Introduction
Fms-like tyrosine kinase 3 (FLT3) is a member
of the class III receptor tyrosine kinase receptor family (1) and is the most frequently mutated gene
(20–30%) in acute myelogenous leukemia (AML) (2–4). Activating
mutations, as well as overexpression of FLT3, are prevalent
in AML, with a role in leukemogenesis (3,5–8). In adult AML, ~24% of such mutations are
internal tandem duplications (ITD) in the juxtamembrane domain
(2,3),
which result in ligand-independent dimerization and tyrosine
phosphorylation of the receptor (9).
In addition, activating point mutations in the FLT3 tyrosine
kinase domain (TKD; FLT3-TKD), mainly at aspartic acid 835,
are identified in ~7% of AML patients (10).
ITD in FLT3 (FLT3-ITD) is associated
with a higher leukocyte count, increased relapse risk, decreased
disease-free survival (DFS) and decreased overall survival (OS)
(11). Furthermore, multivariate
analyses have shown that FLT3-ITD is the most significant
factor for predicting an adverse outcome in AML (11–14). By
contrast, FLT3-TKD mutations have a smaller effect compared
with FLT3-ITD, but tend to worsen the DFS and OS (10), with the differences being statistically
significant for OS in patients aged ≤60 years (15).
FLT3-ITD alters chemotherapy responses in
vitro and in vivo and confers resistance to doxorubicin,
which depends on p53 (16).
Additionally, DNA repair contributes to the FLT3-ITD
drug-resistant phenotype of primary AML (17). Several studies demonstrate drug
resistance conferred by FLT3-ITD (16,18,19); however, thus far, no studies have
demonstrated the effect of mutations in the FLT3 TKD on
anticancer drug resistance.
The present study examined the effects of
FLT3-ITD and FLT3-TKD on cytotoxic drugs by employing
an IL-3 dependent cell line, Ba/F3. In this cell line,
interleukin-3 (IL-3)-independent cell growth occurs in response to
stably transduced oncogenic signaling, such as via FLT3-ITD
and -TKD (20). This system was used
to evaluate the effect of FLT3-ITD and -TKD oncogenic
signals on the cytotoxicity of daunorubicin (DNR) and cytarabine
(Ara-C). As a result, FLT3-ITD and -TKD signals were
observed to alter the response to DNR.
Materials and methods
Generation of Ba/F3 cells expressing
FLT3-ITD and FLT3-TKD
Our previous studies established the Ba/F3 FLT3-WT
and-ITD cells (6,21). To generate stably expressing
Ba/F3-FLT3-TKD cells, pcDNA FLT3-TKD (D835Y) was generated using
pcDNAFLT3-WT (6) and the Quick change
site directed mutagenesis kit XL (Stratagene, La Jolla, CA, USA)
with the following primers: Sense,
5′-CTTTGGATTGGCTCGATATATCATGAGTGATTC-3′ and anti-sense,
5′-GAATCACTCATGATATATCGAGCCAATCCAAAG-3′. The construct was
confirmed by sequence analysis and transfected into Ba/F3 cells
using a CLB-Transfection device (Lonza, Basel, Switzerland). Stably
transfected Ba/F3 clones were isolated by limiting dilution and
selection with 400 µg/ml neomycin in RPMI (Gibco BRL, Thermo Fisher
Scientific, Rockville, MD, USA) containing 10% heat-inactivated
fetal bovine serum, 100 ng/ml recombinant mouse IL-3 (R&D
Systems, Minneapolis, MN, USA) and 50 µmol/l 2-mercaptoethanol.
Cells were cultured at 37°C in a humidified 5% CO2
atmosphere.
mRNA analysis
cDNA was prepared from cells using reverse
transcriptase (Transcriptor First Strand cDNA Synthesis kit; Roche,
Indianapolis, IN, USA). Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was performed using the
Quantitect SYBR-Green PCR reagent (Qiagen, Miami, FL, USA)
according to the manufacturer's protocol and using an Opticon Mini
Real-time PCR Instrument (Bio-Rad, Hercules, CA, USA), as described
previously (22). The primer sequences
were: FLT3 forward, 5′-TCAAGTGCTGTGCATACAATTCCC-3′ and
reverse, 5′-CACCTGTACCATCTGTAGCTGGCT-3′; and GAPDH forward,
5′-GAAGGTGAAGGTCGGAGT-3′ and reverse; 5′-GAAGATGGTGATGGGATTTC-3′.
The thermal cycling conditions for FLT3 and GAPDH
were incubation at 95°C for 15 min, followed by 35 cycles of 95°C
for 30 sec, 55°C for 30 sec and 72°C for 45 sec. The copy number of
each sample was calculated as previously described (21).
Assessment of viable cells
The proportion of viable cells was determined using
a dye reduction assay involving a tetrazolium salt,
2-(2-methoxy-4-nitrophenyl)-3-(4-nitro-
phenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt
(WST-8; Dojindo, Tokyo, Japan), which is a modification of the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolim assay. The
effective dose (ED)50 values were calculated from the
data obtained from the cell growth assays. Exponentially growing
cells were seeded at 1×104 cells per well in
flat-bottomed 96-well plates, with or without IL-3 in the medium.
Seven different doses were selected for DNR (Sigma, St. Louis, MO,
USA) (1.5, 3.1, 6.2, 12.5, 25, 50 and 100 nM) and Ara-C (Sigma)
(0.15, 0.31, 0.62, 1.25. 2.5, 5 and 10 µM). No DNR or Ara-C was
added to the controls cells. Assays were performed 2 days after the
addition of the drugs. For IL-3(−) cells, exponentially growing
cells were washed twice with phosphate-buffered saline and seeded
without IL-3. Viable cells (%) were calculated as the ratio of the
absorbance (490 nm) of DNR or Ara-C-treated cells to the absorbance
of untreated cells. At least three independent experiments were
performed. The calculated ratios were analyzed and the
ED50 values were obtained using tools at http://www.vector.co.jp/soft/win95/edu/se248471.html.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean and P<0.05 (denoted by one asterisk) was considered to
indicate a statistically significant difference. Comparison of the
means was performed using Student's t-test (http://www.physics.csbsju.edu/stats/t-test_bulk_form.html).
Results
Generation of Ba/F3-FLT3-TKD
cells
Our previous study established the generation of
Ba/F3-FLT3-ITD cells (21), and
for the present study, to clarify whether FLT3-ITD or
FLT3-TKD mediate any specific anticancer drug effects,
Ba/F3-FLT3-TKD cells were also generated. The pcDNA
FLT3-TKD (D835Y) vector was electroporated into Ba/F3 cells
and stably transfected lines were isolated by limiting dilution
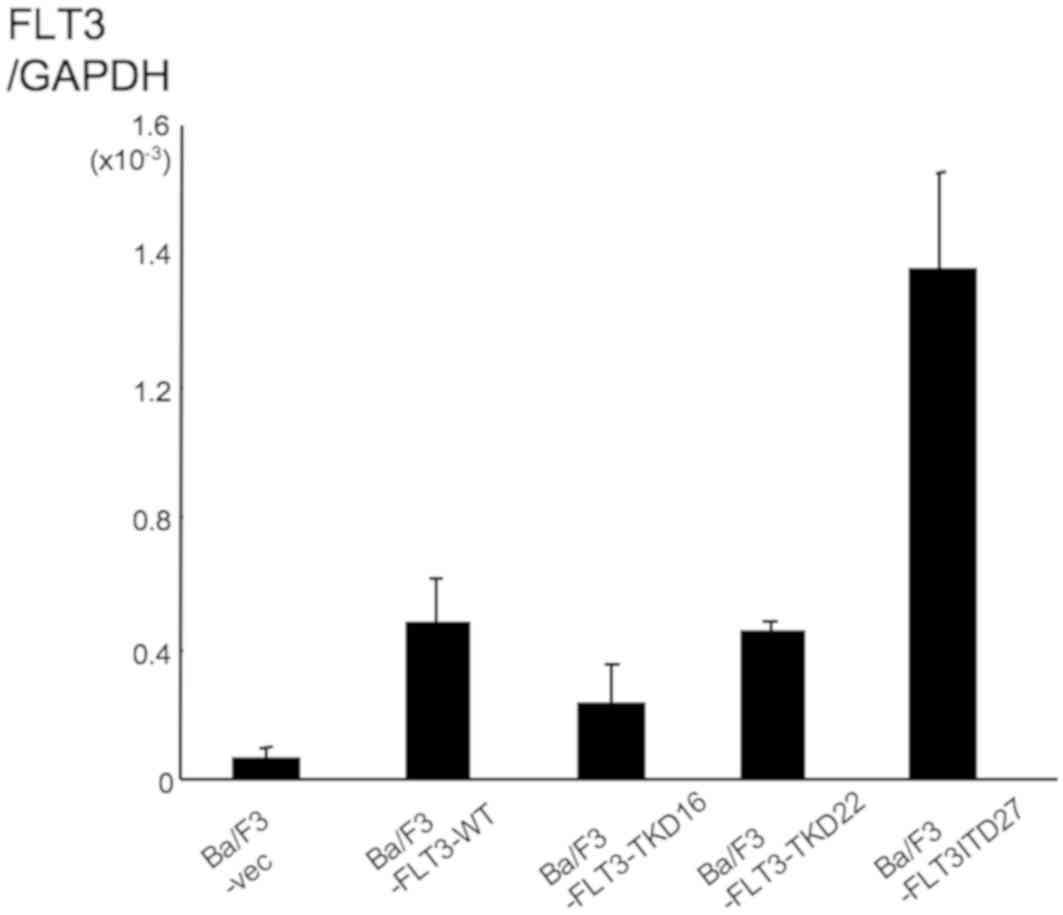
with medium containing neomycin. Among >20 lines obtained, two
clones, Ba/F3-FLT3-TKD16 and Ba/F3-FLT3-TKD22,
exhibited increased levels of FLT3 expression compared with
the parental vector (pcDNA3.1) transfected Ba/F3-vec cells
(Fig. 1). To confirm that the
FLT3-TKD transgene was functionally active,
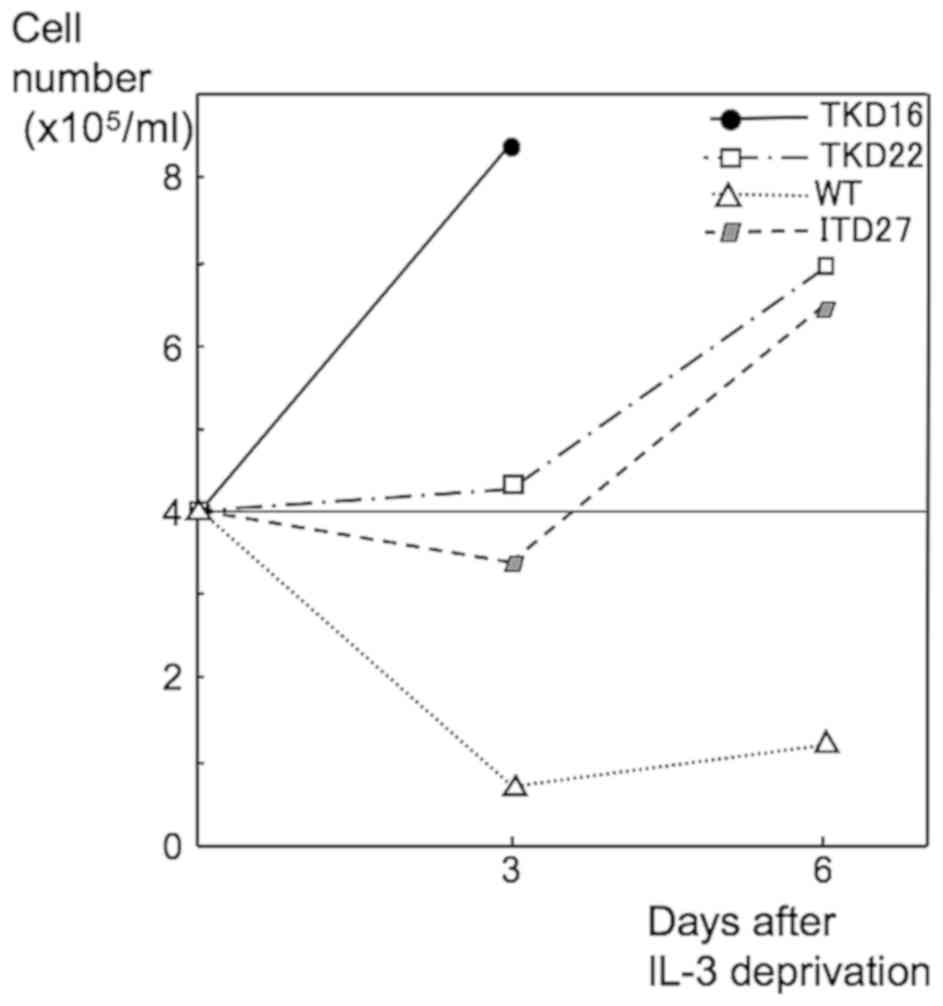
Ba/F3-FLT3-TKD16 and Ba/F3-FLT3-TKD22 cells were
deprived of IL-3. These two lines showed factor-independent growth,
as previously reported (21,23). Similar growth was observed for
Ba/F3-FLT3-ITD (positive control), but not for
Ba/F3-FLT3-WT (negative control) (Fig. 2).
ED50 of DNR increases in IL-3-deprived
Ba/F3-FLT3-ITD27, -ITD29 and -TKD22 cells
Cell viability and ED50 values were
examined in the Ba/F3-FLT3-ITD and Ba/F3-FLT3-TKD
cells. In Ba/F3-FLT3-ITD27, -ITD29 and -TKD22 cells, the
ED50 value was significantly increased in the absence of
IL-3, compared with the controls (Fig.
3A). Additionally, in Ba/F3-FLT3-TKD16 cells, the median value
was higher [IL-3(−), 17.7 nM; IL-3(+), 14.8 nM], although this
difference was not statistically significant (Fig. 3A). By contrast, there were no
differences in the ED50 value for Ara-C between IL-3(−)
and IL-3(+) cells (Fig. 3B). These
results indicate that FLT3-ITD, as well as the mutation of
the FLT3-TKD, may confer DNR resistance to Ba/F3 cells.
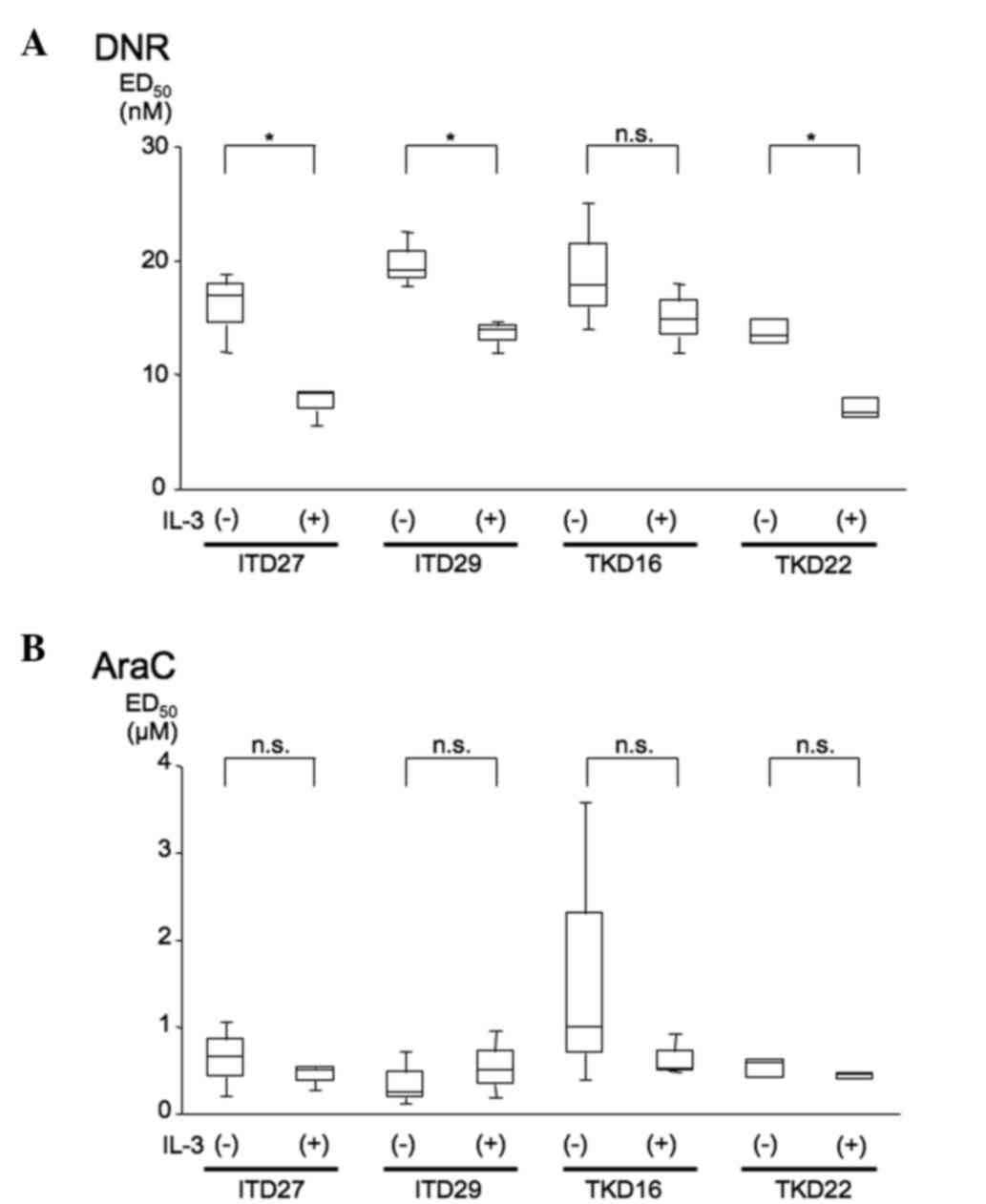 | Figure 3.ED50 values for (A) DNR and
(B) Ara-C. The ED50 values were calculated from the data
obtained from the cell growth assays. WST-8 assays were performed
on cells grown with or without IL-3 in the medium and 2 days after
the addition of drugs: DNR (1.5, 3.1, 6.2, 12.5, 25, 50 or 100 nM)
and Ara-C (0.15, 0.31, 0.62, 1.25. 2.5, 5 or 10 µM). Control cells
were grown without the addition of DNR or Ara-C. Viable cells (%)
were determined by the ratio of the absorbance (490 nm) of DNR or
Ara-C-treated cells relative to the absorbance of untreated cells.
The ratio of viable treated cells to untreated cells was used to
calculate ED50 values. At least three independent
experiments were performed. These data are shown as boxplots
representing the 25 and 75 percentiles, median and 5–95 range. Data
are expressed as the mean ± standard error of the mean and
*P<0.05 was considered to indicate a statistically significant
difference. ED, effective dose; DNR, daunorubicin; IL-3,
interleukin-3; n.s., not significant. |
Discussion
The present data suggest that in AML patients with
FLT3-ITD or FLT3-TKD mutations, DNR is not
efficacious and only causes toxicity. This is consistent with the
findings of a clinical trial showing that while the majority of AML
patients benefit from intensified anthracycline dosing regimens,
high-dose DNR did not provide a significant survival benefit in
patients who had the FLT3-ITD mutation (24). Lee et al (25) also support this notion that
FLT3-ITD causes resistance to doxorubicin; dual treatment of
PML-RARα FLT3-ITD transgenic mice with FLT3 inhibitor
SU11657 and doxorubicin increased sensitivity. Pardee et al
(16) recently reported that
FLT3-ITD confers resistance to doxorubicin in a
p53-dependent manner. In addition to the well-known role of p53 in
apoptosis induction, it has also been shown to induce multiple
prosurvival and DNA repair genes (26). Consistent with this,
FLT3-ITD-expressing AML cell lines and primary patient
samples have increased the levels of reactive oxygen species,
double-strand DNA breaks and increased DNA repair capacity
(17,27).
The present findings are consistent with and support
the above. However, using murine myeloid HF6 and human myeloid K562
cells, our previous study found that FLT3-ITD induced Ara-C
resistance through repression of equilibrative nucleoside
transporter 1 expression (19), which
was not observed in the present study. This discrepancy may be due
to the use of different cell lines. The present study employed
Ba/F3 cells, a murine B-lymphoid cell line, which is distinct from
HF6 and K562 cells. The Ba/F3 cells were used due to their IL-3
deprivation characteristic, which can reveal the effect of FLT3
oncogenic signals. This enabled a specific DNR response to be
identified.
The study also highlights the FLT3-TKD
mutations, which may confer resistance to anthracycline. Although
the data are from a cell line model and are somewhat preliminary,
these results may indicate the benefit of therapy combining
anthracycline and FLT3 inhibitors for patients carrying
FLT3-ITD and FLT3-TKD mutations.
Acknowledgements
The authors would like to thank Mr. Tatsuya Osaka
and Mr. Wataru Nomura for establishing the cell lines and Ms.
Hiroko Nakano for her technical assistance. The study was supported
in part by Grants-in-Aid for Scientific Research (grant no.
26460685) from the Ministry of Education, Science and Culture
(Japan), and the Takeda Sceince Foundation, a foundation from
Kitasato University School of Allied Health Sciences (Grant-in-Aid
for Research Project, no. 2015-1003). Edanz Group Ltd. (www.edanzediting.com) provided editorial
assistance.
References
|
1
|
Rosnet O, Matteï MG, Marchetto S and
Birnbaum D: Isolation and chromosomal localization of a novel
FMS-like tyrosine kinase gene. Genomics. 9:380–385. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yokota S, Kiyoi H, Nakao M, Iwai T, Misawa
S, Okuda T, Sonoda Y, Abe T, Kahsima K, Matsuo Y and Naoe T:
Internal tandem duplication of the FLT3 gene is preferentially seen
in acute myeloid leukemia and myelodysplastic syndrome among
various hematological malignancies. A study on a large series of
patients and cell lines. Leukemia. 11:1605–1609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gilliland DG and Griffin JD: The roles of
FLT3 in hematopoiesis and leukemia. Blood. 100:1532–1542. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takahashi S: Downstream molecular pathways
of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and
therapeutic implications. J Hematol Oncol. 4:132011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ozeki K, Kiyoi H, Hirose Y, Iwai M,
Ninomiya M, Kodera Y, Miyawaki S, Kuriyama K, Shimazaki C, Akiyama
H, et al: Biologic and clinical significance of the FLT3 transcript
level in acute myeloid leukemia. Blood. 103:1901–1908. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takahashi S, Harigae H, Ishii KK, Inomata
M, Fujiwara T, Yokoyama H, Ishizawa K, Kameoka J, Licht JD, Sasaki
T and Kaku M: Over-expression of Flt3 induces NF-kappaB pathway and
increases the expression of IL-6. Leuk Res. 29:893–899. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takahashi S, Harigae H, Kaku M, Sasaki T
and Licht JD: Flt3 mutation activates p21 (WAF1/CIP1) gene
expression through the action of STAT5. Biochem Biophys Res Commun.
316:85–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takahashi S, McConnell MJ, Harigae H, Kaku
M, Sasaki T, Melnick AM and Licht JD: The Flt3 internal tandem
duplication mutant inhibits the function of transcriptional
repressors by blocking interactions with SMRT. Blood.
103:4650–4658. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kiyoi H, Ohno R, Ueda R, Saito H and Naoe
T: Mechanism of constitutive activation of FLT3 with internal
tandem duplication in the juxtamembrane domain. Oncogene.
21:2555–2563. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R,
Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C,
et al: Activating mutation of D835 within the activation loop of
FLT3 in human hematologic malignancies. Blood. 97:2434–2439. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kottaridis PD, Gale RE, Frew ME, Harrison
G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett
AK, et al: The presence of a FLT3 internal tandem duplication in
patients with acute myeloid leukemia (AML) adds important
prognostic information to cytogenetic risk group and response to
the first cycle of chemotherapy: Analysis of 854 patients from the
United Kingdom medical research council AML 10 and 12 trials.
Blood. 98:1752–1759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meshinchi S, Woods WG, Stirewalt DL,
Sweetser DA, Buckley JD, Tjoa TK, Bernstein ID and Radich JP:
Prevalence and prognostic significance of Flt3 internal tandem
duplication in pediatric acute myeloid leukemia. Blood. 97:89–94.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kiyoi H, Naoe T, Nakano Y, Yokota S,
Minami S, Miyawaki S, Asou N, Kuriyama K, Jinnai I, Shimazaki C, et
al: Prognostic implication of FLT3 and N-RAS gene mutations in
acute myeloid leukemia. Blood. 93:3074–3080. 1999.PubMed/NCBI
|
|
14
|
Frohling S, Schlenk RF, Breitruck J,
Benner A, Kreitmeier S, Tobis K, Döhner H and Döhner K: AML Study
Group Ulm. Acute myeloid leukemia: Prognostic significance of
activating FLT3 mutations in younger adults (16 to 60 years) with
acute myeloid leukemia and normal cytogenetics: A study of the AML
Study Group Ulm. Blood. 100:4372–4380. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Thiede C, Steudel C, Mohr B, Schaich M,
Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M,
Neubauer A, et al: Analysis of FLT3-activating mutations in 979
patients with acute myelogenous leukemia: Association with FAB
subtypes and identification of subgroups with poor prognosis.
Blood. 99:4326–4335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pardee TS, Zuber J and Lowe SW: Flt3-ITD
alters chemotherapy response in vitro and in vivo in a
p53-dependent manner. Exp Hematol. 39:473.e4–485.e4. 2011.
View Article : Google Scholar
|
|
17
|
Seedhouse CH, Hunter HM, Lloyd-Lewis B,
Massip AM, Pallis M, Carter GI, Grundy M, Shang S and Russell NH:
DNA repair contributes to the drug-resistant phenotype of primary
acute myeloid leukaemia cells with FLT3 internal tandem
duplications and is reversed by the FLT3 inhibitor PKC412.
Leukemia. 20:2130–2136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zwaan CM, Meshinchi S, Radich JP, Veerman
AJ, Huismans DR, Munske L, Podleschny M, Hählen K, Pieters R,
Zimmermann M, et al: FLT3 internal tandem duplication in 234
children with acute myeloid leukemia: Prognostic significance and
relation to cellular drug resistance. Blood. 102:2387–2394. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin G, Matsushita H, Asai S, Tsukamoto H,
Ono R, Nosaka T, Yahata T, Takahashi S and Miyachi H: FLT3-ITD
induces ara-C resistance in myeloid leukemic cells through the
repression of the ENT1 expression. Biochem Biophys Res Commun.
390:1001–1006. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spiekermann K, Bagrintseva K, Schwab R,
Schmieja K and Hiddemann W: Overexpression and constitutive
activation of FLT3 induces STAT5 activation in primary acute
myeloid leukemia blast cells. Clin Cancer Res. 9:2140–2150.
2003.PubMed/NCBI
|
|
21
|
Takahashi S, Harigae H, Kameoka J, Sasaki
T and Kaku M: AML1B transcriptional repressor function is impaired
by the Flt3 internal tandem duplication. Br J Haematol.
130:428–436. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iseki Y, Nakahara M, Kubo M, Obata F,
Harigae H and Takahashi S: Correlation of PU.1 and signal
regulatory protein α1 expression in PU.1 transgenic K562 cells. Int
J Mol Med. 29:319–323. 2012.PubMed/NCBI
|
|
23
|
Hayakawa F, Towatari M, Kiyoi H, Tanimoto
M, Kitamura T, Saito H and Naoe T: Tandem-duplicated Flt3
constitutively activates STAT5 and MAP kinase and introduces
autonomous cell growth in IL-3-dependent cell lines. Oncogene.
19:624–631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernandez HF, Sun Z, Yao X, Litzow MR,
Luger SM, Paietta EM, Racevskis J, Dewald GW, Ketterling RP,
Bennett JM, et al: Anthracycline dose intensification in acute
myeloid leukemia. N Engl J Med. 361:1249–1259. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee BD, Sevcikova S and Kogan SC: Dual
treatment with FLT3 inhibitor SU11657 and doxorubicin increases
survival of leukemic mice. Leuk Res. 31:1131–1134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Janicke RU, Sohn D and Schulze-Osthoff K:
The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death
Differ. 15:959–976. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sallmyr A, Fan J, Datta K, Kim KT, Grosu
D, Shapiro P, Small D and Rassool F: Internal tandem duplication of
FLT3 (FLT3/ITD) induces increased ROS production, DNA damage and
misrepair: Implications for poor prognosis in AML. Blood.
111:3173–3182. 2008. View Article : Google Scholar : PubMed/NCBI
|