Introduction
Myocardial infarction results in insufficient blood
supply, decreasing the function of myocardial cells and ventricular
remodeling, eventually leading to heart failure (1). Myocardial cell apoptosis is a
critical feature of myocardial infarction (2). Erythropoietin (EPO) is a glycoprotein
hormone produced by the kidney and fetal liver (3). In recent studies, EPO was shown to
have protective effects against ischemic injury in the brain
(4), spinal cord (5), kidney (6), and even in the myocardium (7). Clinical application of EPO to treat
anemia associated with congestive heart failure was found to
significantly improve cardiac function in these patients (8), and increasing evidence suggests that
EPO increases the ability of myocardial cells to endure ischemic
tolerance of hypoxia. Current findings suggest that the protective
effects of EPO on myocardial infarction depend on the
anti-apoptotic effect of certain cytokines, but its exact mechanism
remains unknown (9).
The endoplasmic reticulum (ER) has received much
attention recently for its role in signal transduction relevant to
cell survival and death. Myocardial infarction conditions induce
Ca2+ overload and/or accumulation of unfolding or
misfolding proteins within the ER. ER stress induces multiple
responses, including adaptive changes in translation, protein
folding, secretion and degradation. Prolonged ER stress triggers
apoptosis by induction of C/EBP homologous protein (CHOP),
activation of c-JUN NH2-terminal kinase (JNK) and finally
production of pro-apoptotic factors (caspase-12), which may
contribute to the development of diabetic nephropathy (10). In addition, caspase-12 causes ER
stress-induced apoptosis, which is a feature of
ischemia/reperfusion (I/R) injury, and thus suppressing ER stress
may protect the myocardium from I/R injury (11).
In the present study, we hypothesized that the
protective effect of EPO against ischemic injury in the myocardium
was through the reduction of ER stress-induced apoptosis. We
induced myocardial infarction in a rat model by ligating the left
anterior descending coronary artery and examined systolic and
diastolic function 4 weeks after myocardial infarction, as well as
myocardial fibrosis, apoptosis, angiogenesis and expression of
caspase-12 in myocardial infarction myocardium, non-infarcted
myocardium and myocardial infarction myocardium treated with EPO.
The aim of this study was to determine the relationship between a
decrease in caspase-12 expression in the heart and the protective
effects of EPO on myocardial infarction.
Materials and methods
Materials
Sprague-Dawley male rats were purchased from the
Experimental Animal Centre of Zhejiang University (Hangzhou,
China). Recombinant human erythropoietin (rhEPO) was obtained from
Shenyang Sunshine Pharmaceutical Co., Ltd. (Shenyang, China).
Monoclonal caspase-12 antibody was purchased from Abcam Company
(Cambridge, MA, USA). Langendorff heart perfusion apparatus was
purchased from Nanjing Medease Science and Technology Co., Ltd.
(Nanjing, China).
Animal model induction
Thirty male Sprague-Dawley rats with a body weight
of ∼210 g were used for this study and were randomly divided into
three groups (10 rats/group): sham-operation group, myocardial
infarction group and EPO treatment group. Under pentobarbital
anesthesia, rats were intubated and mechanically ventilated with a
Harvard respirator. Physiological arterial blood gas levels were
maintained by adjusting the respirator and oxygen supplement.
Hearts were exposed via the left thoracotomy, and a coronary snare
was prepared around the left anterior artery. In the myocardial
infarction and EPO treatment groups, the left anterior descending
coronary artery was ligatured between the pulmonary cone and the
left atrial appendage, and the left anterior descending coronary
artery blood supply region became white. Subsequently, the rats
were intraperitoneally injected with 4.0×106 U of
penicillin to prevent infection, and revived at 30°C, while the
sham-operation rats only received thoracotomy. After the operation,
the EPO group rats received an intraperitoneal injection of EPO
(1×103 IU/kg/day), while the sham-operation and
myocardial infarction group rats received an intraperitoneal
injection of normal saline (1 ml/kg/day) for 4 weeks.
Hemodynamic measurements
Coronary perfusion using the Langendorff system with
modified Krebs-Henseleit (KH) solution containing 118.5 NaCl, 4.7
KCl, 1.2 MgSO4, 1.8 CaCl2, 24.8
NaHCO3, 1.2 KH2PO4 and 10 glucose
(in mM) was carried out. After 4 weeks, five isolated hearts in
each group were extracted. A KH buffer-filled latex balloon was
inserted into the left ventricle (LV) and was adjusted to 5–10 mmHg
of the left ventricular end-diastolic pressure (LVEDP) at the
beginning of the experiment. Each isolated heart was perfused with
37°C oxygenated (95% O2-5% CO2) modified KH
buffer. Left ventricular systolic pressure (LVSP) and LVEDP were
recorded with the Langendorff heart perfusion apparatus system by
averaging the values for ten beats. Left ventricular diastolic
pressure (LVDP) was calculated as follows: LVDP = LVSP - LVEDP.
Immunohistochemistry and H&E
staining
Immunohistochemistry was used to localize the
caspase-12 antigen. Five rats from each group were anesthetized
with a lethal dose of pentobarbital. Their thoracic cavities were
then opened and perfused intracardially with normal saline.
Following saline perfusion, the animals were perfused with 300–400
ml of fixative containing 4% paraformaldehyde in 0.1 M phosphate
buffer (pH 7.4). After perfusion, the heart of each rat was
removed, fixed in the same fixative for 4 h and then placed in 30%
phosphate-buffered sucrose until the tissue sank. Tissue sections
(8 μm) were cut on a freezing microtome through the coronary planes
of the heart. Sections were then rinsed in 0.01 M
phosphate-buffered saline (PBS) and mounted onto 0.02%
poly-l-lysine-coated slides.
Tissue sections were washed in PBS, incubated in 1%
bovine serum albumin (BSA) for 30 min, and then incubated overnight
at 4°C in the primary antibody (monoclonal caspase-12 antibody)
plus 1% BSA in PBS. The dilution of the primary antibody was 1:100.
Control sections were incubated in PBS alone. The next day, the
sections were incubated in a biotinylated goat anti-mouse secondary
antibody (diluted to 1:200 in PBS), and subsequently in an
avidin-horseradish peroxidase solution. Immunolabelling was
visualized with 0.05% DAB (Sigma, St. Louis, MO, USA) plus 0.3%
H2O2 in PBS. The sections were then
dehydrated through ethanol and xylene before being cover-slipped
with Permount™. H&E staining was also applied to determine
morphological changes.
Image analysis
For immunohistochemistry, the left ventricular
tissues were selected on each slide and examined at a magnification
of x400 with a UTHSCSA Image Tool 3.0 (University of Texas Medical
School at San Antonio, San Antonio, TX, USA). The number and
optical density of the caspase-12-positive cells were measured.
Statistical analysis
All data are the means ± SD. Statistical analysis
was performed using SPSS® for Windows®
version 12.0 statistical software (SPSS Inc., St. Louis, MO, USA).
The significance of any differences between the groups was
evaluated using the one-way ANOVA. P-values <0.05 were
considered significant.
Results
Hemodynamic parameters
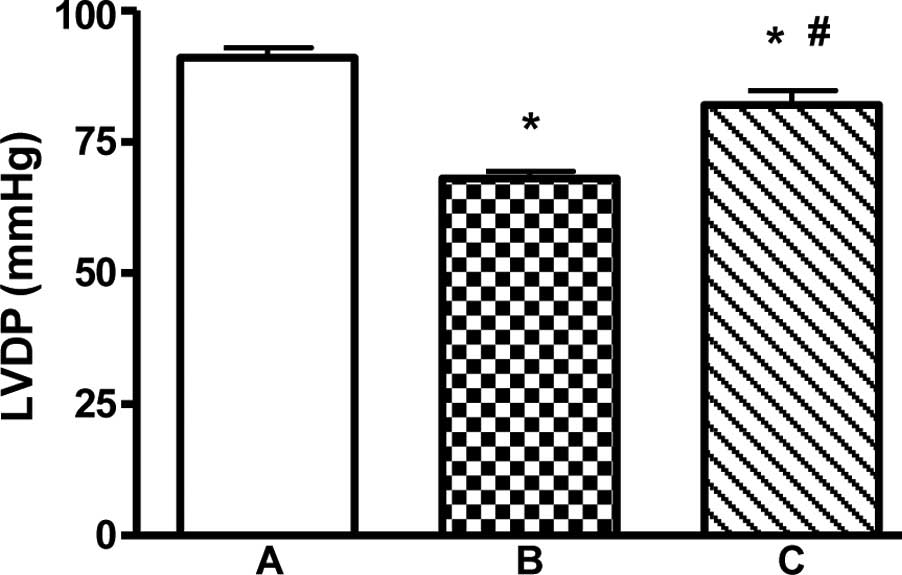
LVDP of the myocardial infarction group rats was
significantly decreased to 68.5±4.1 mmHg, while that in the
sham-operation rats was 91.3±7.9 mmHg. Treatment with EPO for 4
weeks caused a statistically significant increase in cardiac
functional recovery improving the LVDP to 82.9±8.35 mmHg
(P<0.01) in the EPO treatment group when compared to that in the
myocardial infarction group (Fig.
1).
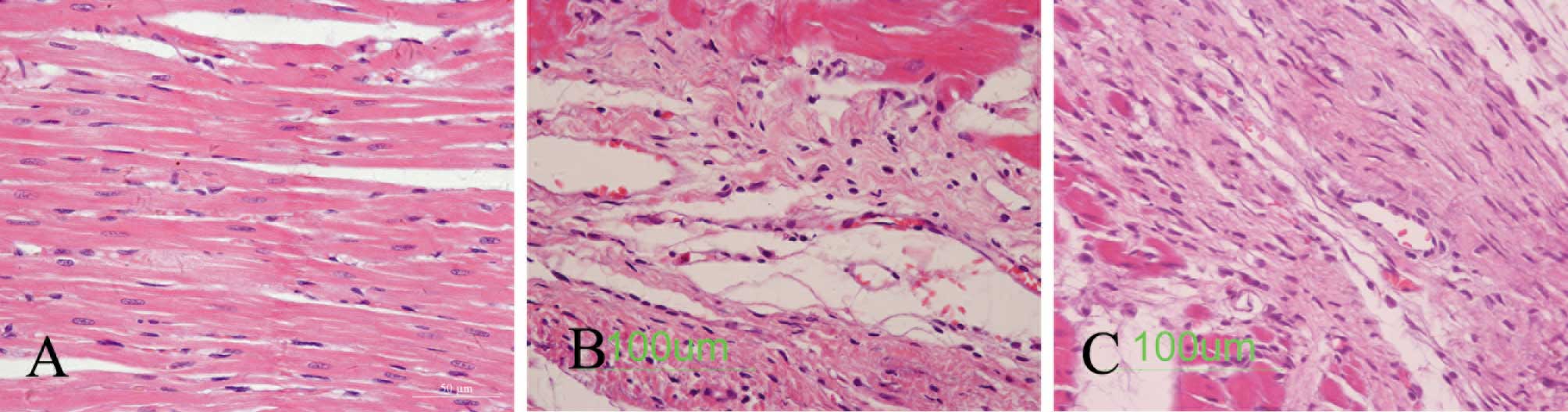
Heart pathological changes
As shown in Fig.
2B, infarction, myocardial fibrosis, the collagen deposition
area and the remnants of cardiac disorder were substantially severe
in the myocardial infarction group rats. Fig. 2C demonstrates a notably higher
number of small vessels around the infarcted myocardium in the EPO
treatment group rats than in the myocardial infarction group rats.
The myocardial interstitial fibrosis area was also improved in the
EPO treatment group rats (Fig.
2).
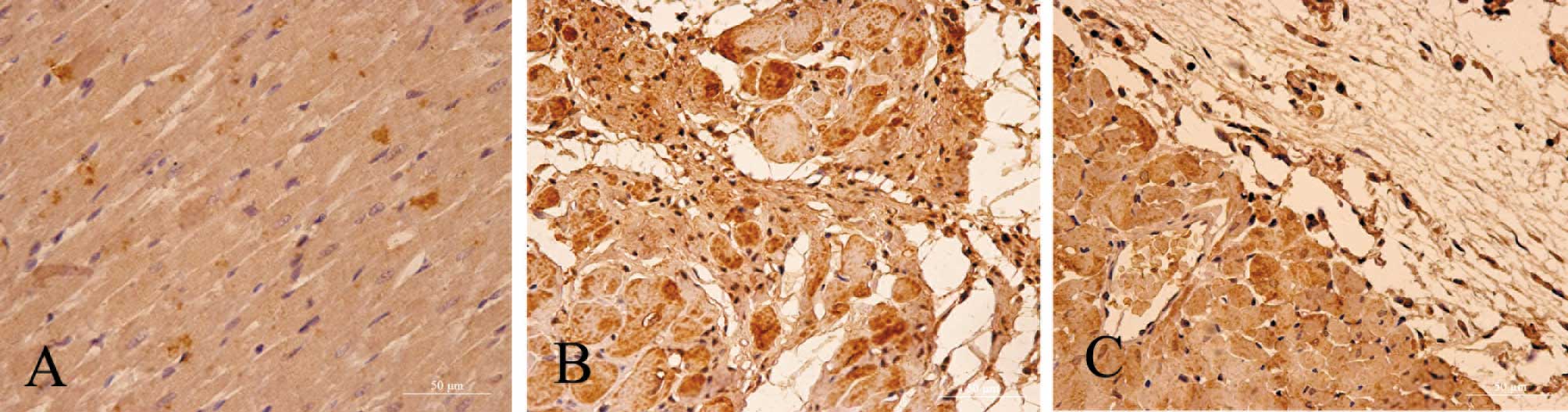
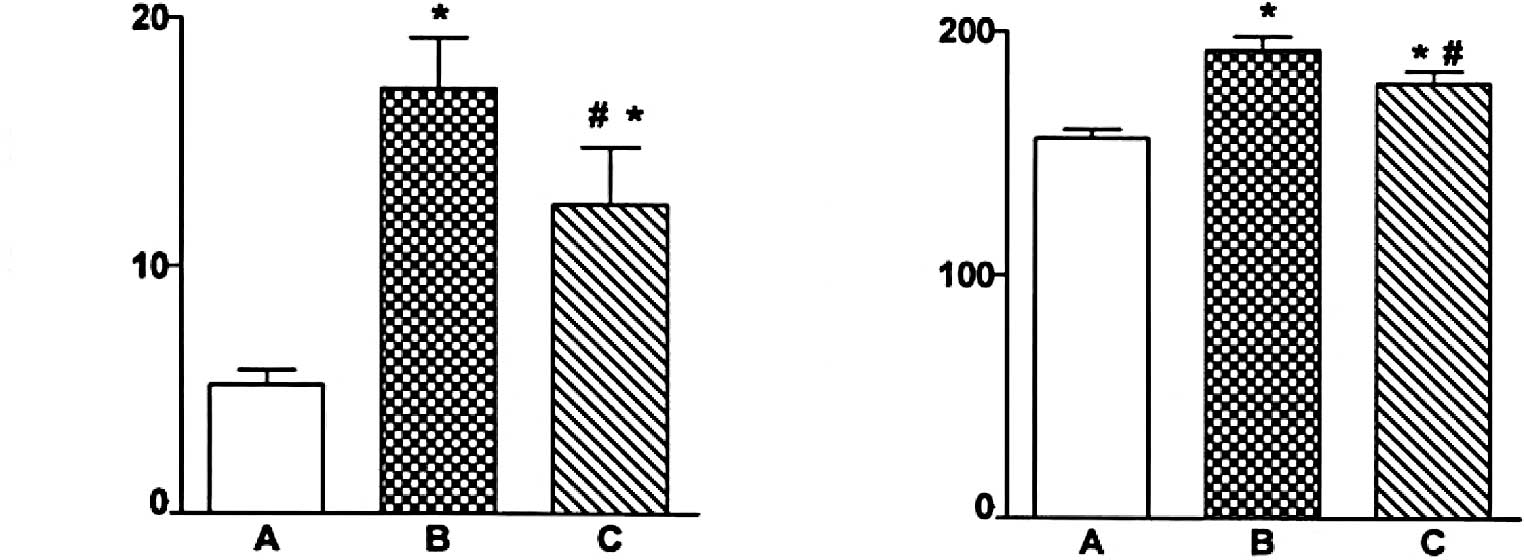
Immunohistochemical staining of
caspase-12 localization
Fig. 2 shows light
photomicrographs of caspase-12 immunohistochemical staining of the
myocardium in the three groups of rats. In heart tissues from the
myocardial infarction rats, the myocardium underwent fibrosis and
structural rarefaction. Myocardium that stained positive for
caspase-12 showed buff-colored granules with DAB staining. The
myocardium showed increased caspase-12 expression in the myocardial
infarction group when compared to the control group. Also, the
EPO-treated rats had a significantly decreased number of
caspase-12-positive cells and decreased optical density of the
caspase-12 immunostained myocardium compared to the myocardial
infarction rats (P<0.01) (Figs.
3 and 4).
Discussion
The ER is the site for the proper folding and
assembly of membrane-localized proteins and those destined for
secretion. It is also a regulator of intracellular calcium
(12). Various pathophysiological
conditions induce Ca2+ overload and/or accumulation of
unfolding or misfolding proteins within the ER, a condition
referred to as ER stress (13). ER
stress-induced apoptosis is believed to be associated with several
pathological processes, including diabetes (14), ischemia-reperfusion injury
(7), Alzheimer's disease and
Parkinson's disease (15,16). Researchers have already found that
myocardial ischemia provokes ER stress which results in the
apoptosis of myocardial and coronary endothelial cells; in
addition, caspase-12 plays a major role in this apoptosis induction
(17,18). Our results demonstrated that the
expression of caspase-12 was significantly elevated in the
myocardial ischemic areas compared to the normal areas. Also,
apoptosis of myocardial cells increased accompanied by myocardial
fibrosis in the myocardial ischemic areas which caused cardiac
dysfunction.
EPO has emerged as a promising candidate for the
treatment of acute ischemic heart disease (7). Apart from its hematopoietic effects,
EPO exhibits anti-apoptotic effects, particularly under ischemic
conditions, and attenuates oxidative stress (19). The binding of EPO to the EPO
receptor leads to a hemodimerization, with subsequent activation of
janus kinase (26). EPO signaling involves multiple pathways,
including activation of STAT 5, activation of proteins with Src
homology, such as PI3 kinase, and activation of ras/MAP kinases
(20,21). In the present study, EPO improved
myocardial contractility, reduced celluar damage and apoptosis,
increased neovascularization, and improved and decreased the
expression of caspase-12 in a rat model of myocardial infarction.
These results suggest that the mechanism of cardiac improvement by
application of EPO may block signal transduction in ER stress via
reduction of the expression of caspase-12, which has a beneficial
effect on myocardial protection.
The present study used the Langendorff system to
record the LVDP and immunohistochemistry to determine the
expression of caspase-12 protein in the heart after 4 weeks of
myocardial infarction induction, and to demonstrate the protective
effect of EPO on myocardial infarction. There was less function
reduction and structural damage to the heart in the EPO-treated
group than in the myocardial infarction group. We demonstrated that
EPO treatment of myocardial infarction prevents the appearance of
subsequent clinical symptoms by reversing the functional and
morphological abnormalities in the myocardial infarction heart in a
rat model through abrogation of the myocardial infarction-induced
increase in caspase-12 expression and prevented ER stress-induced
apoptosis after myocardial infarction. This study provides the
experimental basis for EPO treatment for myocardial infarction,
while the exact mechanism of action will be the subject of further
investigation.
References
|
1.
|
McManus DD, Chinali M, Saczynski JS, et
al: 30-Year trends in heart failure in patients hospitalized with
acute myocardial infarction. Am J Cardiol. 107:353–359. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Takemura G, Ohno M, Hayakawa Y, et al:
Role of apoptosis in the disappearance of infiltrated and
proliferated interstitial cells after myocardial infarction. Circ
Res. 82:1130–1138. 1998. View Article : Google Scholar
|
|
3.
|
Weidemann A and Johnson RS: Nonrenal
regulation of EPO synthesis. Kidney Int. 75:682–688. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Iwai M, Cao G, Yin W, et al:
Erythropoietin promotes neuronal replacement through
revascularization and neurogenesis after neonatal hypoxia/ischemia
in rats. Stroke. 38:2795–2803. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Matis GK and Birbilis TA: Erythropoietin
in spinal cord injury. Eur Spine J. 18:314–323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Breggia AC, Wojchowski DM and Himmelfarb
J: JAK2/Y343/STAT5 signaling axis is required for
erythropoietin-mediated protection against ischemic injury in
primary renal tubular epithelial cells. Am J Physiol Renal Physiol.
295:F1689–F1695. 2008. View Article : Google Scholar
|
|
7.
|
Xu X, Cao Z, Cao B, et al: Carbamylated
erythropoietin protects the myocardium from acute
ischemia/reperfusion injury through a PI3K/AKT-dependent mechanism.
Surgery. 146:506–514. 2009. View Article : Google Scholar
|
|
8.
|
Van der Putten K, Jie KE, Emans ME, et al:
Erythropoietin treatment in patients with combined heart and renal
failure: objectives and design of the EPOCARES study. J Nephrol.
23:363–368. 2010.PubMed/NCBI
|
|
9.
|
Latini R, Brines M and Fiordaliso F: Do
non-hemopoietic effects of erythropoietin play a beneficial role in
heart failure? Heart Fail Rev. 13:415–423. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Liu G, Sun Y, Li Z, et al: Apoptosis
induced by endoplasmic reticulum stress involved in diabetic kidney
disease. Biochem Biophys Res Commun. 370:651–656. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Liu XH, Zhang ZY, Sun S, et al: Ischemic
postconditioning protects myocardium from ischemia/reperfusion
injury through attenuating endoplasmic reticulum stress. Shock.
30:422–427. 2008. View Article : Google Scholar
|
|
12.
|
Hiroi T, Wei H, Hough C, et al: Protracted
lithium treatment protects against the ER stress elicited by
thapsigargin in rat PC12 cells: roles of intracellular calcium,
GRP78 and Bcl-2. Pharmacogenomics J. 5:102–111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Janssen K, Horn S, Niemann MT, et al:
Inhibition of the ER Ca2+ pump forces multidrug-resistant cells
deficient in Bak and Bax into necrosis. J Cell Sci. 122:4481–4491.
2009.
|
|
14.
|
Ali BR: Is cystic fibrosis-related
diabetes an apoptotic consequence of ER stress in pancreatic cells?
Med Hypotheses. 72:55–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Salminen A, Kauppinen A, Suuronen T, et
al: ER stress in Alzheimer's disease: a novel neuronal trigger for
inflammation and Alzheimer's pathology. J Neuroinflammation.
6:412009.
|
|
16.
|
Arduino DM, Esteves AR, Cardoso SM, et al:
Endoplasmic reticulum and mitochondria interplay mediates apoptotic
cell death: relevance to Parkinson's disease. Neurochem Int.
55:341–348. 2009.PubMed/NCBI
|
|
17.
|
Xin W, Li X, Lu X, et al: Involvement of
endoplasmic reticulum stress-associated apoptosis in a heart
failure model induced by chronic myocardial ischemia. Int J Mol
Med. 27:503–509. 2011.PubMed/NCBI
|
|
18.
|
Kumar S, Kasseckert S, Kostin S, et al:
Ischemic acidosis causes apoptosis in coronary endothelial cells
through activation of caspase-12. Cardiovasc Res. 73:172–180. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Yada T, Shimokawa H, Hiramatsu O, et al:
Erythropoietin enhances hydrogen peroxide-mediated dilatation of
canine coronary collateral arterioles during myocardial ischemia in
dogs in vivo. Am J Physiol Heart Circ Physiol. 299:H1928–H1935.
2010. View Article : Google Scholar
|
|
20.
|
Hirata A, Minamino T, Asanuma H, et al:
Erythropoietin just before reperfusion reduces both lethal
arrhythmias and infarct size via the phosphatidylinositol-3
kinase-dependent pathway in canine hearts. Cardiovasc Drugs Ther.
19:33–40. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Shi Z, Hodges VM, Dunlop EA, et al:
Erythropoietin-induced activation of the JAK2/STAT5, PI3K/AKT, AND
RAS/ERK pathways promotes malignant cell behavior in a modified
breast cancer cell line. Mol Cancer Res. 8:615–626. 2010.
View Article : Google Scholar : PubMed/NCBI
|