Introduction
Ubiquitin-specific processing enzyme 22
(USP22) is a novel deubiquitinating enzyme that can cleave
ubiquitin (Ub) from Ub-conjugated protein substrates (1). As the subunit of the human SAGA
coactivator complex, USP22 is linked to the regulation of
gene transcription by deubiquitinating histones H2A and H2B
(2,3). In
addition, USP22 deubiquitinates intracellular protein,
including the shelterin protein telomeric repeat binding factor 1
(4), the histone deacetylase sirtuin 1
(5) and the far upstream
element-binding protein 1 fructose-1,6-bisphosphatase 1 (4), and therefore performs an extensive
physiological function. A murine study showed that USP22
also regulates embryonic stem cell differentiation (6). In humans, the USP22 gene is
located on chromosome 17, consists of 14 exons, and is transcribed
and produced broadly across various tissues (7). Of note, elevated levels of USP22
have been identified in numerous types of human cancer, including
colorectal (8) lung (9) and breast cancer (10). USP22 has been indicated in
tumorigenesis. Deletion of USP22 leads to the accumulation
of cells in the G1 phase of the cell cycle (2). For these reasons, USP22 is a
putative cancer stem cell marker. Reducing the rate of USP22
expression may be a suitable target for cancer therapy (11). However, the mechanisms that lead to
USP22 transcriptional activation, particularly in the human
tumor cells, remain unknown.
Previously, USP22 transcription was activated
by mitogen stimulation or viral infection in normal T and B
lymphocytes (12), suggesting the
regulation of USP22 gene expression occurs mainly at the
transcriptional level. However, the mechanism in which signal
transduction pathways regulate USP22 transcription is
unclear. It is well-known that activation of the mitogen-activated
protein kinase (MAPK) pathways is the main downstream event in
response to mitogen stimulation. Three activated subgroups of
MAPKs: Extracellular signal-regulated kinases (ERKs), p38 MAPK and
c-Jun N-terminal kinases (JNKs), regulate diverse cellular
responses, including cell proliferation, differentiation, survival,
the inflammatory response and even cell death (13).
In the present study, p38 MAPK was involved in the
regulation of USP22 transcription, but ERKs and JNKs were
not. The chemotherapeutic agent cisplatin suppressed the
USP22 gene partly through p38 MAPK. These results provide
novel insights on the molecular mechanisms underlying USP22
expression.
Materials and methods
Cell cultures
Human cervical carcinoma (HeLa) cells were obtained
from the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China) and were cultured in Dulbecco's modified Eagle's
medium from Invitrogen (Carlsbad, CA, USA) supplemented with 10%
fetal bovine serum at 37°C in a 5% CO2 incubator.
Cisplatin and MAPK inhibitors U0126, SB203580 and SP600125 were
from Sigma-Aldrich (St. Louis, MO, USA).
Plasmid constructs
The USP22 promoter fragments inserted into
pGL-3 were constructed as described previously (14). Site-directed mutagenesis was carried
out within the USP22 basic promoter p-210/+52 according to
the manufacturer's instructions for the MutanBEST kit (Takara Bio
Inc., Otsu, Shiga, Japan). Mutagenic primer pairs used for the
polymerase chain reaction (PCR) amplification included
5′-GTAGCGTAATCTCCGTCCGC-3′ for the
CREB/ATF-binding site mutagenesis, 5′-CCTGTAGGCTCTGGGTAGAC-3′ for the
MYB-binding site mutagenesis, 5′-GGATCGGTGCCTGCCTTGCA-3′ for the
Sp1-binding site (−7/−12) mutagenesis (complementary reverse
primers are not shown, and mutated nucleotides are underlined). All
the mutations were confirmed by DNA sequencing. The MAPK kinase 6
(MKK6) expression plasmid and dominant negative MKK6 (DN MKK6)
plasmid were provided by Professor Jiahuai Han of Xiamen University
(Xiamen, Fujian, China).
Transfections and dual luciferase
reporter assay
Cells (1×104) were plated in 24-well
plates 12 h before transfection with 0.5 µg of various USP22
promoter constructs and 0.1 µg pRL-TK (Promega Corp., Madison, WI,
USA) using Lipofectamine 2000 (Invitrogen) in each well.
Twenty-four hours after transfection, cells were was hed in
phosphate-buffered saline and lysed for 30 min at room temperature
using the passive lysis buffer (Promega Corp.). Luciferase activity
was determined using the dual luciferase reporter assay system
(Promega Corp.). The normalized luciferase activity was expressed
as the ratio of firefly luciferase activity to Renilla luciferase
for each sample. All the transfection experiments were repeated
four times.
Total RNA isolation and quantitative
PCR (qPCR)
Total RNA from cells treated with agents was
prepared using TRIzol according to the manufacturer's instructions
(Invitrogen). RNA was reverse-transcribed with oligo-dT primers
using an RNA PCR kit (AMV) ver. 3.0 (Takara Bio Inc.), and the cDNA
fragments were analyzed by qPCR using the SYBR-Green PCR Master mix
(Toyobo Co., Ltd., Osaka, Japan) on an ABI 7500 real-time PCR
System (Applied Biosystems, Foster City, CA, USA). USP22
primer pairs were: Forward, 5′-ACCACCACGCTCACGGACTG-3′; and
reverse, 5′-TTGGCTGAGTGTTCAAATCG-3′; P21 primer pairs were:
Forward, 5′-GCAGATCCACAGCGATATCC-3′; and reverse,
5′-CAACTGCTCACTGTCCACGG-3′. GAPDH primers were: Forward,
5′-AGAAGGCTGGGGCTCATTTG-3′; and reverse,
5′-AGGGGCCATCCACAGTCTTC-3′.
Western blot analysis
Cells were lysed with 1X SDS sample buffer. Protein
was separated by 10% SDS-PAGE and electroblotted onto
nitrocellulose membrane. The membrane was subsequently incubated
with anti-USP22 antibody or anti-GAPDH antibody (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and developed using the
electrochemiluminescence system (Pierce Biotechnology, Inc.,
Rockford, IL, USA), according to the manufacturer's
instructions.
Flow cytometry
Cells (1×105) were seeded in 6-well
plates overnight and subsequently SB203580 or dimethyl sulfoxide
(DMSO) was added. After 12 h, the medium was changed for medium
containing cisplatin for a further 12 or 24 h incubation. The cells
were harvested by trypsinization, was hed with 1X PBS and
resuspended in 50 µl of 1X PBS. Cell death was assessed using flow
cytometry (BD Biosciences, Franklin Lakes, NJ, USA) following
staining with fluorescein isothiocyanate-Annexin V and propidium
iodide.
Statistical analysis
Numerical data are presented as the mean ± standard
error of the mean. Statistical differences between sample means
were determined using the unpaired, two-tailed Student's t-test.
The significance level was set at α<0.05, and therefore,
P<0.05 was considered to indicate a statistically significant
difference.
Results
p38 MAPK regulates USP22
promoter activity
Pharmacological reagents that inhibit MAPK pathways
with different specificity were used to determine whether MAPK
signaling is involved in the regulation of USP22 expression:
U0126, SB203580 and SP600125. These selectively inhibit ERK1/2, p38
MAPK and JNK, respectively. HeLa cells were transfected with the
reporter construct, pGL-210/+52, which contains the USP22
basic promoter region from −210 to +52 (14). The transfected cells were cultured for
a further 12 h in the presence of 2 µM U0126, 2 µM SB203580, 10 µM
SP600125 and 0.1% DMSO as blank controls, and luciferase activity
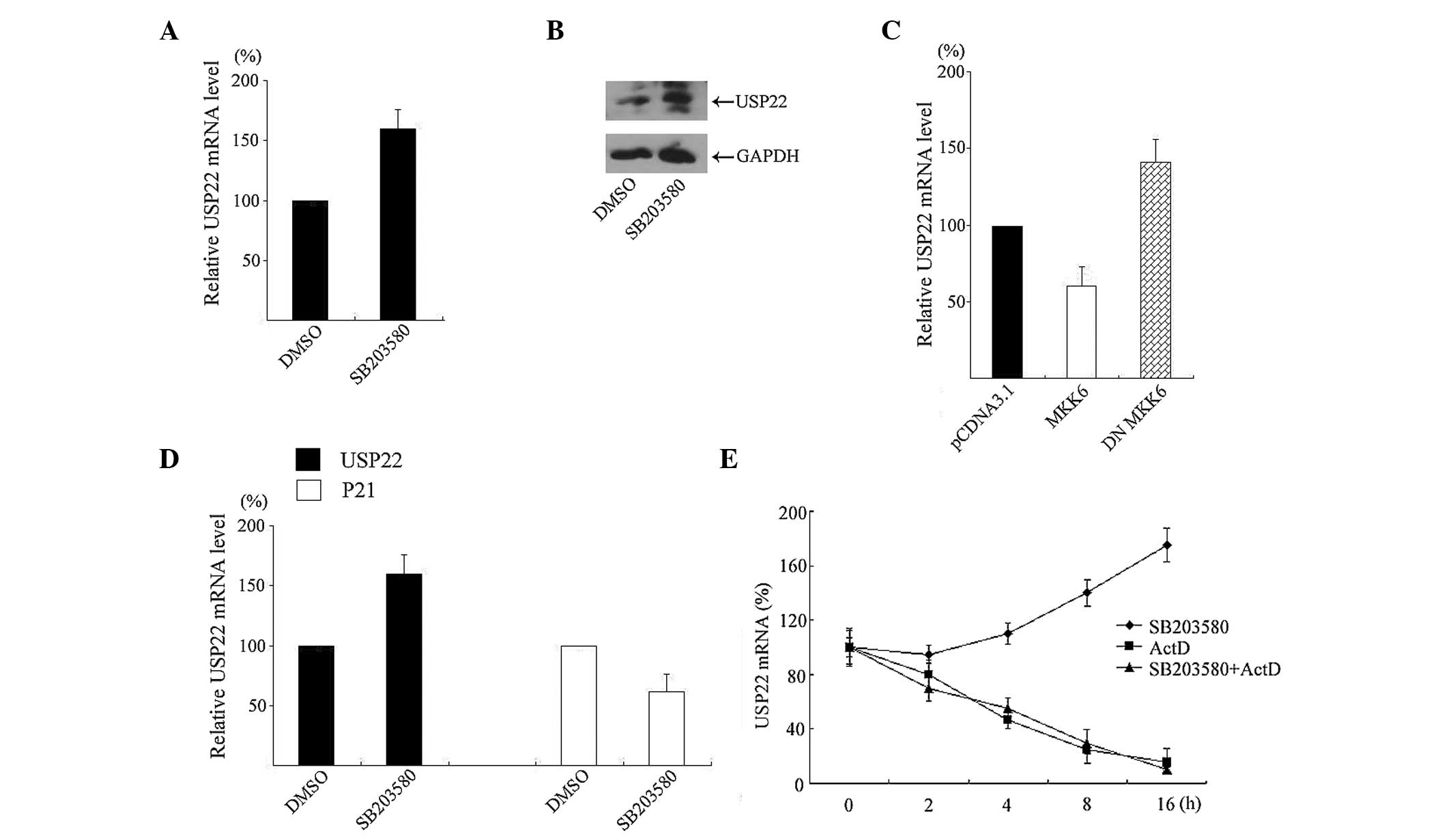
was measured. As shown in Fig. 1A,
incubation of 2 µM SB203580 resulted in an ~150% induction of
luciferase activity relative to DMSO (P<0.05). However,
treatment of SP600125 and U0126 did not affect the luciferase
activity in the tested cells (P>0.05).
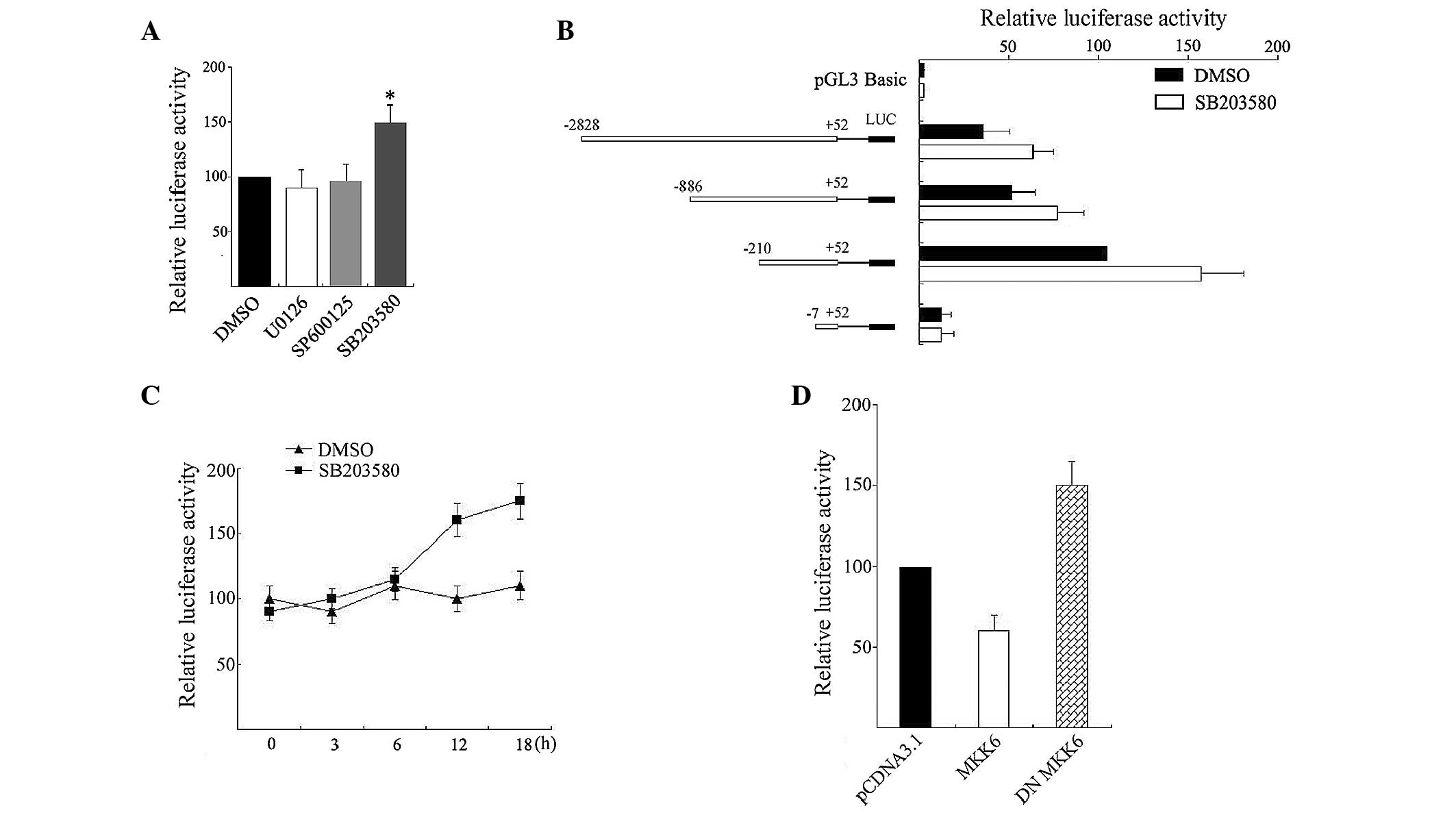 | Figure 1.p38 MAPK regulated USP22
promoter activity. (A) Treatment with 2 µM U0126, 2 µM SB203580, 10
µM SP600125 or vehicle DMSO (−) was added to HeLa cells transfected
with 0.5 µg USP22 promoter construct pGL-210/+52 and 0.1 µg
pRL-TK. The cells were harvested and assayed for luciferase. The
normalized relative luciferase activity for pGL-210/+52 treated
with DMSO was set at 100. The p38 inhibitor SB203580 markedly
enhanced (*P<0.05 compared to DMSO treated) pGL3–210/+52
activity. (B) HeLa cells were transiently transfected with reporter
vectors containing series of 5′ terminal deletion constructs of the
USP22 promoter and treated with 2 µM SB203580. After 12 h,
cells were harvested and assayed for luciferase. The normalized
relative luciferase activity for pGL-210/+52 treated with DMSO was
set at 100. (C) HeLa cells were transfected with pGL-210/+52 and
incubated with SB203580 for different periods. Cells were harvested
and assayed for luciferase. The normalized relative luciferase
activity for pGL-210/+52 treated with DMSO for 0 h was set at 100.
(D) Effects of MKK6 and DN MKK6 on pGL-210/+52 reporter construct.
Cells were transiently transfected with 0.5 µg of reporter
construct plus 1.0 µg of pCDNA3.1 (empty vector), or MKK6 or DN
MKK6. After 24 h, the transfected cells were analysed for
luciferase activity. The normalized relative luciferase activity
obtained in cells transfected with pGL-210/+52 and pCDNA3.1 was set
at 100. MAPK, mitogen-activated protein kinase; USP22,
ubiquitin-specific processing enzyme 22; HeLa, human cervical
carcinoma; MKK6, MAPK kinase 6; DN MKK6, dominant negative
MKK6. |
To further confirm the induction effects and
localize the region in the promoter responsible for this enhancing
effect by SB203580, reporter vectors containing a series of 5′
terminal deletion constructs of the USP22 promoter were
transfected into HeLa cells and treated with 2 µM SB203580 for 12
h. Luciferase activity showed that SB203580 enhancing USP22
promoter activity was observed with constructs pGL-2828/+52,
pGL-886/+52 and pGL-210/+52, but not with construct pGL-7/+52
(Fig. 1B), suggesting a certain degree
of promotion by SB203580 may be mediated through response elements
located within the −210/−7 domain. Subsequently, pGL-210/+52 was
transfected and incubated with SB203580 for different periods. As
shown in Fig. 1C, SB203580 induced
USP22 promoter activity but this was not an early event.
Significant increases in promoter activity (~150%) occurred after
12 h incubation.
MKK6 phosphorylates and activates p38 MAPK (15). To confirm whether MKK6 regulates
USP22 expression, DN MKK6 and pGL-210/+52 were transiently
co-transfected into HeLa cells. A luciferase assay showed that
expression of DN MKK6 increased transcription of USP22 by
150%, which was similar to the action of SB203580. By contrast,
forced expression of a constitutively active MKK6 had a significant
inhibitory effect on USP22 promoter activity (Fig. 1D). All these results demonstrated that
p38 MAPK is involved in the regulation of USP22 promoter
activity.
p38 MAPK regulates endogenous
USP22 expression
As the p38 MAPK pathway is involved in regulating
USP22 promoter activity, the effects of p38 MAPK on
endogenous USP22 expression were assessed. As shown in
Fig. 2A, treatment of HeLa cells with
2 µM SB203580 for 12 h significantly increased USP22 mRNA
expression by ~160% as examined using qPCR. Consistent with the PCR
results, USP22 protein levels were also increased by
SB203580 incubation (Fig. 2B).
MKK6 and DN MKK6 were transfected into HeLa cells
and USP22 mRNA expression was quantitated. As shown in
Fig. 2C, forced expression of MKK6
decreased endogenous USP22 mRNA and DN MKK6 increased the
expression.
p21, a cyclin-dependent kinase inhibitor, is
repressed by USP22 (4). For
this reason, levels of p21 mRNA were measured in response to
SB203580. As shown in Fig. 2D,
SB203580 treatment for 12 h decreased p21 mRNA expression in
HeLa cells.
To further determine whether p38 MAPK influences
USP22 mRNA stability in HeLa cells, actinomycin D was used
to block de novo mRNA transcription. The level of mRNA was
determined at different points in time using qPCR. When cells were
treated with actinomycin D, the half-life of USP22 mRNA was
between 4 and 5 h. When SB203580 was added 12 h before actinomycin
D treatment, the half-life of USP22 mRNA did not change
significantly and was similar to that in the cells without SB203580
treatment (Fig. 2E), excluding the
possibility that p38 MAPK influences USP22 mRNA
stability.
p38 MAPK regulates USP22 via
the Sp1 binding site
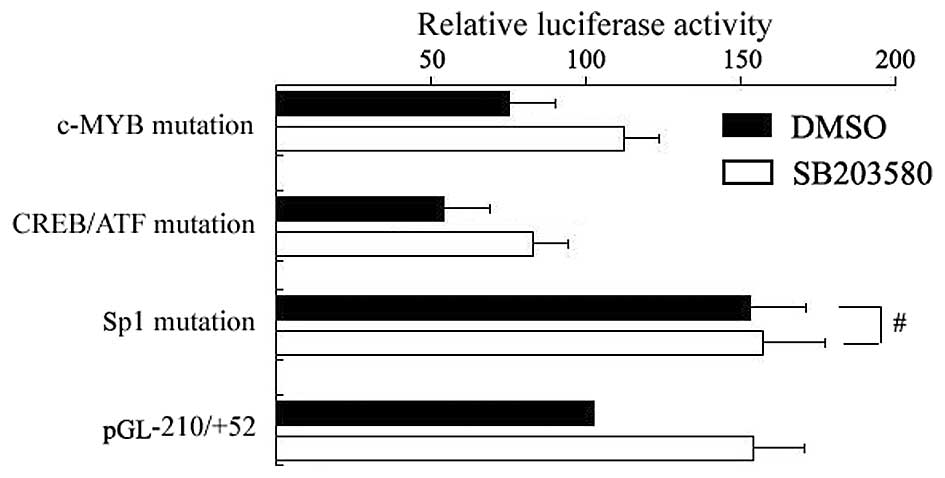
As mentioned above, the region responsible for
enhancing USP22 promoter activity using SB203580 is located
within the −210/−7 domain. TFSEARCH analysis showed there to be
several potential transcription factor binding sites within this
region, including an MYB, a CREB/ATF and a Sp1 binding site. To
characterize the involvement of particular cis elements in
response to SB203580, wild-type and mutant reporter gene constructs
of the USP22 promoters were generated. After plasmids were
transfected into HeLa cells, SB203580 was incubated for another 12
h. As shown in Fig. 3, mutation of the
MYB and CREB/ATF binding sites did not disrupt the enhancing effect
of USP22 promoters by SB203580. However, mutation of the Sp1
binding site (−7/−13) resulted in increased basal USP22
promoter activity and abrogated the enhancing promoter activity
treated by SB203580 (Fig. 3),
suggesting SB203580 elicits promotion of USP22 transcription
through the Sp1 binding site.
Cisplatin represses USP22
expression through p38 MAPK
A previous study has shown that p38 MAPK can be
activated by several anticancer reagents, such as cisplatin
(16). Whether cisplatin can suppress
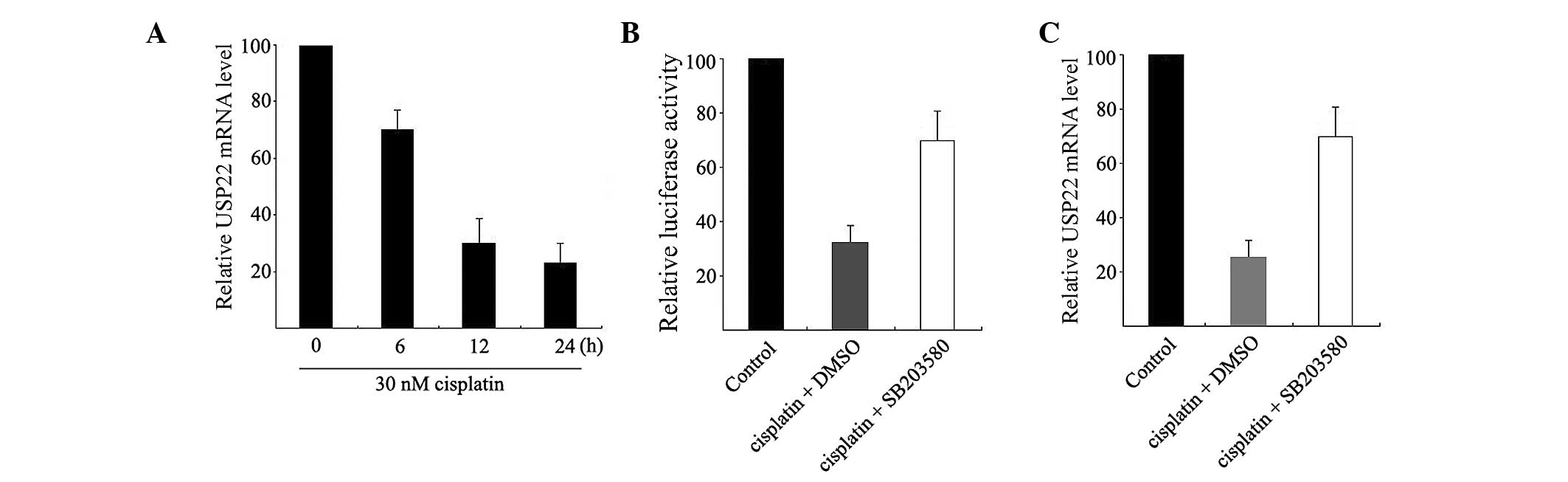
USP22 expression was explored. HeLa cells were treated with
30 nM cisplatin for 0, 6, 12 and 24 h and USP22 expression
at the mRNA level was analyzed using qPCR. As shown in Fig. 4A, cisplatin can induce an extremely
significant decrease in USP22 mRNA expression in HeLa cells.
Significant changes can be observed after 6 h of treatment (52%
inhibition).
Subsequently, HeLa cells were co-treated with
cisplatin and SB203580 for 12 h. As shown in Fig. 4B, cisplatin decreased USP22
promoter activity and this suppression was mostly restored in HeLa
cells by treatment with 2 µM SB203580. Similarly, SB203580
partially restored the cisplatin-induced decrease in USP22
mRNA (Fig. 4C). These results indicate
that cisplatin can suppress USP22 expression at the
transcriptional level and this suppression is possibly mediated by
p38 MAPK.
SB203580 did not protect HeLa cells
from cisplatin-induced apoptosis
As overexpression of USP22 may be a factor
for therapeutic resistance, the role of p38 MAPK/USP22
signaling was determined in the survival of cancer cells exposed to
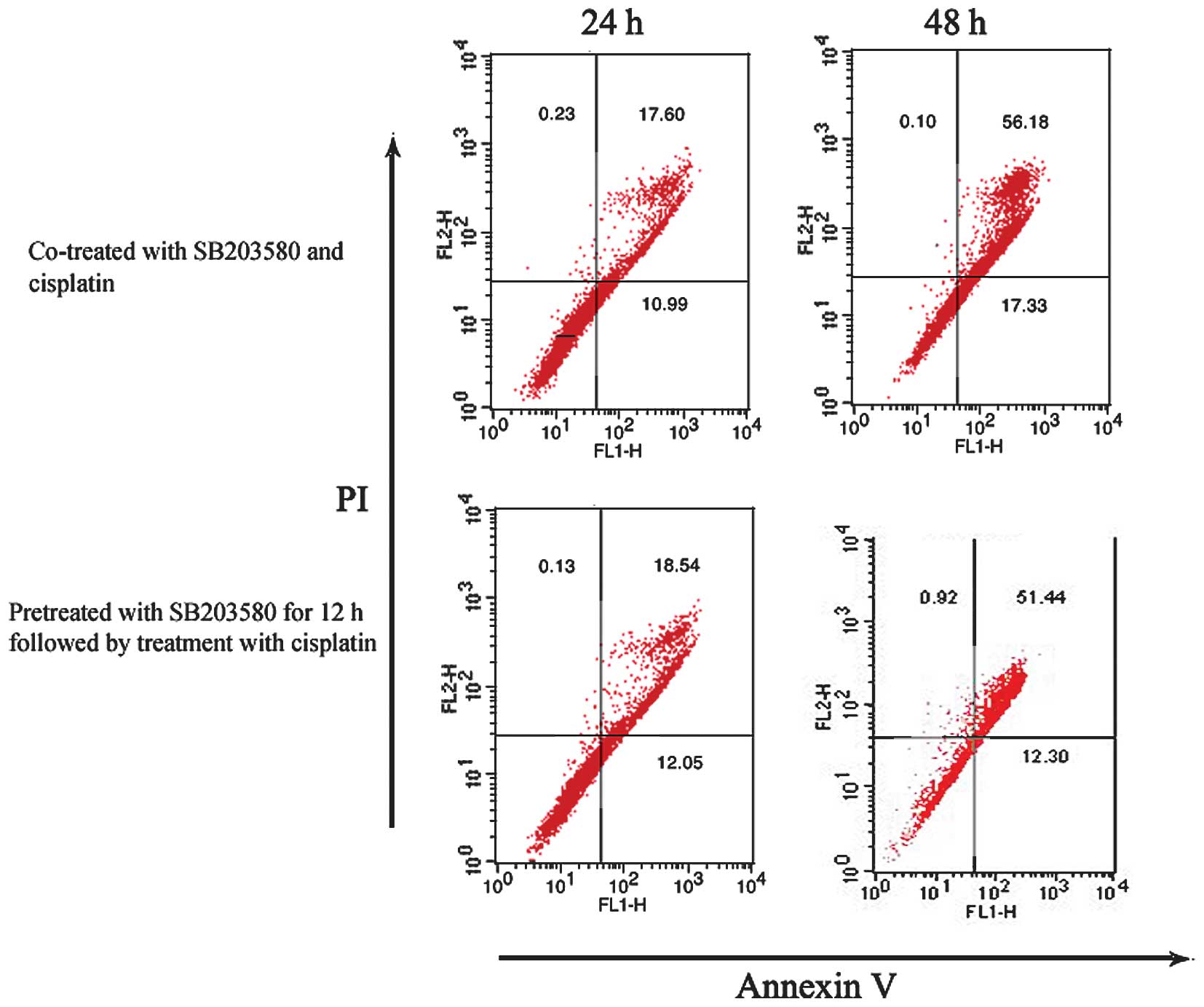
cisplatin. HeLa cells were treated with cisplatin and SB203580 in
two ways and were subsequently analyzed by flow cytometry. One
group of cells was pretreated with SB203580 for 12 h and
subsequently cisplatin was added and allowed to incubate for
another 12 or 24 h. The other was co-treated with SB203580 and
cisplatin for 12 or 24 h. As shown in Fig.
5A, pretreatment or co-treatment of HeLa cells with SB203580
did not result in less cisplatin-induced apoptosis as determined by
Annexin V staining. These results suggest that SB203580-induced
promotion of USP22 expression did not protect HeLa cells
against cisplatin-induced apoptosis.
Discussion
Considering the crucial role of USP22 in
carcinogenesis and tumor progression, p38 MAPK regulation of
USP22 may have important implications. Studying signaling
pathways that regulate USP22 expression may lead to
identification of new therapeutic targets for cancer therapy. The
present study reported that p38 MAPK acts upstream of USP22
and plays a negative role in USP22 transcription. p38 MAPK
was found to regulate USP22 transcription via a Sp1 binding
site. In addition, cisplatin suppressed USP22 expression in
part through p38 MAPK.
One of the findings of the present study is that
USP22 transcription is controlled by p38 MAPK.
Pharmacological inhibitors were used and results showed that the
inhibitor of p38 MAPK enhanced USP22 promoter activity under
culture conditions but ERKs and JNKs did not. The further multiple
experimental results leading to this conclusion are as follows: i)
SB203580 promoted endogenous USP22 mRNA expression, ii)
transfection of the p38 activator MKK6 repressed USP22
promoter activity and endogenous mRNA expression, and iii)
transfection of the p38 inhibitor DN MKK6 enhanced USP22
promoter activity and endogenous mRNA expression. All these results
indicated that p38 MAPK acts as the upstream regulator in
regulation of USP22 transcription. The p38 MAPK pathway is
an important regulator of numerous cellular responses and the
target genes regulated by activated p38 MAPK are complex.
Initially, cytokine genes, such as tumor necrosis factor-α and
interleukin-10 (IL-10), have been described as target genes
promoted by p38 MAPK in T cells and monocytes following stimulation
(17,18). Recently, p38 MAPK has received
increasing attention as a tumor suppressor and its activation
suppresses oncogenesis in cancer cells with different patterns
(19). For example, BMI-1, one
of 11 death signature genes as USP22, is reported to be
downregulated by p38 MAPK by posttranscription modification
(7). Cyclin D1, the oncogene, is also
negatively regulated by p38 MAPK at the transcription level
(20). In the present study, the
possibility that p38 MAPK affects the stability of USP22
mRNA was excluded and p38 MAPK was confirmed to regulate
USP22 at the transcriptional level. Results showed that in
response to SB203580 treatment, expression of USP22 and the
key regulator of cell cycle, p21, exhibited the inverse trend. A
previous study has demonstrated that p38 MAPK participates in
p21 upregulation, subsequently inhibiting cell growth
(21). It is not fully understood how
activated p38 MAPK can upregulate p21 expression. One study
showed that p38 MAPK stabilizes p21 mRNA (22). Another study reported that p38 MAPK can
enhance p21 expression by promoting the transcriptional
elongation (23). USP22 acts
upstream of p21 and represses p21 expression
(4) and the present results indicate
that expression of the p38 MAPK promoter p21 may take place
partially through USP22 downregulation.
p38 MAPK regulation of gene expression is mediated
by the activation of a wide range of protein kinases, transcription
factors and other proteins. The present study showed that p38
MAPK-induced repression of USP22 is mediated by a Sp1
binding site. Bioinformatic analysis showed that the 5′ flank
region of USP22 is a typical feature of the TATA-less
promoter and several DNA motifs are within the basic USP22
promoter, including an Sp1, a CREB/ATF and a c-MYB binding site.
Mutation of these sites has been confirmed to lead to USP22
promoter activity changes (data not shown). The present study
showed only the Sp1 binding site (GGGCGG) ahead of the
transcription start site (−7 to −13) to be responsible for SB203580
treatment, as mutations at this site enhance USP22 promoter
activity and abolish the effects of SB203580 on USP22
promoter activity. Previous studies have shown that the GGGCGG
sequence is involved in regulation of USP22 expression and
is specifically bound by Sp1. Despite its history, transcription
factor Sp1 is activated by phosphorylation in response to p38 MAPK.
For example, it has been reported that p38 MAPK regulation of IL-10
promoters activity is via Sp1. The mechanisms by which p38 MAPK
activates Sp1 to regulate USP22 transcription will be
discussed in a future study.
p38 MAPK activation is also necessary for the
suppression of cancer cell growth as initiated by a variety of
anticancer agents, including cisplatin (24). In the present study, cisplatin was used
to activate p38 MAPK in HeLa cells. Treatment of these cells with
cisplatin suppressed production of USP22 mRNA, which was
mostly accounted for by decreased promoter activity. Cisplatin
exerts its cytotoxic properties by DNA strand-cross links,
therefore activating downstream signaling cascades, which
ultimately induce apoptosis. In the present study, cisplatin was
found to repress USP22 expression via the p38 MAPK pathway;
SB203580 treatment antagonized cisplatin to repress USP22
expression, and the decrease of the USP22 promoter by
cisplatin was also abolished by disruption of the Sp1 binding site.
It has been proved that p38 MAPK is the universal sensor for
cisplatin presence and the activation of p38 MAPK is required for
apoptosis. Considering the p38 MAPK downregulation of USP22,
it is not difficult to understand that cisplatin treatment
suppresses USP22 expression.
USP22 is considered as the putative cancer
stem cell marker and its overexpression has been associated with
therapy resistance. Therefore, it appears that enhanced
USP22 expression by SB203580 may exert anti-apoptotic
effects in theory. However, previous studies showed that the role
of SB203580 on cisplatin-induced apoptosis in tumor cells remains
controversial. Numerous studies showed that the p38 MAPK inhibitor,
SB203580, protected cells from cisplatin-induced apoptosis.
However, several studies showed that SB203580 did not reverse cell
apoptosis but sensitized cells to apoptosis. In the present study,
pretreatment or co-treatment of SB203580 was not observed to
protect HeLa cells from cisplatin-induced apoptosis, suggesting
that although SB203580 enhances USP22 expression, this is
not sufficient to antagonize the cisplatin-induced apoptosis.
In conclusion, the present study provides evidence
that p38 MAPK regulates USP22 promoter activity and its
expression in a human cancer cell line, and cisplatin represses
USP22 expression partly through the p38 MAPK pathway. These
findings provided new insights on the molecular mechanisms
underlying USP22 expression and may have indications for
developing novel therapeutic strategies.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (grant nos. 31000581 and
81460172).
References
|
1
|
Lee HJ, Kim MS, Shin JM, Park TJ, Chung HM
and Baek KH: The expression patterns of deubiquitinating enzymes,
USP22 and Usp22. Gene Expr Patterns. 6:277–284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang XY, Varthi M, Sykes SM, Phillips C,
Warzecha C, Zhu W, Wyce A, Thorne AW, Berger SL and McMahon SB: The
putative cancer stem cell marker USP22 is a subunit of the human
SAGA complex required for activated transcription and cell-cycle
progression. Mol Cell. 29:102–111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao Y, Lang G, Ito S, Bonnet J, Metzger
E, Sawatsubashi S, Suzuki E, Le Guezennec X, Stunnenberg HG,
Krasnov A, et al: A TFTC/STAGA module mediates histone H2A and H2B
deubiquitination, coactivates nuclear receptors, and counteracts
heterochromatin silencing. Mol Cell. 29:92–101. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Atanassov BS and Dent SY: USP22 regulates
cell proliferation by deubiquitinating the transcriptional
regulator FBP1. EMBO Rep. 12:924–930. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao
B, Dong H, Wei J, Song J, Zhang DD, et al: USP22 antagonizes p53
transcriptional activation by deubiquitinating Sirt1 to suppress
cell apoptosis and is required for mouse embryonic development. Mol
Cell. 46:484–494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sussman RT, Stanek TJ, Esteso P, Gearhart
JD, Knudsen KE and McMahon SB: The epigenetic modifier
ubiquitin-specific protease 22 (USP22) regulates embryonic stem
cell differentiation via transcriptional repression of
sex-determining region Y-box 2 (SOX2). J Biol Chem.
288:24234–24246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim S, Kang JK, Kim YK, Seo DW, Ahn SH,
Lee JC, Lee CH, You JS, Cho EJ, Lee HW, et al: Histone deacetylase
inhibitor apicidin induces cyclin E expression through Sp1 sites.
Biochem Biophys Res Commun. 342:1168–1173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiong J, Xu X, Zhou X, Liu J, Gong Z, Wu P
and Li W: USP22 transcriptional activity is negatively regulated by
the histone deacetylase inhibitor trichostatin A. Mol Med Rep.
10:3343–3347. 2014.PubMed/NCBI
|
|
9
|
Qi X, Tang J, Pramanik R, Schultz RM,
Shirasawa S, Sasazuki T, Han J and Chen G: p38 MAPK activation
selectively induces cell death in K-ras-mutated human colon cancer
cells through regulation of vitamin D receptor. J Biol Chem.
279:22138–22144. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang G, Zhang Y, Wu J, Xiong J, Deng H,
Wang J, Yang C and Zhu Z: Osteopontin regulates growth and
migration of human nasopharyngeal cancer cells. Mol Med Rep.
4:1169–1173. 2011.PubMed/NCBI
|
|
11
|
Glinsky GV: Death-from-cancer signatures
and stem cell contribution to metastatic cancer. Cell Cycle.
4:1171–1175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ovaa H, Kessler BM, Rolen U, Galardy PJ,
Ploegh HL and Masucci MG: Activity-based ubiquitin-specific
protease (USP) profiling of virus-infected and malignant human
cells. Proc Natl Acad Sci USA. 101:2253–2258. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Boutros T, Chevet E and Metrakos P:
Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase
regulation: Roles in cell growth, death, and cancer. Pharmacol Rev.
60:261–310. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong J, Che X, Li X, Yu H, Gong Z and Li
W: Cloning and characterization of the human USP22 gene promoter.
PLoS One. 7:e527162012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dashti SR, Efimova T and Eckert RL: MEK6
regulates human involucrin gene expression via a p38alpha - and
p38delta -dependent mechanism. J Biol Chem. 276:27214–27220. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hernandez Losa J, Parada Cobo C, Guinea
Viniegra J, Sanchez-Arevalo Lobo VJ, Ramony Cajal S and
Sanchez-Prieto R: Role of the p38 MAPK pathway in cisplatin-based
therapy. Oncogene. 22:3998–4006. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cain BS, Meldrum DR, Meng X, Dinarello CA,
Shames BD, Banerjee A and Harken AH: p38 MAPK inhibition decreases
TNF-alpha production and enhances postischemic human myocardial
function. J Surg Res. 83:7–12. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma W, Lim W, Gee K, Aucoin S, Nandan D,
Kozlowski M, Diaz-Mitoma F and Kumar A: The p38 mitogen-activated
kinase pathway regulates the human interleukin-10 promoter via the
activation of Sp1 transcription factor in
lipopolysaccharide-stimulated human macrophages. J Biol Chem.
276:13664–13674. 2001.PubMed/NCBI
|
|
19
|
Bulavin DV and Fornace AJ Jr: p38 MAP
kinase's emerging role as a tumor suppressor. Adv Cancer Res.
92:95–118. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hamidouche Z, Fromigue O, Nuber U, Vaudin
P, Pages JC, Ebert R, Jakob F, Miraoui H and Marie PJ: Autocrine
fibroblast growth factor 18 mediates dexamethasone-induced
osteogenic differentiation of murine mesenchymal stem cells. J Cell
Physiol. 224:509–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alderton F, Humphrey PP and Sellers LA:
High-intensity p38 kinase activity is critical for p21(cip1)
induction and the antiproliferative function of G(i)
protein-coupled receptors. Mol Pharmacol. 59:1119–1128.
2001.PubMed/NCBI
|
|
22
|
Lafarga V, Cuadrado A, Lopez de Silanes I,
Bengoechea R, Fernandez-Capetillo O and Nebreda AR: p38
Mitogen-activated protein kinase- and HuR-dependent stabilization
of p21(Cip1) mRNA mediates the G(1)/ S checkpoint. Mol Cell Biol.
29:4341–4351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumari G, Ulrich T and Gaubatz S: A role
for p38 in transcriptional elongation of p21 (CIP1) in response to
Aurora B inhibition. Cell Cycle. 12:2051–2060. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Olson JM and Hallahan AR: p38 MAP kinase:
A convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|