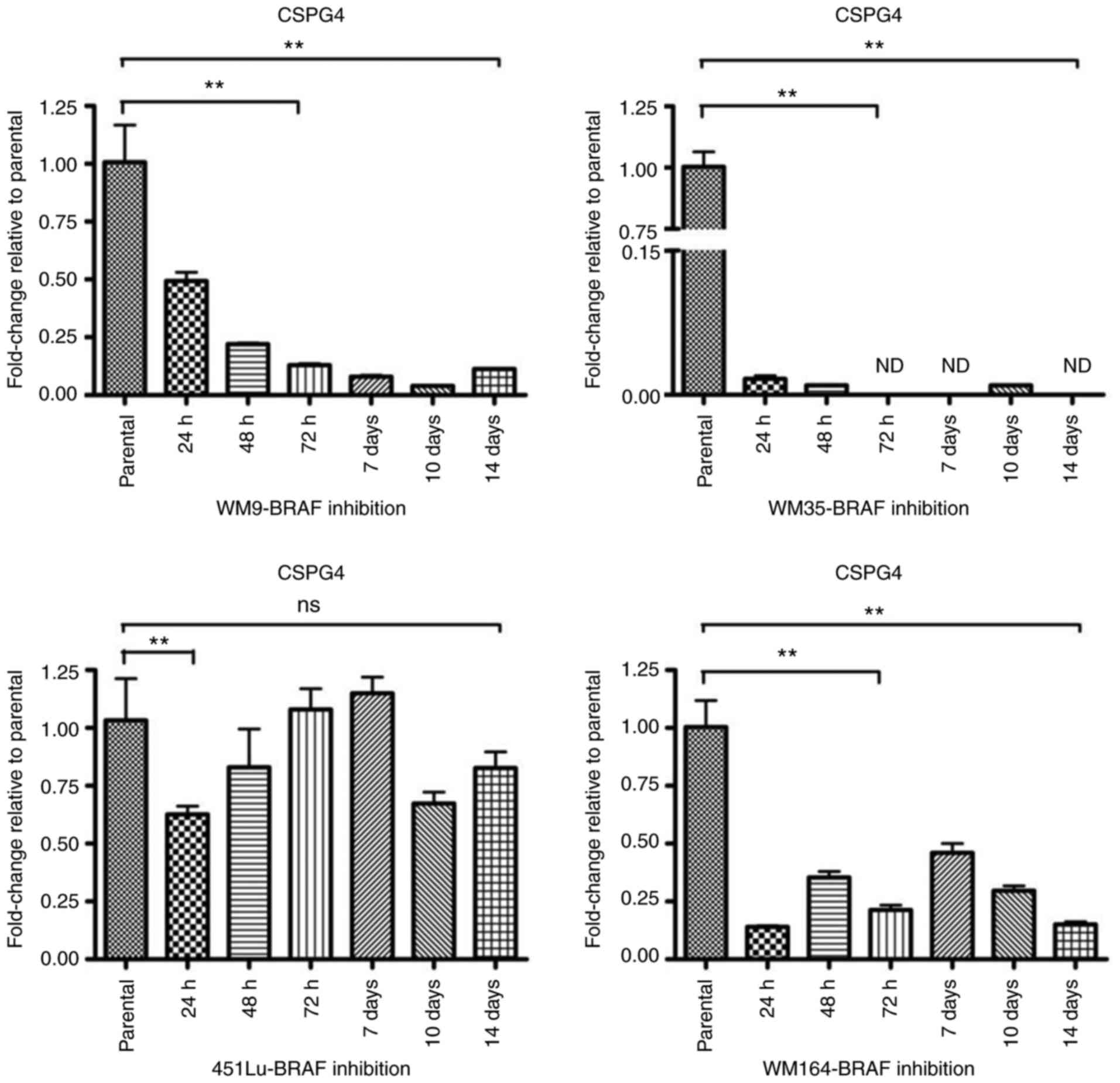
upon BRAF inhibition, RT-qPCR was performed. The relative messenger
RNA levels of CSPG4 in PLX4032-exposed cells were decreased
as compared to the parental cells (Fig.
5). As already observed at the protein level (Fig. 3A), changes in CSPG4 mRNA levels
over time were specific for each cell line. In WM9 cells,
CSPG4 mRNA levels decreased gradually over time (to
0.49±0.07 after 24 h, 0.22±0.01 after 48 h, 0.13±0.01 after 72 h,
0.08±0.01 after 7 days, 0.04±0.00 after 10 days) and started to
increase slightly after day 14 (0.11±0.01). The most noteworthy
CSPG4 downregulation was observed in the WM35 cell line. The
PLX4032 treatment decreased the mRNA level of CSPG4 to
0.02±0.01 already after 24 h. The gene expression remained low for
up to 14 days of treatment and was not detected (ND) on days 3, 7
and 14. CSPG4 mRNA levels in the 451Lu cell line were
markedly reduced after 24 h to 0.63±0.06. No notable changes were
observed after 3 and 7 days of PLX4032 treatment, as compared to
parental cells. Reduced levels of CSPG4 mRNA were also
detected after 48 h, 10 days and 14 days of PLX4032 exposure. In
WM164 cells, CSPG4 mRNA levels fluctuated upon BRAF
inhibition, but stayed lower than in parental cells, up to 14 days
(Fig. 5).
CSPG4 expression is downregulated in
vivo after tumor progression under BRAF/MEK inhibitor
treatment
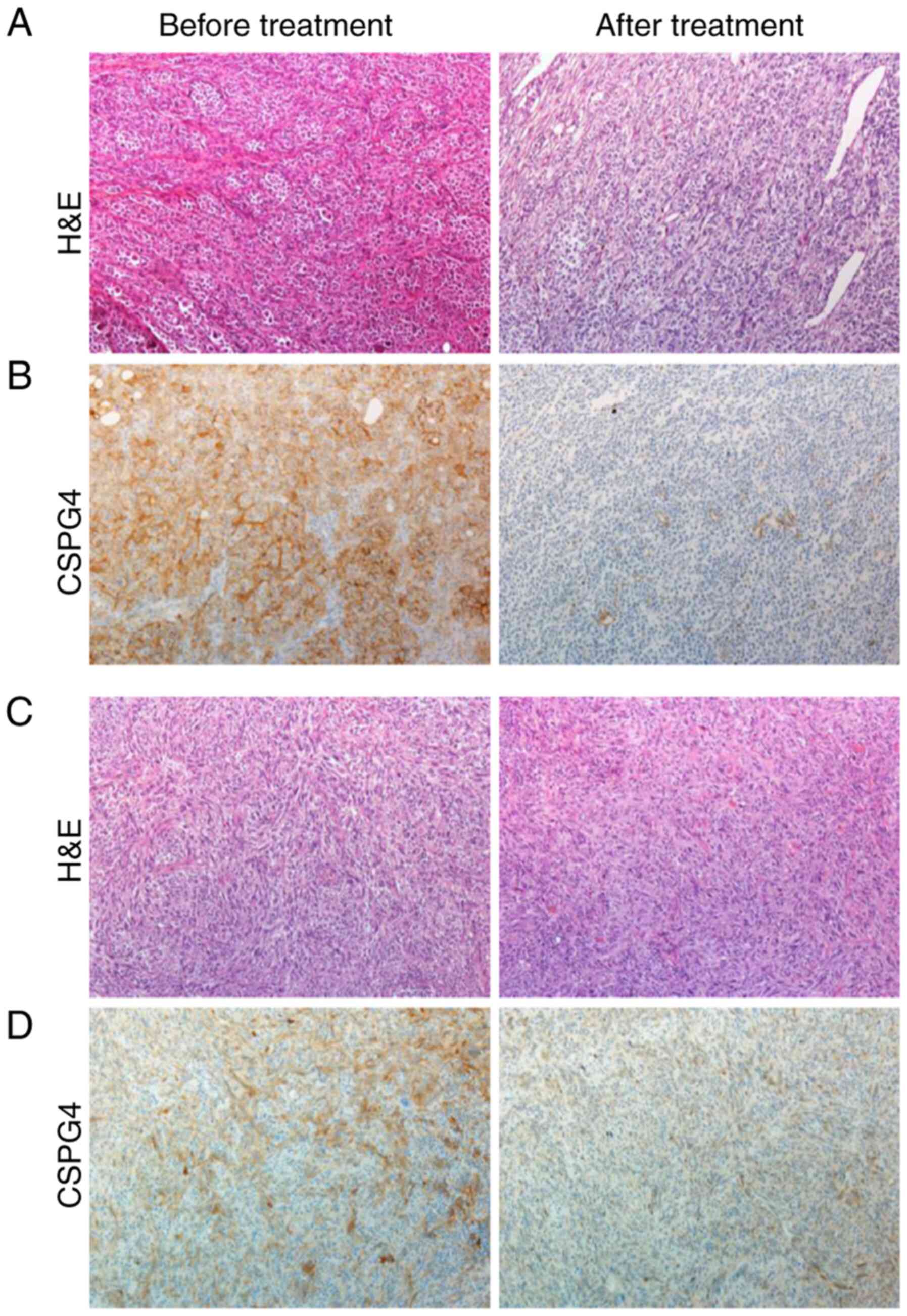
To support our in vitro data for the clinical
relevance of CSPG4 expression before and after therapy with kinase
inhibitors, patient-derived tumor samples were retrospectively
analyzed by immunohistochemistry (Fig.
6). Four out of five pretreatment melanoma tissue samples
showed a 2+ moderate positive staining for CSPG4 (Fig. 6B, left panel), and one pretreatment
sample showed a 1+ weak positive staining for CSPG4 (Fig. 6D, left panel). In contrast, four out
of five posttreatment melanoma samples stained negative for CSPG4,
as shown in Fig. 6B (right panel),
one sample stained 1+ for CSPG4 (Fig.
6D, right panel).
Discussion
Chondroitin sulfate proteoglycan 4 (CSPG4) is a
multifunctional transmembrane proteoglycan involved in migration,
spreading and invasion of melanoma (20). As reviewed by Ampofo et al,
expression of CSPG4 is known to be influenced by inflammation and
hypoxia and is regulated by methyltransferases, transcription
factors and miRNAs (32).
Identification of additional regulatory factors of CSPG4 expression
may contribute to the understanding of CSPG4-mediated cellular
functions and help design more effective treatments against
melanoma. The present study demonstrated that BRAF inhibition is
another important regulatory factor of CSPG4 expression.
We confirmed the presence of CSPG4 both at the
protein and the gene level in a panel of seven out of eight
melanoma cell lines. Regardless of the origin of the tumor cells
(primary tumor or metastasis) and BRAF or NRAS mutation statuses,
the majority of cell lines (WM9, WM35, 451Lu, WM164, D24 and CJM)
displayed a uniform pattern of CSPG4 expression, as detected by
anti-CSPG4 antibodies in western blotting. In line with other
reports, within the same population of cells, CSPG4 was expressed
both with a chondroitin sulfate (CS) chain and as an unmodified
core protein (19,35). As anticipated, the M14 cell line,
previously described as CSPG4-negative (30), did not show any CSPG4 expression.
The introduction of targeted therapies, such as BRAF
and MEK inhibitors, has improved treatment options for metastatic
melanoma patients. However, despite remarkable early therapeutic
effects, a complete response is rarely observed due to acquired
resistance (7).
Induced-drug tolerant cells (IDTCs), which
constitute the step preceding the development of permanent drug
resistance, are extremely important from a therapeutic perspective
(15). Under hostile conditions (such
as drug exposure, hypoxia or nutrient starvation), a fraction of
tumor cells survives and over time can re-populate the tumor. Thus,
targeting IDTCs in order to prevent or delay permanent drug
resistance may enhance overall therapeutic efficacy. Since CSPG4 is
a candidate target molecule, we examined whether its expression is
altered in IDTCs and drug-resistant melanoma cells during exposure
to kinase inhibitors. The amount of CSPG4 in IDTCs of all tested
CSPG4-positive cell lines was markedly lower, as compared to
parental cells. However, the presence of CSPG4 may not be required
for a cell to become multidrug resistant since the CSPG4-negative
cell line M14 underwent the same morphological and functional
changes. After IDTCs escaped their slow cycling semiquiescent state
and became permanently resistant, the expression of CSPG4 changed
as well, in a cell line-specific manner. We demonstrated that
different melanoma cell lines require different time spans to
restore CSPG4 expression upon treatment with PLX4032, or that the
proteoglycan in some cell lines is completely lost upon drug
exposure. This effect did not change in a long-term experiment (up
to 90 days) in all cell lines tested (data not shown). For a
detailed understanding of CSPG4 expression dynamics, it is
important to continuously follow its expression not only in short
term cultures, but also in long-term experiments. Nevertheless,
upregulated CSPG4 expression in permanent resistant melanoma cells
could be a sign of the onset of additional resistance and survival
mechanisms for cancer cells that will require further investigation
(6).
Detailed analysis of cell lysates for CSPG4
expression upon BRAF inhibition within a time frame of up to 14
days revealed cell line-specific patterns of CSPG4 downregulation.
In parental cells, CSPG4 was expressed both as a 280 kDa core
protein and a 450 kDa chondroitin sulfate modified component. Upon
BRAF inhibition, both forms gradually disappeared in WM9 cells,
while in WM35 only the amount of the CS-decorated component
decreased. In 451Lu cells, both CSPG4 forms decreased over time,
and only the core protein appeared again. In WM164, both components
initially decreased, but the CS-modified protein remained
detectable for a much longer period of 14 days than the core
protein. Previous studies showed that chondroitin sulfate
decoration of the CSPG4 core protein may confer different
attributes. First, CS chains may influence the CSPG4 distribution
on the cell surface, focusing the core protein and proteoglycan
into different microdomains in the plasma membrane (21). Second, CSPG4-linked chondroitin
sulfate is critical for enhancement of matrix metalloproteinase
(MMP) activity and thus extracellular matrix (ECM) degradation and
increased tumor invasion (22).
Finally, CS decoration facilitates an interaction of CSPG4 with
fibronectin and α4β1-integrin, thereby modulating melanoma cell
proliferation, adhesion, migration and invasion (20,23).
Considering all these features, we speculate that the modifications
of CSPG4 expression, and especially alterations in CS decoration,
observed in our study, influence the overall behavior of the cell.
As changes in CSPG4 upon BRAF inhibition are individual for
different cell lines, it is likely that each cell line would
exhibit different capabilities of CSPG4-dependent cellular
functions.
CSPG4, as a membrane-spanning molecule, serves as a
key proteoglycan that participates in signaling between the
extracellular and intracellular compartments of the cell. In fact,
CSPG4 was shown to promote MAPK signaling by mediating the
growth-factor induced activation of receptor tyrosine kinases
located on the cell surface. In addition, the cytoplasmic domain of
CSPG4 contains a phosphoacceptor site at Thr2314,
phosphorylated by ERK1/2, resulting in stimulation of cell
proliferation (36). It was also
described that the presence of CSPG4 is required for constitutive
activation of the ERK1/2 pathway in melanoma cells possessing the
BRAF V600E mutation (25). As
demonstrated in 451Lu cells CSPG4 was downregulated upon BRAF
inhibition. In addition, we observed the inhibition of cell
proliferation and ERK1/2 phosphorylation at the same time points.
In contrast, CS decoration seemed to play a role in AKT
phosphorylation. The observed influence of the CS chain on AKT
activation, independently from ERK1/2, is in line with studies
showing that enhanced activation of FAK and ERK1/2 signaling by
CSPG4 involves autonomous mechanisms (24).
The membrane proximal portion of extracellular
domain of CSPG4 contains a number of putative proteolytic cleavage
sites, leading to the release of CSPG4 from the cell surface
(34). In our study, BRAF inhibition
did not trigger shedding of CSPG4 in all tested cell lines. In
fact, higher CSPG4 synthesis correlated significantly with greater
CSPG4 release into the culture medium. This finding provided a
rationale for investigating whether CSPG4 expression was regulated
at the genomic level. Indeed, our results indicated markedly lower
CSPG4 mRNA levels upon exposure to PLX4032 in all melanoma
cell lines tested. Further studies are however required to fully
describe the precise mechanism of CSPG4 downregulation. This
process might involve DNA methylation, as it was shown that the
promoter of CSPG4 contains many 5′CpG methylation sites
(37). Another possibility is a
regulatory mechanism involving a specific miRNA - miR129-2
(38). In addition to these
mechanisms, CSPG4 can be regulated by several transcription
factors, such as Sp1, Pax3 and Egr1 (32). The transcription factor Egr-1 is a
crucial mediator in ERK-dependent signaling. In our investigation
of the Egr-1 gene expression upon BRAF inhibition, no clear
conclusion about a possible involvement of Egr-1 in
CSPG4 downregulation could be made (data not shown).
CSPG4 has been found to be expressed in the majority
(70% or greater) of superficial spreading and nodular human
melanomas (20). It has not yet been
examined whether its expression in tumors vary before and after
treatment with kinase inhibitors. Our retrospective analysis of
CSPG4 expression in melanoma tissue samples support the
observations obtained in vitro, showing that CSPG4 is
downregulated also in vivo after BRAF/MEK inhibition. The
limited number of matched tumor samples did not permit to calculate
a mathematical correlation of the CSPG4 expression between
BRAF-mutant and drug-resistant melanoma samples. It would still be
of great importance to perform a large, prospective, systemic
quantitative study assessing CSPG4 expression before and after
treatment with kinase inhibitors.
In conclusion, our results indicate that BRAF
inhibition is a regulatory factor of CSPG4 expression in melanoma
cells. Since CSPG4 plays a central role in coordinating cell
proliferation, adhesion, migration and survival, downregulation of
this proteoglycan may have a significant effect on the overall
behavior of melanoma cells. The outcomes of this study provide the
basis for further investigation of the underlying mechanisms and
the role of CSPG4 in the development of drug-resistance in
melanoma.
Acknowledgements
The authors would like to thank Helmut Schaider
(Dermatology Research Centre, The University of Queensland
Diamantina Institute, Translational Research Institute, The
University of Queensland, Brisbane, Australia) for critical
discussions throughout the project and for providing melanoma cell
lines; Diana Mechtcheriakova and Anastasia Meshcheryakova
(Department of Pathophysiology and Allergy Research, Center for
Pathophysiology, Infectiology and Immunology, Medical University of
Vienna, Austria) for technical assistance in qPCR experiments;
Isabella Ellinger, Felicitas Mungenast and Martina Salzmann
(Department of Pathophysiology and Allergy Research, Center for
Pathophysiology, Infectiology and Immunology, Medical University
Vienna, Austria) for support in immunofluorescence experiments;
Johan Skaar, Rita Seeboeck (Department of Pathology, University
Hospital St. Poelten, Karl Landsteiner University of Health
Sciences, St. Poelten, Austria) and Vanessa Mayr (Department of
Pathophysiology and Allergy Research, Center for Pathophysiology,
Infectiology and Immunology, Medical University of Vienna, Austria)
for technical assistance in the immunohistochemical analysis.
Funding
This project was funded by the NÖ Forschungs- und
Bildungsges.m.b.H. (NFB), grant number: LSC15-007.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
KU and CH conceived the concept, designed the study,
and analyzed and interpreted the data. TK and HB participated in
designing the study and analyzing the data. KU and VV performed all
experiments. MK contributed to the IHC analysis. KU, CH and HB
wrote the manuscript. All authors read and revised the manuscript,
and approved the final version. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated.
Ethics approval and consent to
participate
All clinical samples were obtained from the
collection of patients at the Department of Dermatology at the
University Hospital St. Poelten, Karl Landsteiner University of
Health Sciences. The collection and storage of samples were
performed according to local ethical guidelines. Due to the
retrospective nature of the study informed consent was waived by
the Ethics Committee of the Karl Landsteiner University. The study
was conducted in accordance with the Declaration of Helsinki and
was approved by the Ethics Committee of the Karl Landsteiner
University (EK number: 1011/2019).
Patient consent for publication
Not required according to local ethical
guidelines.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AKT
|
protein kinase B
|
|
CS
|
chondroitin sulfate
|
|
CSPG4
|
chondroitin sulfate proteoglycan 4
|
|
ECM
|
extracellular matrix
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FAK
|
focal adhesion kinase
|
|
GAG
|
glycosaminoglycan
|
|
IDTCs
|
induced drug-tolerant cells
|
|
mAb
|
monoclonal antibody
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
PBS
|
phosphate-buffered saline
|
|
RTK
|
receptor tyrosine kinase
|
References
|
1
|
Schadendorf D, van Akkooi ACJ, Berking C,
Griewank KG, Gutzmer R, Hauschild A, Stang A, Roesch A and Ugurel
S: Melanoma. Lancet. 392:971–984. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spagnolo F, Ghiorzo P and Queirolo P:
Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic
melanoma. Oncotarget. 5:10206–10221. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ascierto PA, Kirkwood JM, Grob JJ, Simeone
E, Grimaldi AM, Maio M, Palmieri G, Testori A, Marincola FM and
Mozzillo N: The role of BRAF V600 mutation in melanoma. J Transl
Med. 10:852012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pratilas CA, Taylor BS, Ye Q, Viale A,
Sander C, Solit DB and Rosen N: (V600E)BRAF is associated with
disabled feedback inhibition of RAF-MEK signaling and elevated
transcriptional output of the pathway. Proc Natl Acad Sci USA.
106:4519–4524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al: Improved survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med. 364:2507–2516. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wagle N, Emery C, Berger MF, Davis MJ,
Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE,
Hahn WC, et al: Dissecting therapeutic resistance to RAF inhibition
in melanoma by tumor genomic profiling. J Clin Oncol. 29:3085–3096.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Griffin M, Scotto D, Josephs DH, Mele S,
Crescioli S, Bax HJ, Pellizzari G, Wynne MD, Nakamura M, Hoffmann
RM, et al: BRAF inhibitors: Resistance and the promise of
combination treatments for melanoma. Oncotarget. 8:78174–78192.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Flaherty KT, Infante JR, Daud A, Gonzalez
R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N,
et al: Combined BRAF and MEK inhibition in melanoma with BRAF V600
mutations. N Engl J Med. 367:1694–1703. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Johnson DB, Flaherty KT, Weber JS, Infante
JR, Kim KB, Kefford RF, Hamid O, Schuchter L, Cebon J, Sharfman WH,
et al: Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib)
in patients with BRAFV600-mutant melanoma experiencing progression
with single-agent BRAF inhibitor. J Clin Oncol. 32:3697–3704. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Robert C, Grob JJ, Stroyakovskiy D,
Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V,
Schachter J, Garbe C, Bondarenko I, et al: Five-year outcomes with
dabrafenib plus trametinib in metastatic melanoma. N Engl J Med.
381:626–636. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sharma SV, Lee DY, Li B, Quinlan MP,
Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach
MA, et al: A chromatin-mediated reversible drug-tolerant state in
cancer cell subpopulations. Cell. 141:69–80. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roesch A, Fukunaga-Kalabis M, Schmidt EC,
Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T
and Herlyn M: A temporarily distinct subpopulation of slow-cycling
melanoma cells is required for continuous tumor growth. Cell.
141:583–594. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arozarena I and Wellbrock C: Phenotype
plasticity as enabler of melanoma progression and therapy
resistance. Nat Rev Cancer. 19:377–391. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Das Thakur M, Salangsang F, Landman AS,
Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M and Stuart
DD: Modelling vemurafenib resistance in melanoma reveals a strategy
to forestall drug resistance. Nature. 494:251–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Menon DR, Das S, Krepler C, Vultur A,
Rinner B, Schauer S, Kashofer K, Wagner K, Zhang G, Rad EB, et al:
A stress-induced early innate response causes multidrug tolerance
in melanoma. Oncogene. 34:45452015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rolih V, Barutello G, Iussich S, De Maria
R, Quaglino E, Buracco P, Cavallo F and Riccardo F: CSPG4: A
prototype oncoantigen for translational immunotherapy studies. J
Transl Med. 15:1512017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ilieva KM, Cheung A, Mele S, Chiaruttini
G, Crescioli S, Griffin M, Nakamura M, Spicer JF, Tsoka S, Lacy KE,
et al: Chondroitin sulfate proteoglycan 4 and its potential as an
antibody immunotherapy target across different tumor types. Front
Immunol. 8:19112017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ross AH, Cossu G, Herlyn M, Bell JR,
Steplewski Z and Koprowski H: Isolation and chemical
characterization of a melanoma-associated proteoglycan antigen.
Arch Biochem Biophys. 225:370–383. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campoli MR, Chang CC, Kageshita T, Wang X,
McCarthy JB and Ferrone S: Human high molecular
weight-melanoma-associated antigen (HMW-MAA): A melanoma cell
surface chondroitin sulfate proteoglycan (MSCP) with biological and
clinical significance. Crit Rev Immunol. 24:267–296. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Price MA, Colvin Wanshura LE, Yang J,
Carlson J, Xiang B, Li G, Ferrone S, Dudek AZ, Turley EA and
McCarthy JB: CSPG4, a potential therapeutic target, facilitates
malignant progression of melanoma. Pigment Cell Melanoma Res.
24:1148–1157. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stallcup WB and Dahlin-Huppe K:
Chondroitin sulfate and cytoplasmic domain-dependent membrane
targeting of the NG2 proteoglycan promotes retraction fiber
formation and cell polarization. J Cell Sci. 114:2315–2325.
2001.PubMed/NCBI
|
|
22
|
Iida J, Wilhelmson KL, Ng J, Lee P,
Morrison C, Tam E, Overall CM and McCarthy JB: Cell surface
chondroitin sulfate glycosaminoglycan in melanoma: Role in the
activation of pro-MMP-2 (pro-gelatinase A). Biochem J. 403:553–563.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Iida J, Meijne AM, Oegema TR Jr, Yednock
TA, Kovach NL, Furcht LT and McCarthy JB: A role of chondroitin
sulfate glycosaminoglycan binding site in alpha4beta1
integrin-mediated melanoma cell adhesion. J Biol Chem.
273:5955–5962. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang J, Price MA, Neudauer CL, Wilson C,
Ferrone S, Xia H, Iida J, Simpson MA and McCarthy JB: Melanoma
chondroitin sulfate proteoglycan enhances FAK and ERK activation by
distinct mechanisms. J Cell Biol. 165:881–891. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Price MA, Li GY, Bar-Eli M, Salgia
R, Jagedeeswaran R, Carlson JH, Ferrone S, Turley EA and McCarthy
JB: Melanoma proteoglycan modifies gene expression to stimulate
tumor cell motility, growth, and epithelial-to-mesenchymal
transition. Cancer Res. 69:7538–7547. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jordaan S, Chetty S, Mungra N, Koopmans I,
van Bommel PE, Helfrich W and Barth S: CSPG4: A target for
selective delivery of human cytolytic fusion proteins and TRAIL.
Biomedicines. 5:372017. View Article : Google Scholar
|
|
27
|
Hoffmann RM, Crescioli S, Mele S, Sachouli
E, Cheung A, Chui CK, Andriollo P, Jackson PJM, Lacy KE, Spicer JF,
et al: A novel antibody-drug conjugate (ADC) delivering a DNA
mono-alkylating payload to chondroitin sulfate proteoglycan
(CSPG4)-expressing melanoma. Cancers (Basel). 12:10292020.
View Article : Google Scholar
|
|
28
|
Wang Y, Geldres C, Ferrone S and Dotti G:
Chondroitin sulfate proteoglycan 4 as a target for chimeric antigen
receptor-based T-cell immunotherapy of solid tumors. Expert Opin
Ther Targets. 19:1339–1350. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hafner C, Breiteneder H, Ferrone S,
Thallinger C, Wagner S, Schmidt WM, Jasinska J, Kundi M, Wolff K,
Zielinski CC, et al: Suppression of human melanoma tumor growth in
SCID mice by a human high molecular weight-melanoma associated
antigen (HMW-MAA) specific monoclonal antibody. Int J Cancer.
114:426–432. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pucciarelli D, Lengger N, Takacova M,
Csaderova L, Bartosova M, Breiteneder H, Pastorekova S and Hafner
C: Anti-chondroitin sulfate proteoglycan 4-specific antibodies
modify the effects of vemurafenib on melanoma cells differentially
in normoxia and hypoxia. Int J Oncol. 47:81–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu L, Favoino E, Wang Y, Ma Y, Deng X and
Wang X: The CSPG4-specific monoclonal antibody enhances and
prolongs the effects of the BRAF inhibitor in melanoma cells.
Immunol Res. 50:294–302. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ampofo E, Schmitt BM, Menger MD and
Laschke MW: The regulatory mechanisms of NG2/CSPG4 expression. Cell
Mol Biol Lett. 22:42017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Campoli M, Ferrone S and Wang X:
Functional and clinical relevance of chondroitin sulfate
proteoglycan 4. Adv Cancer Res. 109:73–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang J, Price MA, Wanshura LEC, He J, Yi
M, Welch DR, Li G, Conner S, Sachs J, Turley EA and McCarthy JB:
Chondroitin sulfate proteoglycan 4 enhanced melanoma motility and
growth requires a cysteine in the core protein transmembrane
domain. Melanoma Res. 29:365–375. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Makagiansar IT, Williams S, Mustelin T and
Stallcup WB: Differential phosphorylation of NG2 proteoglycan by
ERK and PKCalpha helps balance cell proliferation and migration. J
Cell Biol. 178:155–165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luo W, Wang X, Kageshita T, Wakasugi S,
Karpf AR and Ferrone S: Regulation of high molecular
weight-melanoma associated antigen (HMW-MAA) gene expression by
promoter DNA methylation in human melanoma cells. Oncogene.
25:2873–2884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yadavilli S, Scafidi J, Becher OJ,
Saratsis AM, Hiner RL, Kambhampati M, Mariarita S, MacDonald TJ,
Codispoti KE, Magge SN, et al: The emerging role of NG2 in
pediatric diffuse intrinsic pontine glioma. Oncotarget.
6:12141–12155. 2015. View Article : Google Scholar : PubMed/NCBI
|