Introduction
Many of the genetic alterations that occur during
cancer development include point mutations in oncogenes and tumor
suppressor genes. Changes in ras family oncogenes at codons
12, 13 and 61 are some of the most common mutational events in
human carcinogenesis (1). Such
point mutations are used as molecular markers for the detection of
cancer cells in clinical samples. In addition, the same approach
may be utilized in carcinogenicity bioassays.
Several techniques are now available to detect point
mutations. Single-strand conformation polymorphism (2), RNase protection assay (3), denatured gel electrophoresis
(4) and allele-specific
oligonucleotides (5) are most
commonly used for this purpose. However, when the level of the
mutant sequence in a mixed population of alleles is 5% or less,
these methods are not applicable. Mutant allele-specific
amplification (MASA) (6–8) is a more powerful method that is used
for detecting a small number of mutant alleles in a large number of
normal alleles, and in one case proved capable of detecting one CC
to TT mutation among 107 wild-type genes in
mitochondrial DNA (9). However,
the reported sensitivity of this method is frequently
10−4 or less (10–12).
Moreover, when using this assay, it is necessary to optimize PCR
conditions such as the cycle number, annealing temperature and
Mg2+ concentration for each primer sequence to avoid
mismatched amplification (13,14).
Heterocyclic amine mutagens are formed in fried or
grilled meats as products of protein pyrolysis or Maillard
reactions (15,16). The human daily intake of one of
these, 2-amino-3, 8-dimethylimidazo[4,5-f]quinoxaline
(MeIQx), is estimated to be 0.2–2.6 μg/person (17). MeIQx can be detected in the urine
of healthy volunteers after eating cooked meat (18–20),
and MeIQx-DNA adducts have been found in kidney and colon tissues
in humans (21). In rats, MeIQx
induces DNA adduct formation in the liver (22) and the development of hepatocellular
carcinomas with treatment at high doses (23).
In the present study, we established a highly
sensitive quantitative method for the detection of the G to T base
substitution mutations in codon 12 of the rat H-ras gene in order
to improve in vivo carcinogenicity bioassays of presumptive
carcinogens. The sensitivity of the assay is 10−5, which
means detection of one mutation in 105 wild-type genes.
Using this method, we here examined H-ras mutations in the
liver DNA of rats treated with the genotoxic carcinogen, MeIQx.
Materials and methods
Preparation of a mutant plasmid
To generate a plasmid with a portion of the rat
H-ras gene containing codon 12, a 463-bp fragment from exon 1,
amplification was performed by PCR using two primers (RHras-146S:
5′-CCACTGGCTTGCTTGCCTACT-3′ and RHras317A:
5′-TTCCGGTAGGAGTCCTGCAA-3′), and the PCR product was subcloned into
the SmaI site of the pKF18 vector (Takara Bio Inc., Shiga,
Japan). A mutant standard with G to T at the second nucleotide of
codon 12 was prepared using a Mutant-Super Express Km kit (Takara
Bio Inc.), and the presence of the mutation was confirmed by DNA
sequencing.
Preparation of standard DNA
To prepare the standard for calibration, plasmid DNA
containing mutant H-ras at 10−2 to 0 was
amplified by high fidelity PCR. Plasmid DNA (5 fmols) was amplified
in a total volume of 100 μl containing 50 pmol each of the
RHras-146S and RHras317A primers, 20 mM Tris-HCl (pH 7.5), 0.2 mM
dNTPs, 1 mM MgCl2, 7.5 mM DTT, 2.5 μg BSA, and 2 units
KOD-Plus-DNA polymerase (Toyobo Co. Osaka, Japan). Following
denaturation at 95°C for 3 min, amplification was performed in a
GeneAmp 9600 thermal cycler (Applied Biosystems, CA, USA) with the
following protocol: 30 cycles for 30 sec at 95°C, 30 sec at 55°C
and 60 sec at 68°C. After amplification, to remove the PCR primers
and dNTPs completely, the PCR products were gel-filtrated on
ProbeQuant G-50 Micro Columns (Amersham Biosciences Co., NJ, USA),
incubated with 10 units exonuclease I and 2 units shrimp alkaline
phosphatase (Amersham Biosciences Co.) at 37°C for 30 min, and then
purified using a GFX PCR DNA Purification kit (Amersham
Biosciences).
Thermosequenase Cycle End Labeling
(TCEL)
The PCR product (100 fmol) was used as a template
for TCEL reactions in a total volume of 10 μl containing 26 mM
Tris-HCl (pH 9.5), 6.51 mM MgCl2, 2 pmol of the primer
for mutation detection (RHrasc12bA: 5′-GGGCACTCTTTCCCACGCCT-3′), 30
pmol ddCTP, 70 fmol ddATP, 70 fmol ddGTP, 70 fmol ddTTP, 30 fmol
[α-33P]ddCTP or [α-33P]ddATP (1,500 Ci/mmol)
(Amersham Biosciences), 0.5 units Thermosequenase (Amersham
Biosciences) and 10% DMSO. Non-labeled dideoxynucleotides were
added to prevent a wrong addition of labeled dideoxynucleotides at
the 3′-end of a detection primer. TCEL reactions were performed in
a GeneAmp 9600 thermal cycler with the following protocol: 50
cycles for 15 sec at 95°C and 15 sec at 55°C. The reaction products
were concentrated with a vacuum concentrator down to a 1/10 volume,
mixed with 5 μl of deionized formamide containing 0.05% BPB and
0.05% XC, heated at 95°C for 2 min, placed on ice and loaded on a
12% (W/V) denaturing polyacrylamide gel containing 7 M urea. Gels
were run at 1,000 V constant voltage for 3.5 h. The amounts of
33P-labeled primers were measured using a bio-imaging
analyzer (BAS 2500; Fuji Photo Film Co., Tokyo, Japan).
Animals and chemicals
MeIQX (purity 99.9%) was purchased from the Nard
Institute (Nishinomiya, Japan). A total of 110 male 21-day-old F344
rats [70 for mutation frequency analysis, 20 for the analysis of
cell proliferation using 5-bromodeoxyuridine (BrdU) labeling and 20
for apoptosis detection] were obtained from Charles River Japan,
Inc. (Atugi, Japan). The animals were housed in an animal facility
room maintained at a 12-h light/dark cycle at a constant
temperature of 25±1°C and a relative humidity of 55±5% and observed
daily.
Animal experiment 1
Seventy rats were employed in this experiment for
measurement of H-ras mutation frequency. They received MeIQx
at doses of 0 (group 1, control), 0.001 (group 2), 0.01 (group 3),
0.1 (group 4), 1 (group 5), 10 (group 6) and 100 ppm (group 7) in a
powdered basal diet for 1 or 2 weeks, continuously. The MeIQx diets
were prepared by Oriental Yeast Co. (Tokyo, Japan), and the
concentration in each diet was confirmed by HPLC. After 1–2 weeks
of MeIQx administration, all rats in each group were sacrificed
under anesthesia with diethyl ether, and the livers were quickly
removed and frozen in liquid nitrogen for molecular assessment. Rat
liver genomic DNA was prepared with a QIAamp DNA Mini kit according
to the manufacturer's instructions (Qiagen, Hilden, Germany), and 1
μg of genomic DNA was amplified by high fidelity PCR using
KOD-Plus-DNA polymerase.
Animal experiment 2
Forty rats were used in the experiment for the
immunohistochemical demonstration of BrdU labeling and apoptosis.
They were fed an MeIQx diet at doses of 0, 1, 10 and 100 ppm
continuously for 2 weeks. Twenty rats each were sacrificed for BrdU
and apoptosis measurement; in the former case 1 h after receiving
an i.p. injection of BrdU (100 mg/kg body weight). At autopsy,
livers were excised and 3-μm slices were cut with a razor blade and
fixed in 10% buffered formalin solution for subsequent
immunohistochemical identification of BrdU-positive cells. After
deparaffinization, H2O2 pre-treatment and
microwaving, liver sections were treated sequentially with normal
horse serum, anti-mouse BrdU antibody (Dako Japan Co., Ltd., Tokyo,
Japan; 1:1000) at 4°C overnight, horse anti-mouse IgG (1:400) for
30 min and the avidin-biotin peroxidase complex reagent for 30 min.
Analysis of ∼5,000 hepatocytes in each sample was performed, and
BrdU labeling indices for rat liver were calculated as percentages
of positively stained cells among ∼1,000 hepatocytes. To detect
apoptotic cells in sections of fixed, paraffin-embedded tissue, the
TUNEL method was applied using an ApopTag Peroxidase kit for in
situ apoptosis detection according to the manufacturer's
instructions (Intergen Co., NY, USA).
Statistical analysis
Comparisons of the observed values for statistically
significant differences were performed using the Student's t-test,
with the aid of the StatView ver. 5.0 statistical package (Abacus
Concepts, Inc., CA, USA).
Results
The principle of the TCEL method
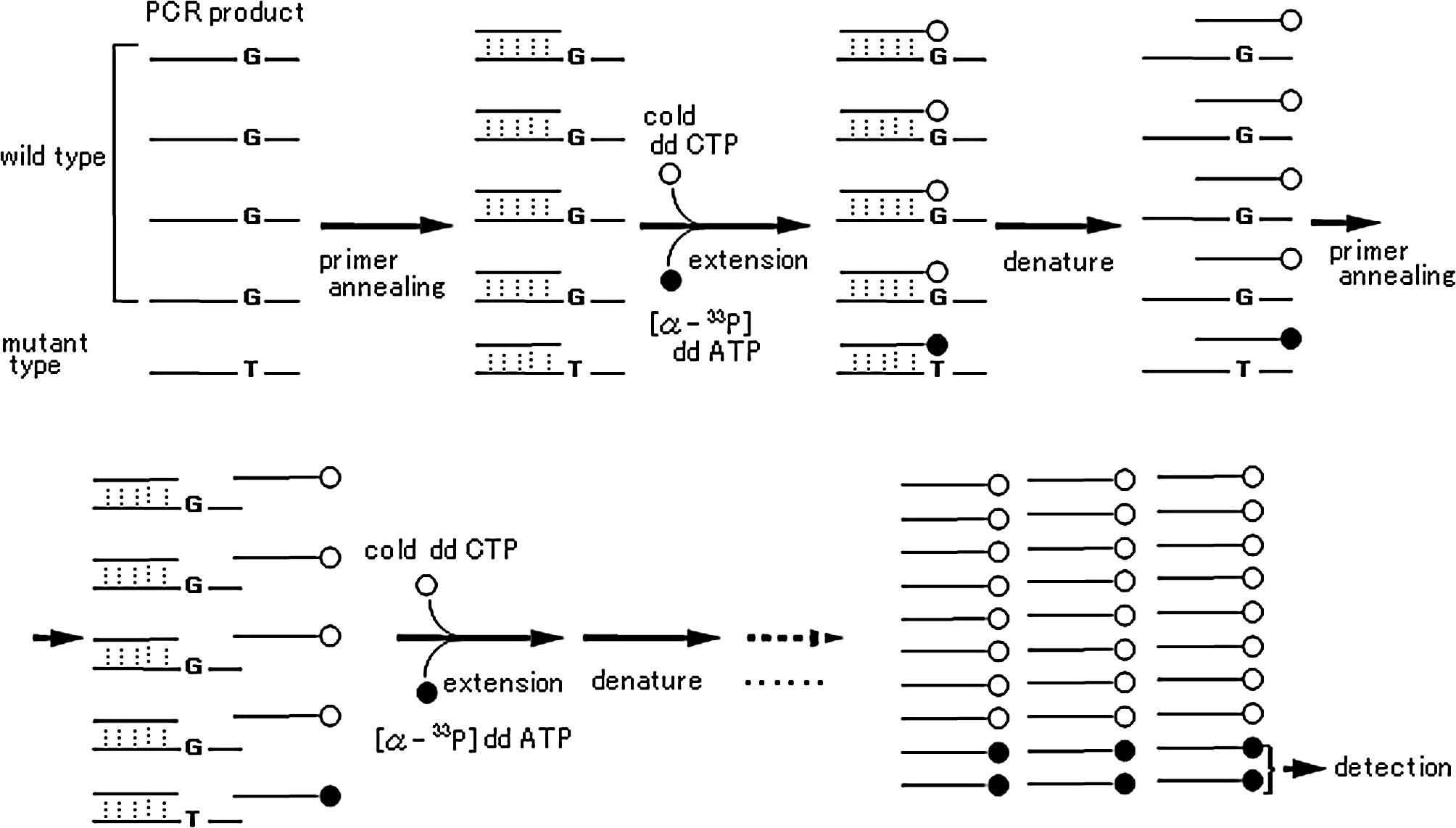
The principle of the TCEL method based on single
nucleotide primer extension is as follows: i) a detection primer is
synthesized to end at one nucleotide 5′ of the mutation being
probed for in the PCR product; ii) a cycle sequencing reaction is
performed using Thermosequenase and a mixture of the non-labeled
terminator (ddCTP) and mutant 33P-labeled terminator
([α-33P]ddATP), so that extension by a single nucleotide
occurs with the primer by either normal or mutant terminators
according to the mutation rate (Fig.
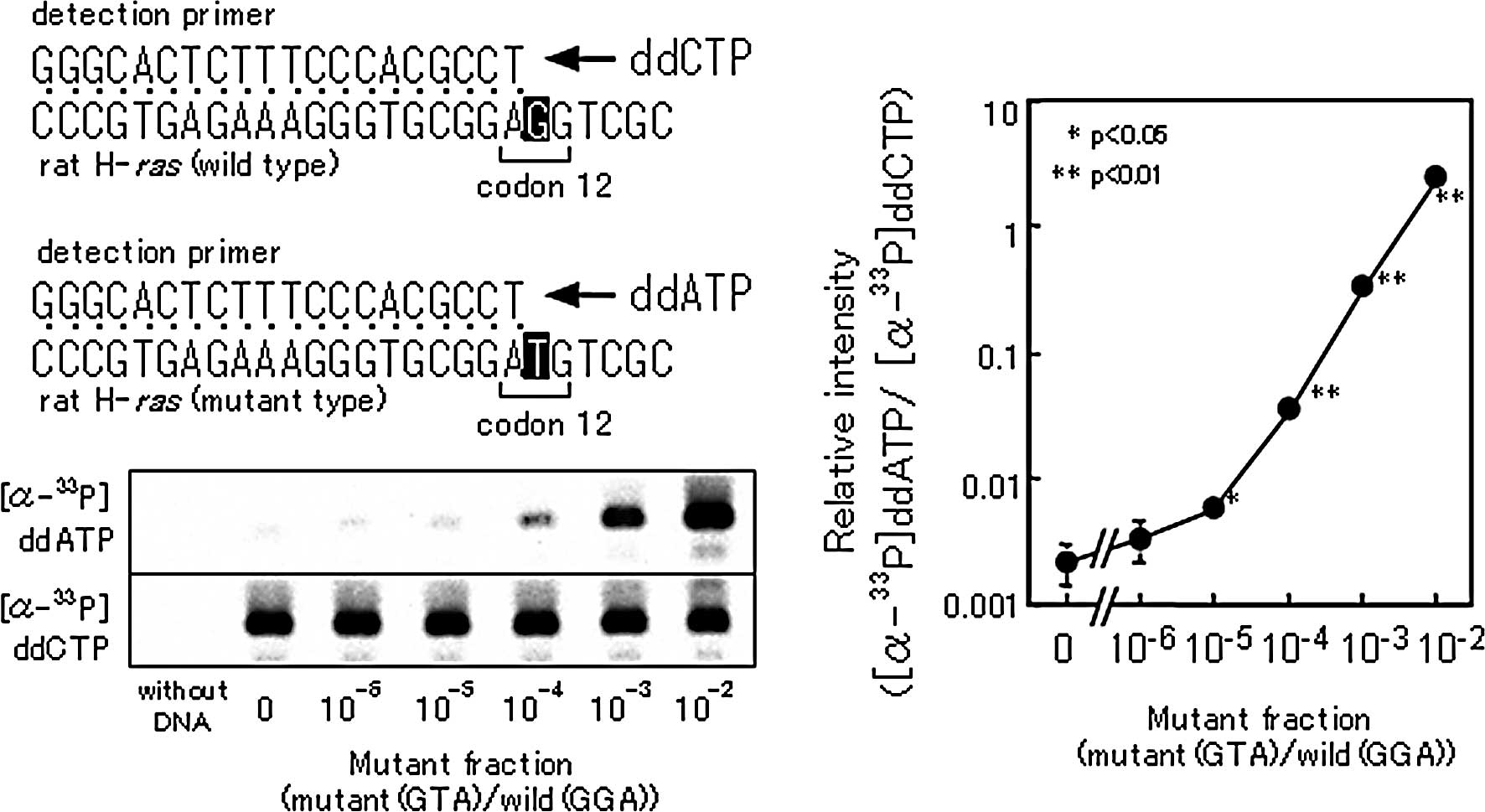
1). Fig. 2 shows a
representative standard curve for the TCEL assay, which allows
detection of one mutant allele among 105 normal
alleles.
Animal experiments
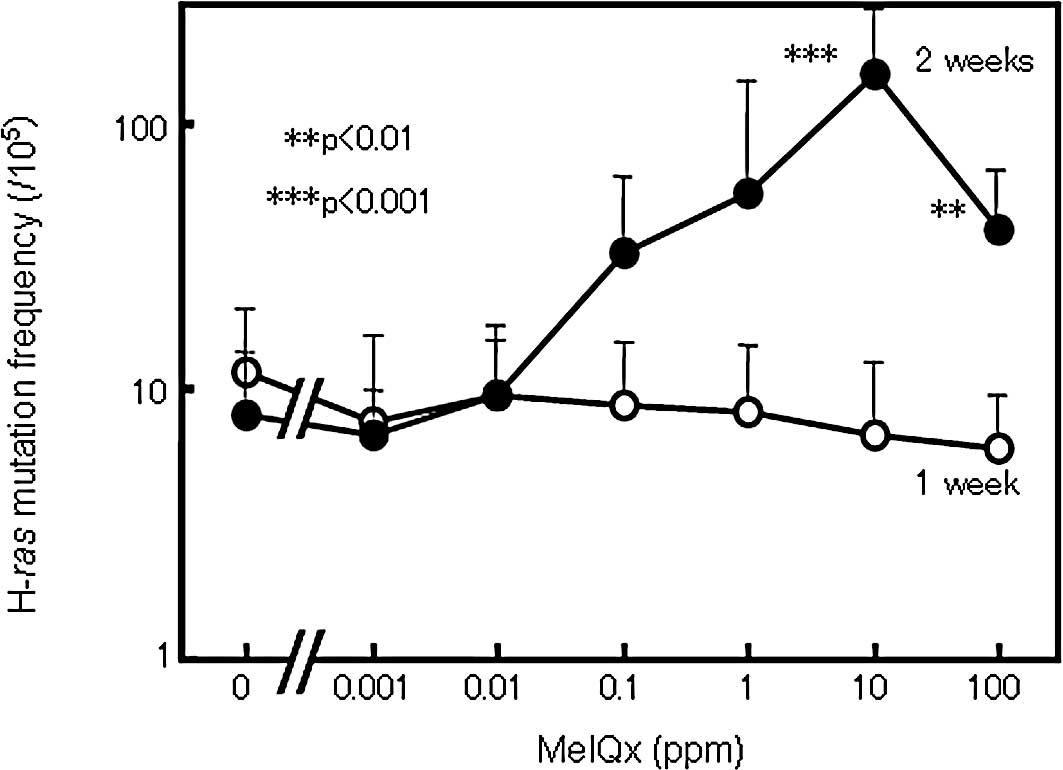
The frequency of H-ras mutations detected by
TCEL assay in the rat livers of all groups receiving from 0.001 to
100 ppm of MeIQx in the diet for 1 week did not differ from the
control value (Fig. 3). On the
other hand, in the livers of rats treated with MeIQx for 2 weeks,
the H-ras mutation frequency was elevated generally in a
dose-dependent manner with statistical significance at 10 and 100
ppm (Fig. 3). However, the level
in the group treated with 100 ppm MeIQx was non-significantly lower
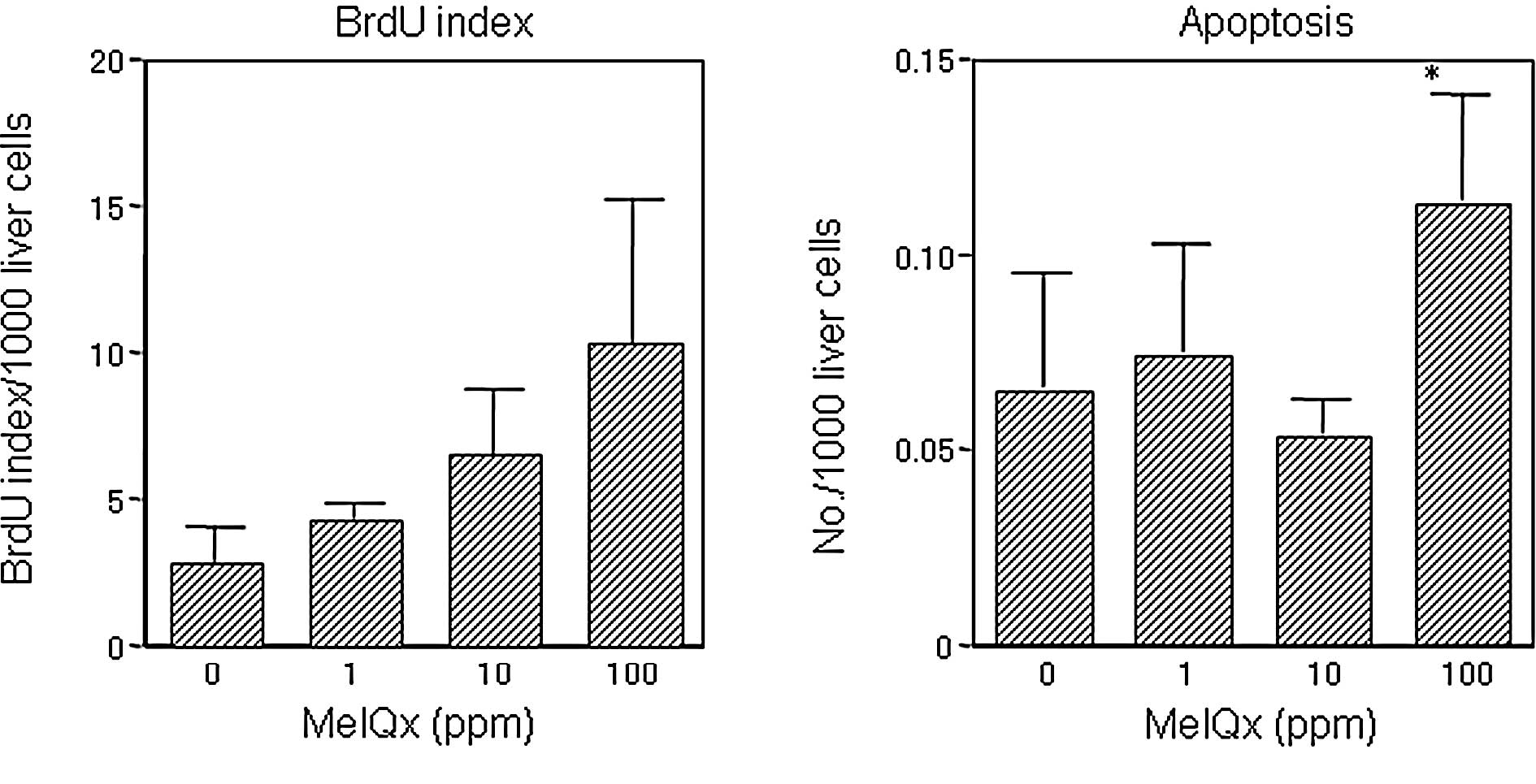
than the level in the group treated with 10 ppm (Fig. 3). Liver BrdU and apoptotic indices
for the groups receiving 1 and 10 ppm MeIQx did not differ from the
controls (Fig. 4), in contrast to
the significant elevation apparent in the 100-ppm MeIQx group.
Discussion
The MASA method is one of the most highly sensitive
methods to detect mutation alleles in the presence of a very large
excess of normal alleles (6–8). It
is based on the inability of Taq DNA polymerase to elongate a
primer when the 3′ end does not match the template DNA. However, in
many cases, substantial modification is necessary to achieve
reliable results (13,14). We previously examined the
sensitivity, quantitativeness and reproducibility of the MASA
method for the detection of rat H-ras mutations, but we did
not obtain satisfactory results. In our investigation, the limit of
detection for MASA was one mutant allele for a background of
thousands of normal alleles (data not shown), and the sensitivity
level was equal to that previously reported (10).
In the present study, we established a highly
sensitive quantitative method, the TCEL assay, based on single
nucleotide primer extension and established a quantitative range of
1 in 10−2 to 10−5. To confirm the
reliability, blind tests with standard samples were performed by
two workers independently, and the obtained results were reliable
(data not shown). One of the purposes of the present investigation
was to check the feasibility of adding somatic mutations to
parameters for assessment of in vivo carcinogenicity. Using
our method, we quantified H-ras mutations in the livers of
rats treated with a genotoxic carcinogen, MeIQx. Thus, while the
administration of MeIQx for 1 week did not influence the mutation
frequency, presumably since this does not provide sufficient time
for fixation, in line with previous observations (8), the treatment with 10 and 100 ppm
MeIQx for 2 weeks induced appreciable H-ras mutations. When
administration of MeIQx for 2 weeks was repeated, similar results
were obtained (unpublished data). It has been reported that a high
dose of MeIQx causes a decrease in the transformation rate in the
in vitro cell transformation assay using C3H/M2 fibroblasts
(24). In the present
investigation, a significant elevation in liver apoptotic and BrdU
indices in the 100-ppm MeIQx group was found and this partially
explains the lack of dose dependence, with a decrease in the
mutation rate in the high dose group resulting from the
cytotoxicity of MeIQx, while the BrdU index reflects compensatory
regeneration.
In summary, we developed a highly sensitive,
quantitative and reproducible method for detecting small amounts of
mutant H-ras DNA. Furthermore, our method may offer another
approach to the assessment of in vivo carcinogenicity.
Moreover, it should be applicable to various genetic targets with
appropriate simple changes in primer sequences. The technique may
also be used for the analysis of clinical samples and may provide a
useful tool for cancer screening or early diagnosis of human
cancer.
Acknowledgements
This study was supported by a grant
from the Japan Science and Technology Corporation, included in the
Project of Core Research for Evolutional Science and Technology
(CREST) and Regional Science Promotion Program (RSP). We thank Dr
Shinji Hirotsune for the help and support during the development of
the methodology.
References
|
1.
|
Sistonen L and Alitalo K: Activation of
c-ras oncogenes by mutations and amplification. Ann Clin Res.
18:297–303. 1986.PubMed/NCBI
|
|
2.
|
Orita M, Suzuki Y, Sekiya T, et al: Rapid
and sensitive detection of point mutations and DNA polymorphisms
using the polymerase chain reaction. Genomics. 5:874–879. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Winter E, Yamamoto F, Almoguera C, et al:
A method to detect and characterize point mutations in transcribed
genes: amplification and overexpression of the mutant c-Ki-ras
allele in human tumor cells. Proc Natl Acad Sci USA. 82:7575–7579.
1985. View Article : Google Scholar
|
|
4.
|
Myers RM, Lumelsky N, Lerman LS, et al:
Detection of single base substitutions in total genomic DNA.
Nature. 313:495–498. 1985. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Conner BJ, Reyes AA, Morin C, et al:
Detection of sickle cell beta S-globin allele by hybridization with
synthetic oligonucleotides. Proc Natl Acad Sci USA. 80:278–282.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Ehlen T and Dubeau L: Detection of ras
point mutations by the polymerase chain reaction using
mutation-specific, inosine-containing oligonucleotide primers.
Biochem Biophys Res Commun. 160:441–447. 1989. View Article : Google Scholar
|
|
7.
|
Ichikawa T, Yano Y, Uchida M, et al: The
activation of K-ras gene at an early stage of lung tumorigenesis in
mice. Cancer Lett. 107:165–170. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yano T, Yuasa M, Murakami A, et al: The
detection of chemically initiated cells having the mutation of
K-ras gene at an early stage of lung carcinogenesis in mice. Anal
Biochem. 244:187–189. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kawasaki K, Suzuki T, Ueda M, et al: CC to
TT mutation in the mitochondrial DNA of normal skin: relationship
to ultraviolet light exposure. Mutat Res. 22:35–43. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Takeda S, Ichii S and Nakamura Y:
Detection of K-ras mutation in sputum by mutant-allele-specific
amplification (MASA). Hum Mutat. 2:112–117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Liang TJ, Hasegawa K, Munoz SJ, et al:
Hepatitis B virus precore mutation and fulminant hepatitis in the
United States. A polymerase chain reaction-based assay for the
detection of specific mutation. J Clin Invest. 93:550–555. 1994.
View Article : Google Scholar
|
|
12.
|
Horikoshi T, Lenz HJ, Danenberg K, et al:
Quantitative determination of the ratio of mutated to normal ras
genes in the blood of leukemia patients by allele-specific PCR.
Leuk Res. 18:693–702. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Etoh T, Ueo H, Inoue H, et al: Clinical
significance of K-ras mutations in intraoperative tumor drainage
blood from patients with colorectal carcinoma. Ann Surg Oncol.
8:407–412. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Hashimoto T, Kobayashi Y, Ishikawa Y, et
al: Prognostic value of genetically diagnosed lymph node
micrometastasis in non-small cell lung carcinoma cases. Cancer Res.
60:6472–6478. 2000.PubMed/NCBI
|
|
15.
|
Felton JS and Knize MG: Occurrence,
identification, and bacterial mutagenicity of heterocyclic amines
in cooked food. Mutat Res. 259:205–217. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Snyderwine EG: Some perspectives on the
nutritional aspects of breast cancer research. Food-derived
heterocyclic amines as etiologic agents in human mammary cancer.
Cancer. 74:1070–1077. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
IARC Working Group on the evaluation of
carcinogenic risk to humans: MeIQx
(2-amino-3,8-demethylimidazo[4,5-f]quinoxaline). IARC
Monographs on the Evaluation of Carcinogenic Risks to Humans – Some
Naturally Occurring Substances: Food Items and Constituents,
Heterocyclic Aromatic Amines and Mycotoxins. 56. Armstrong B: IARC
Publisher; Lyon: pp. 221–228. 1993
|
|
18.
|
Murray S, Gooderham NJ, Boobis AR, et al:
Detection and measurement of MeIQx in human urine after ingestion
of a cooked meat meal. Carcinogenesis. 10:763–765. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ushiyama H, Wakabayashi K, Hirose M, et
al: Presence of carcinogenic heterocyclic amines in urine of
healthy volunteers eating normal diet, but not of inpatients
receiving parenteral alimentation. Carcinogenesis. 12:1417–1422.
1991. View Article : Google Scholar
|
|
20.
|
Tannenbaum SE, Stillwell WG, Ji H, et al:
MeIQx (2-amino-3,8-demethylimidazo [4,5-f]quinoxaline):
metabolism in humans and urinary metabolites in human populations.
Heterocyclic Amines in Cooked Foods, Possible Human Carcinogens.
Asamson RH, Gustafsson JA, Ito N, Nagao M, Sugimura T, Wakabayashi
K and Yamazoe Y: Princeton Scientific Publishing Co. Inc.;
Princeton: pp. 197–206. 1993
|
|
21.
|
Totsuka Y, Fukutome K, Takahashi M, et al:
Presence of
N2-(deoxyguanosin-8-yl)-2-amino-3,8-dimethylimidazo[4,5-f]
quinoxaline (dG-C8-MeIQx) in human tissues. Carcinogenesis.
17:1029–1034. 1996.PubMed/NCBI
|
|
22.
|
Yamashita K, Adachi M, Kato S, et al: DNA
adducts formed by
2-amino-3,8-dimethylimidazo[4,5-f]quinoxaline in rat liver:
dose-response on chronic administration. Jpn J Cancer Res.
81:470–476. 1990.
|
|
23.
|
Kato T, Ohgaki H, Hasegawa H, et al:
Carcinogenicity in rats of a mutagenic compound,
2-amino-3,8-dimethylimidazo[4,5-f] quinoxaline.
Carcinogenesis. 9:71–73. 1998.PubMed/NCBI
|
|
24.
|
Pfau W, Martin FL, Cole KJ, et al:
Heterocyclic aromatic amines induce DNA strand breaks and cell
transformation. Carcinogenesis. 20:545–551. 1999. View Article : Google Scholar : PubMed/NCBI
|