Introduction
Pancreatic cancer is one of the most common
malignant tumors. At the time of diagnosis, less than 10% of
pancreatic cancer can be resected (1). Its 5-year survival rate is the lowest
one among all cancer types (2).
Because of its high degree of malignancy, rapid progression, late
stage diagnosis, early metastasis and poor prognosis, pancreatic
cancer is known as the ‘king of cancer’. According to the clinical
guidelines of NCCN and ESMO, gemcitabine is the first-line
chemotherapeutic drug for the treatment of metastatic pancreatic
cancer. However, the major impediment in successful treatment is
the limited effect of gemcitabine. The median survival time after
the treatment of gemcitabine was found to be only 5.65 months,
whereas the 1-year survival rate was merely 16–19% (3). Thus, the chemotherapy for pancreatic
cancer has reached a plateau. In recent years, molecular-targeted
therapy is regarded as an exciting research hotspot in the
treatment of tumors because of its high specificity and minimal
adverse effects. Epidermal growth factor receptor (EGFR), a member
of the ErbB family, promotes tumor cell proliferation, inhibits
apoptosis, improves migratory ability and thereby increases tumor
invasion and distant metastasis. Previous studies have shown that
the EGFR is overexpressed in pancreatic cancer cells. Erlotinib is
one of the tyrosine kinase inhibitors of EGFR (EGFR-TKIs). A phase
III clinical trial (4) showed that
the combined use of erlotinib with gemcitabine prolonged median
survival time (6.24 vs. 5.91 months, P=0.034) and 1-year survival
rates (23 vs. 17%, P=0.023) in patients with advanced pancreatic
cancer. Thus, erlotinib was approved by the US-FDA as a first-line
treatment for advanced pancreatic cancer in November 2005. Although
the combined use of erlotinib with gemcitabine was found to extend
the survival period of cancer patients, the improvement of the
median survival time and the 1-year survival rate is limited.
Therefore, it is of great importance to identify novel combinations
of anticancer drugs with erlotinib for the treatment of advanced
pancreatic cancer.
Pemetrexed is a novel antifolate that enters tumor
cells rapidly via several membrane transporters, where it is
metabolized to polyglutamate derivatives that are potent inhibitors
of thymidylate synthase and, to a much lesser extent, glycinamide
ribonucleotide formyltransferase (5,6).
Pemetrexed arrests cells mainly in the S phase and induces
apoptosis in a broad spectrum of solid tumors. Previous basic
research has also shown that pemetrexed inhibits the proliferation
of PANC-1 pancreatic cancer cells and exerts an additive effect
when it is combined with gemcitabine (7). Phase II clinical studies have shown
that pemetrexed significantly improves advanced pancreatic cancer
treatment with few adverse side effects (8).
A timing effect was noted when erlotinib was
combined with pemetrexed for the treatment of non-small cell lung
cancer (NSCLC) (9), but reports on
the alteration of cytotoxicity by a combined regimen of pemetrexed
and erlotinib on pancreatic carcinoma are limited. Hence, the
purpose of this study was to investigate the sequence-dependent
effects of pemetrexed and erlotinib on BXPC-3 and PANC-1 cells and
to probe the possible cellular mechanism. In the present study, the
molecular mechanisms underlying the synergistic cytotoxicity
between erlotinib and pemetrexed in vitro were first
investigated. Furthermore, several factors, including modulation of
EGFR, HER3 and Akt phosphorylation, which may contribute to this
synergistic interaction when used in combination were
characterized. We further hope that this study will aid in
exploring combination treatment options for the cure of advanced
pancreatic cancer.
Materials and methods
Chemicals and reagents
Erlotinib (Tarceva®) was obtained from
Roche (Nonnenwald, Penzberg, Germany) and dissolved in DMSO as a
stock solution of 10 mmol/l. Pemetrexed (Alimta®) was
obtained commercially from Eli Lilly and Co. and dissolved in 0.9%
NaCl to a final concentration of 40 g/l. Both compounds were stored
at −20°C in tightly sealed sterile tubes and diluted to the desired
concentrations in PBS within 24 h of each experiment. Antibodies
and their sources were as follows: anti-EGFR antibody (Santa Cruz)
and anti-phosphorylated EGFR (Tyr1068) antibody (Cell Signaling);
anti-AKT antibody (Santa Cruz) and anti-phosphorylated AKT (Ser473)
antibody (Cell Signaling); anti-HER antibody (Santa Cruz) and
anti-phosphorylated HER (Tyr1289) antibody (Cell Signaling);
anti-MET antibody and anti-phosphorylated MET (Tyr1003) antibody
(Cell Signaling).
Cell lines
BXPC-3 and PANC-1 cells were cultured in RPMI-1640
medium supplemented with 10% fetal calf serum at 37°C in a
humidified atmosphere of 95% air and 5% CO2.
Growth cytotoxicity assay
The effect of erlotinib and pemetrexed on cell
survival using different exposure schedules in vitro was
assessed by the MTT colorimetric assay carried out in 96-well
plates. Cells were then treated with increasing concentrations of
erlotinib or pemetrexed for a duration of 72 h. To determine the
proliferative effects of simultaneous or sequential administration
of erlotinib and pemetrexed, cells were incubated for 72 h after
concurrent or sequential treatment with erlotinib and pemetrexed
for a 24-h interval. At the end of the 72-h incubation, viable cell
numbers were measured using MTT assay. MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] was
added at a final concentration of 0.5 mg/ml to each well. Cells
were then incubated at 37°C in a humidified atmosphere of 95% air
and 5% CO2 for 4 h, the supernatant was removed, and the
converted dye was solubilized with 150 μl DMSO. The absorbance was
measured at 540 nm. Growth inhibition was expressed as a percentage
of surviving cells in drug-treated vs. PBS-treated control cells
(which was considered as 100% viability). The IC50 value
was the concentration resulting in 50% cell growth inhibition after
the 72 h exposure to the drug(s) compared to the untreated control
cells and was calculated using CalcuSyn software (Biosoft,
Inc.).
Mutation analysis of EGFR and K-ras
genes
Genomic DNA was extracted from each cell line with
the TIANamp DNA extraction kit (Tiangen Biotech). Exons 18–21 of
the EGFR and exons 2 and 3 of the K-ras showed
mutational changes by quantitative PCR high-resolution melting
(qPCR-HRM) analysis (Suzhou Microdiag Biomedicine Tech. Co.,
Ltd.).
Cell cycle distribution analysis
Exponentially growing BXPC-3 cells were seeded in
6-well plates and treated with erlotinib and pemetrexed alone or in
combination concurrently or sequentially for a defined time
interval. The cells were trypsinized and pelleted by
centrifugation. After washing the pellet with PBS, the cells were
counted, and 1×106 cells were fixed in 70% ethanol at
−20°C for 24 h. These fixed cells were then washed with PBS and
incubated with RNase A (0.25 mg/ ml) for 30 min at 37°C; 5 μl of
propidium iodide (KeyGen, China) was then added to the cell
suspension. The mixture was incubated at room temperature (RT) for
30 min in the dark. The suspended cells were then analyzed for cell
cycle distribution using the FACSCalibur Flow Cytometer (BD,
USA).
Immunoblot analysis
Whole-cell extracts were obtained by lysis of cells
in a 2X sodium dodecyl sulfate (SDS)-polyacrylamide gel
electrophoresis sample buffer [125 mmol/l Tris-HCl (pH 6.8), 6%
SDS, 10% glycerol, 10 mmol/l of 2-mercaptoethanol]. Whole-cell
extracts were separated by electrophoresis on 8% SDS-polyacrylamide
gels and transferred onto nitrocellulose membranes in 25 mmol/l
Tris-HCl (pH 8.3) containing 90 mmol/l glycine and 20% methanol for
2 h at 100 mA. The membranes were blocked with Tween Tris-buffered
saline (TBS) with 0.1% Triton X-100 and 5% nonfat milk for 2–3 h at
RT. Primary antibodies were diluted in a blocking solution (1:200
for EGFR, AKT and HER3; and 1:5,000 for p-EGFR, p-HER3, p-AKT, MET
and p-MET) Membranes were incubated overnight at 4°C with the
primary antibodies. After washing with TBS Tween-20 for 30 min,
membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies at RT for 1 h. The membranes were then washed
for 1 h with TBS Tween-20. Autoradiography was performed using
chemiluminescent detection reagents according to the manufacturer’s
instructions, and signal detection and quantification were carried
out with an ECL system (Amersham Biosciences, Piscataway, NJ, USA)
and Image Quant analysis software (Quantity One; Bio-Rad, Hercules,
CA, USA).
Statistical analysis
All assays were performed in triplicate. Data are
expressed as the means ± SD of values. Statistical analyses were
performed using the Student’s t-test. A value of P<0.05 was
considered statistically significant.
Results
Cytotoxicity of pemetrexed and erlotinib
and correlation with genetic background
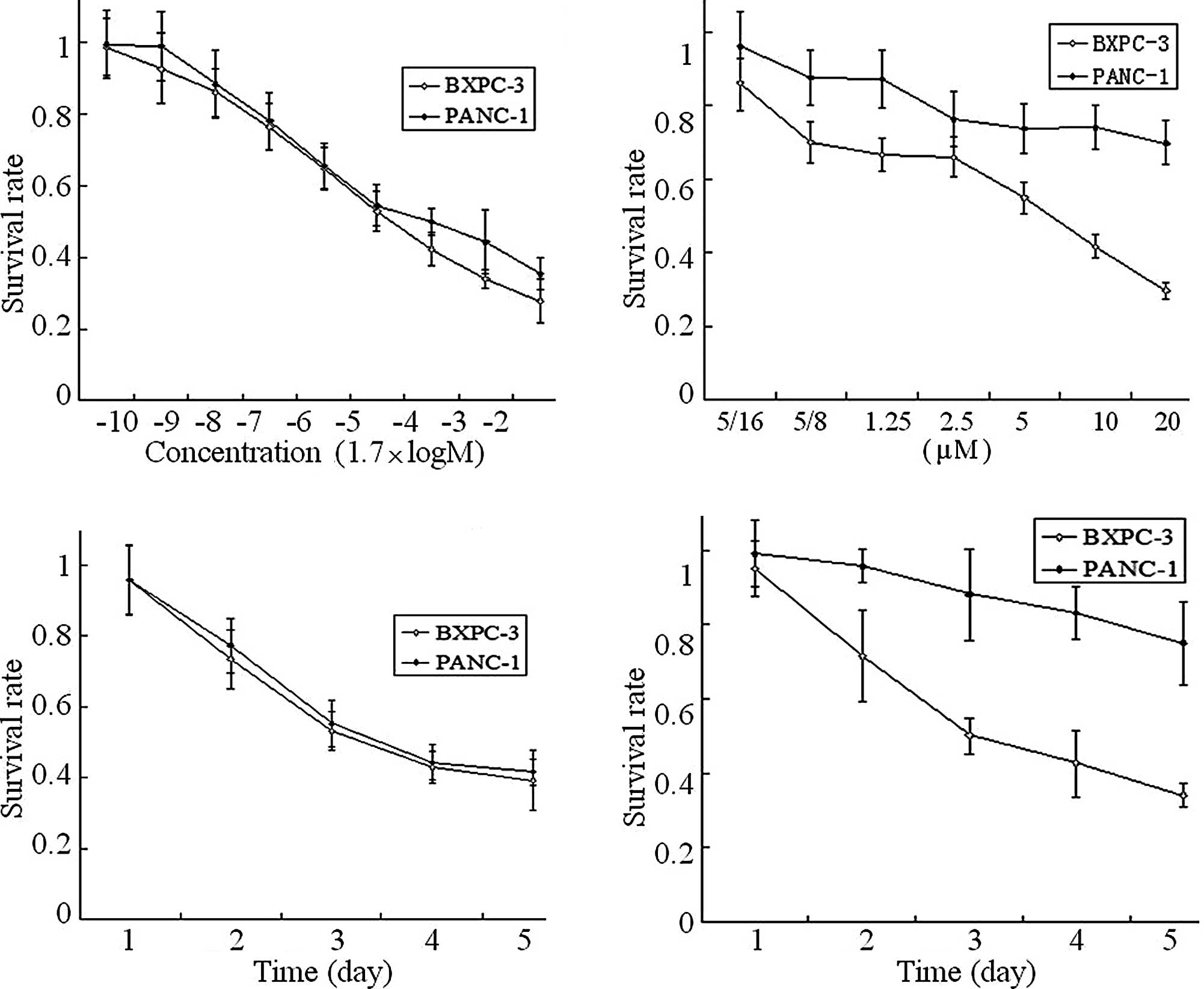
Table I and
Fig. 1 summarize the genetic
background and cytotoxicity of erlotinib and pemetrexed on the
human pancreatic cancer BXPC-3 and PANC-1 cells. Exons 18–21 of the
EGFR and exons 2 and 3 of the K-ras gene by qPCR-HRM
and mutational analysis showed that BXPC-1 cells were wild-type and
PANC-1 cells harbored a specific mutation in K-ras exon 2.
The cytotoxicity results showed that erlotinib (0.3125-20 μmol/l)
and pemetrexed (1.7×10−7-17 mmol/l) suppressed BXPC-3
and PANC-1 cell proliferation, and the effect was
concentration-dependent (Fig. 1A and
B) and time-dependent (Fig. 1C and
D). The IC50 of pemetrexed in the BXPC-3 and PANC-1
cells was 39.86±1.68 and 83.76±0.19 μmol/l, respectively. The
IC50 of erlotinib in the BXPC-3 and PANC-1 cells was
8.86±1.68 and >20 μmol/l, respectively. Concentrations of
erlotinib >20 μmol/l could not be achieved due to its low
solubility in culture medium. Accordingly, 10 μmol/l was chosen as
the concentration of erlotinib used in the PANC-1 cells for
subsequent studies. At the IC50 of pemetrexed and
erlotinib, the inhibitory effects were time-dependent and reached
the maximal effect at day 5.
 | Table I.Cytotoxicities of erlotinib or
pemetrexed and the mutation status of the EGFR and
K-ras genes in the BXPC-3 and PANC-1 cell lines. |
Table I.
Cytotoxicities of erlotinib or
pemetrexed and the mutation status of the EGFR and
K-ras genes in the BXPC-3 and PANC-1 cell lines.
| Cell line | EGFR gene | K-ras
gene | Erlotinib
IC50 (μm) | Pemetrexed
IC50 (μm) |
|---|
| BXPC-3 | WT | WT | 8.86±0.19 | 39.86±1.68 |
| PANC-1 | WT | Mut (2 exon) | >20 | 83.76±1.75 |
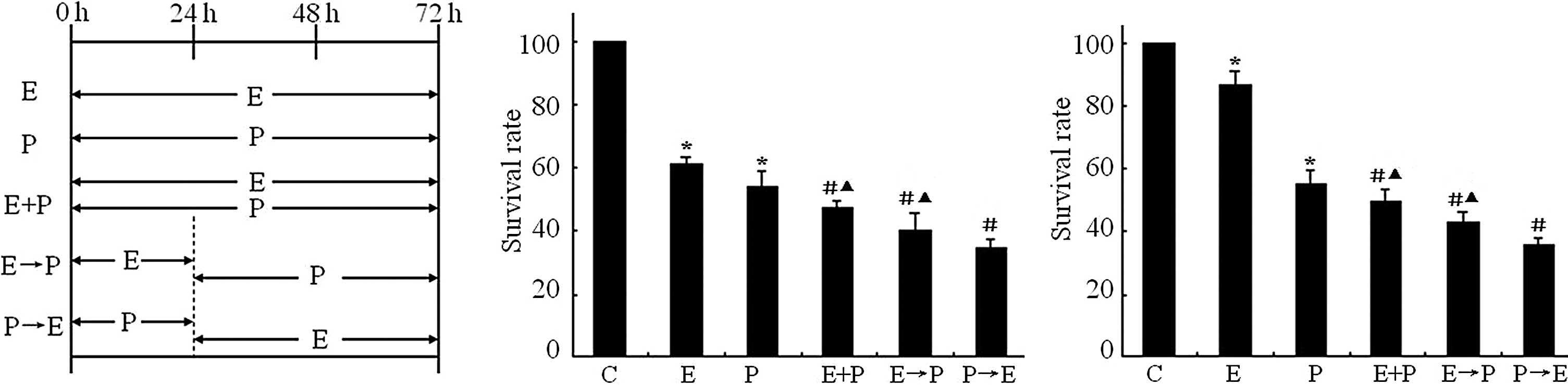
Effects of different schedules of
erlotinib and pemetrexed on cell proliferation
We observed the growth inhibitory effects of
erlotinib in combination with pemetrexed concurrently or
sequentially at a 24-h interval. Fig.
2A illustrates the five exposure schedules tested that mimic
possible clinical scenarios: i) erlotinib or pemetrexed alone for
72 h (E and P), ii) erlotinib and pemetrexed concurrently (E+P) for
72 h, iii) erlotinib for 24 h followed by pemetrexed for 48 h
(E→P), and iv) pemetrexed for 24 h followed by erlotinib for 48 h
(P→E). At IC50, the effects of pemetrexed in combination
with erlotinib on cell proliferation depended on the sequence. The
results showed that in the BXPC-3 cells (erlotinib-sensitive
cells), the cell survival rate was 60.83% for erlotinib alone and
54.01% for pemetrexed alone, 47.31% for erlotinib and pemetrexed
concurrently (E+P), 40.15% for erlotinib followed by pemetrexed
(E→P) and 34.56% for pemetrexed followed by erlotinib (P→E)
(Fig. 2B). The results showed that
in the PANC-1 cells (erlotinib-resistant cells), the cell survival
rate was 86.46% for erlotinib alone and 55.29% for pemetrexed
alone, 49.38% for erlotinib and pemetrexed concurrently (E+P),
42.75% for erlotinib followed by pemetrexed (E→P) and 35.84% for
pemetrexed followed by erlotinib (P→E) (Fig. 2C). Compared to cells treated with
erlotinib or pemetrexed alone, significant additive effects on cell
proliferation were found when erlotinib was added concurrently or
sequentially with pemetrexed (P<0.05). Furthermore, sequential
administration of erlotinib following pemetrexed significantly
enhanced the cell growth inhibition in BXPC-3 and PANC-1 cells,
compared to pemetrexed administered concurrently or prior to
erlotinib (P+E or P→E) (P<0.05).
Cell cycle effects of pemetrexed and
erlotinib
To probe the possible mechanism of growth inhibition
of erlotinib in combination with pemetrexed in BXPC-3 cells, cell
cycle analysis was performed. The BXPC-3 cells were treated for 24
h with 8.86 μmol/l erlotinib, 39.86 μmol/l pemetrexed alone and in
combination concurrently or sequentially at a 24-h interval. The
concentrations of erlotinib or pemetrexed used were the
IC50 concentrations, respectively. As shown in Fig. 3, 48 h of exposure to erlotinib
alone induced G0/G1 phase arrest (P<0.05)
and decreased the number of cells in the S phase (P<0.05),
resulting in a 20% cell increase in the G0/
G1 phase compared to the untreated control cells.
However, pemetrexed alone induced S arrest (P<0.05) and
decreased the number of cells in the G2/M phase
(P<0.05). Compared to pemetrexed alone, both concurrent
cotreatment of erlotinib with pemetrexed and sequential treatment
of erlotinib followed by pemetrexed resulted in an accumulation of
cells in the G0/ G1 phase (P<0.05) and a
decrease in the G2/M phase (P<0.05). By contrast, sequential
administration of pemetrexed followed by erlotinib resulted in S
arrest (P<0.05) and a decrease in cells in the G2/M
phase (P<0.05) compared to erlotinib alone.
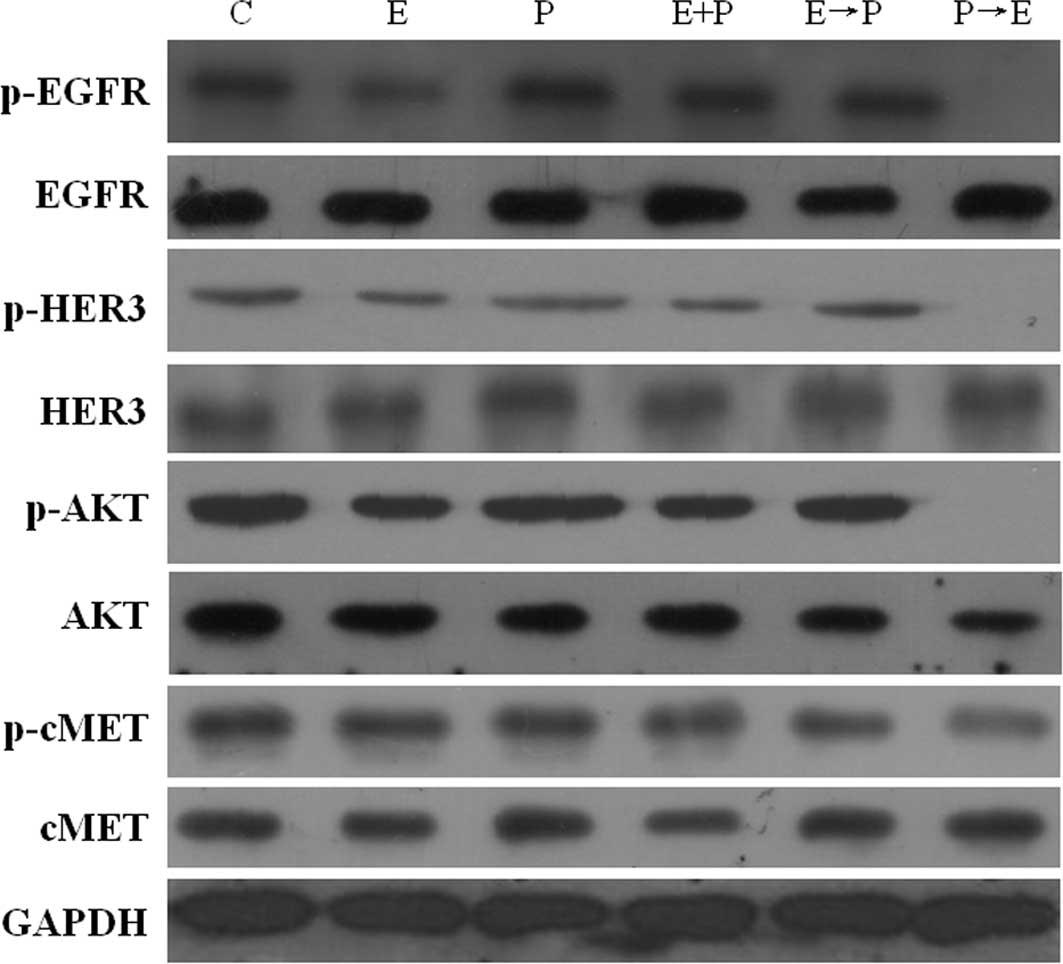
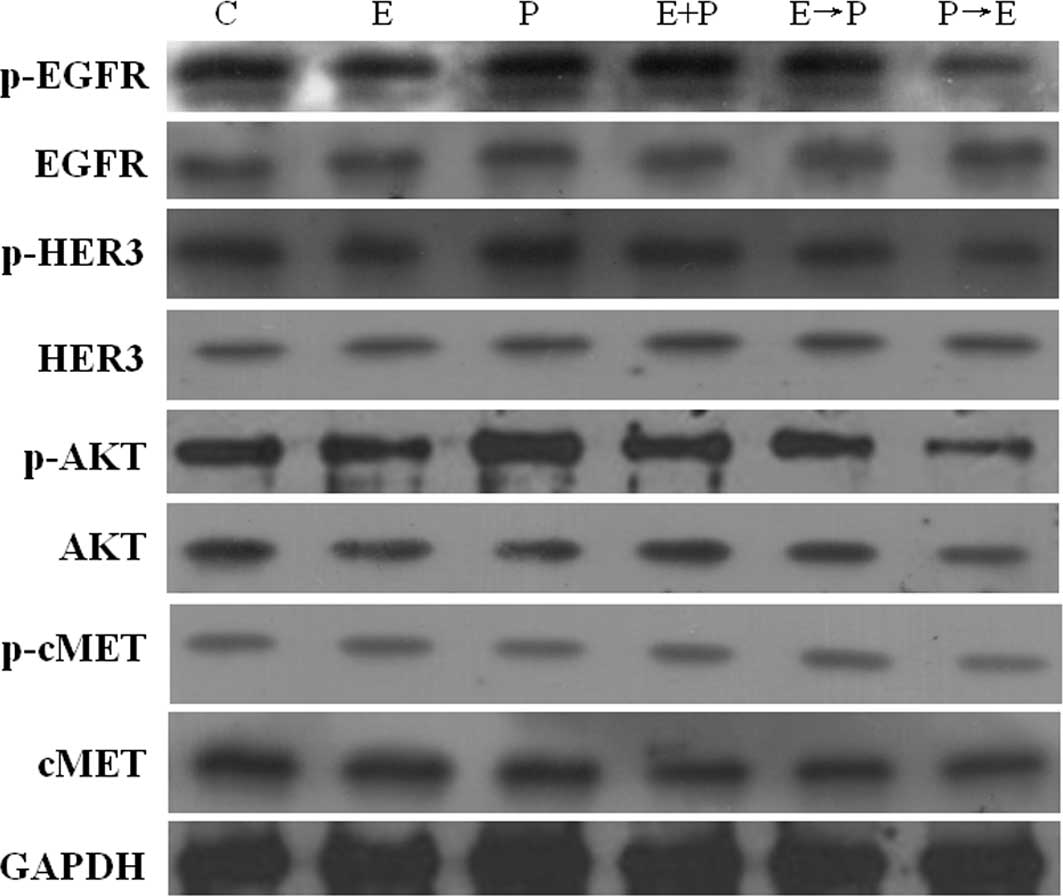
Pemetrexed activates the EGFR, HER3 and
AKT signaling pathway in human pancreatic BXPC-3 and PANC-1
cells
To gain insight into the mechanism(s) underlying the
cytotoxic synergism between pemetrexed and erlotinib, the effect of
pemetrexed on the EGFR pathway in the BXPC-3 and PANC-1 cells was
further examined by Western blot analysis (Figs. 4 and 5). As expected, erlotinib induced a
significant suppression of EGF-induced phosphorylation of EGFR in
the BXPC-3 and PANC-1 cells; the percent reductions in
EGFR-phosphorylated protein were 47.4 and 30% in BXPC-3 and PANC-1
cells, respectively. Conversely, pemetrexed significantly enhanced
EGFR phosphorylated levels, and the protein levels were 26.3 and
13.6% higher compared to the control cells, respectively. Moreover
the administration schedule (P→E) significantly reduced the
phosphorylation status of EGFR when compared to treatment with
erlotinib alone.
Since EGFR signaling is transduced mainly through
the HER3/AKT pathways, we investigated the phosphorylation status
of HER3 and AKT to determine their activity after drug treatment.
Erlotinib resulted in the inhibition of p-HER3 and p-AKT in the
BXPC-3 and PANC-1 cell lines. The p-HER3 levels were potently (∼17
and 14%) down-regulated by erlotinib in the BXPC-3 and PANC-1
cells, respectively. p-AKT levels were potently (∼45%)
down-regulated by erlotinib in the BXPC-3 cells. Conversely,
pemetrexed significantly enhanced EGFR, HER3 and AKT
phosphorylation levels. p-EGFR levels were up-regulated (∼26 and
14%) by pemetrexed in the BXPC-3 and PANC-1 cells, respectively.
p-HER3 levels were up-regulated (∼58 and 26%) by pemetrexed in the
BXPC-3 and PANC-1 cells, respectively. p-AKT levels were
up-regulated (∼15 and 26%) by pemetrexed in the BXPC-3 and PANC-1
cells. As a mechanism of escape of cancer from anti-EGFR therapy,
the phosphorylation level of MET was determined by immunoblotting.
The results conclusively showed that no significant change in the
phosphorylation level of MET was noted for the different schedules
of erlotinib and pemetrexed exposure (Figs. 4 and 5).
Discussion
The aim of the present study was to investigate the
cytotoxic activity of erlotinib and pemetrexed in combination and
to define the optimal schedule and the cellular mechanism involved
in drug interaction against human pancreatic cancer cells. Two
significant findings of this study are as follows: i) Synergistic
effects on cell proliferation were found when pemetrexed was used
in combination with erlotinib. The degree of the synergistic
effects depended on the treatment sequence, which was most
significant when erlotinib was sequentially administrated at a 24-h
interval following pemetrexed. ii) These synergistic effects may be
related to the activation of the EGFR/HER3/AKT pathway induced by
pemetrexed.
In the present study, a set of experiments was
designed to elucidate the combination effects and possible cellular
mechanism underlying the interaction between pemetrexed and
erlotinib in PANC-1 and BXPC-3 cells. PANC-1 and BXPC-3 cells were
exposed to pemetrexed and erlotinib using five treatment schedules.
In agreement with previous studies (10,11),
the results of the present study showed that pemetrexed and
erlotinib alone significantly inhibited cell proliferation. We
found that the synergistic effects of pemetrexed and erlotinib on
cell proliferation were sequence-dependent. Furthermore, the
cytotoxic synergism was observed in both erlotinib-sensitive and
erlotinib-resistant human pancreatic cell lines. This was
independent of the mutation status of the EGFR or
K-ras gene. These results were in accord with previous
pre-clinical findings that the existence of a synergistic
interaction between EGFR-TKIs and chemotherapeutic agents in NSCLC
cell lines was schedule-dependent (9,12).
Previous reports have illustrated the importance of
modulating the cell cycle to exploit the optimal effect of drug
combinations. Furthermore, there is growing laboratory evidence of
a possible sequence-dependent antagonism between EGFR-TKIs and
cytotoxic agents as a result of the well-known G1-phase
arrest of tumor cells by EGFR-TKIs, which protect tumor cells from
cell cycle-specific cytotoxic agents (13,14).
In the present study, flow cytometry demonstrated that both
pemetrexed and erlotinib caused an accumulation of cells in the S
and G1 phases, respectively. Compared to pemetrexed
alone, concurrent treatment of both agents as well as erlotinib
followed by pemetrexed resulted in an accumulation of cells in the
G1 phase. Compared to erlotinib alone, concurrent
treatment resulted in an accumulation of cells in the S phase. Our
result also showed that sequential administration of pemetrexed
followed by erlotinib resulted in S phase arrest when compared to
erlotinib treatment alone. These results may explain why the
concomitant or sequential treatment of pemetrexed followed by
erlotinib exerted significant additive effects on cell
proliferation. By contrast, sequential administration of erlotinib
following pemetrexed induced G1 arrest. The increase in
cells in the G1 phase could not provide a plausible
explanation for discordant results of sequence-dependent effects on
cell proliferation when erlotinib followed pemetrexed. It has been
reported that modulation of EGFR activity following
chemotherapeutic agents determines the response to pemetrexed in
NSCLC cells (9), and erlotinib
strengthens the cytotoxic effect of pemetrexed in NSCLC cells by
harboring specific molecular characteristics (15). Further studies are required to
explore the possible cellular mechanism.
Furthermore, the possible molecular mechanism of the
cell signaling pathways involved was determined. The dependency of
NSCLC and pancreatic cancer cells on the EGFR pathway for growth
and survival was found to be an important determinant of
sensitivity towards EGFR-TKI monotherapy (16,17).
Previous reports have shown that the mammalian target of rapamycin
inhibitor rapamycin activates the phosphatidylinositol 3-kinase
(PI3K)/AKT pathway and enhances cytotoxicity of the PI3K/AKT
inhibitor, LY294002 (18). In
pre-clinical studies, the combination of pemetrexed and erlotinib
yielded conflicting results in various NSCLC tumor cell lines. Li
et al discovered that schedule-dependent synergism of
pemetrexed and erlotinib was associated with pemetrexed-induced
EGFR phosphorylation and AKT phosphorylation in NSCLC tumors
(9). On the contrary, a previous
study demonstrated that the effect of pemetrexed reduced Akt
phosphorylation in NSCLC tumors (15). However, the contradictory findings
may be related to the discrepancy between drug exposure conditions
and sensitivity of the experimental methods. In agreement with
these findings, in vitro experimental data obtained in this
study indicate enhanced phosphorylation of EGFR/Akt after exposure
to pemetrexed. Our results suggest that pemetrexed enhances the
antitumor activity of erlotinib by activation of the EGFR-AKT
pathway.
The EGFR family comprises four distinct receptors:
EGFR/ ErbB1, HER-2/ErbB2, HER-3/ErbB3 and HER-4/ErbB4. Pancreatic
cancer cells frequently overexpress EGFR, HER-2, HER-3 and, less
frequently, HER-4, as well as six ligands that bind directly to
EGFR (19). Overexpression of
EGFR, HER-2 and HER-3 has also been implicated in the development
and progression of pancreatic cancer (20,21).
Giovannetti et al found that 60–70% of pancreatic cancer
cases have an amplification of HER-3 (22). Previous reports have shown that
phosphorylated HER-3 acts as a scaffold to recruit signaling
proteins, including PI3K (23,24).
Phosphorylated HER3 binds the regulatory subunit of PI3K (p85)
leading to activation of PI3K (p110α) and its downstream kinase
AKT, essential for cell growth and survival (25,26).
The EGFR kinase inhibitor gefitinib prevents phosphorylation of
both EGFR and HER3, as a result, the PI3K-AKT pathway is uncoupled
from HER3 and inactivated. A recent study suggests that HER-3 may
be a useful biomarker for the selection of patients who are most
likely to respond to the EGFR inhibitor erlotinib (27). The phosphorylation status of HER3
and AKT was studied here to determine their activity after the
scheduled drug treatment. The data showed that pemetrexed
significantly enhanced HER3 and AKT phosphorylation levels. The
p-HER3 levels were potently (∼17 and 14%) down-regulated by
erlotinib in the BXPC-3 and PANC-1 cells, respectively. Our
findings in this study indicate that pemetrexed enhances the
antitumor activity of erlotinib by activating the EGFR/HER3/AKT
pathway, thus rendering the cells more dependent on the EGFR
pathway and more susceptible to the inhibition by erlotinib.
Although the above-mentioned results justify the
synergistic effects of sequential administration of pemetrexed
followed by erlotinib, it cannot explain the synergistic effects
noted upon the concomitant exposure of both agents or erlotinib
followed by pemetrexed. The expression of MET phosphorylation was
also observed in this study. In gefitinib-resistant cells, Engelman
et al (28) found that the
oncogenic receptor tyrosine kinase MET could also phosphorylate
HER3, leading to activation of the PI3K/AKT pathway, and
amplification of MET caused gefitinib resistance by driving ERBB3
(HER3)-dependent activation of PI3K, a pathway thought to be
specific to EGFR/ERBB family receptors (29). Pharmacological inhibition of either
EGFR or MET alone was insufficient to inactivate the HER3-PI3K-AKT
axis or stall resistant cell growth (30,31).
However, our data showed that there was no significant change in
MET phosphorylation.
In conclusion, the present study characterized
several molecular mechanisms and determinants involved in the
synergistic effect between erlotinib and pemetrexed against
pancreatic cancer BXPC-3 and PANC-1 cells, regardless of their
genetic signature. Our results suggest that pemetrexed enhances the
antitumor activity of erlotinib by activating the EGFR/HER3/AKT
pathway, thus rendering the cells more dependent on the EGFR
pathway and more susceptible to the inhibition by erlotinib.
Although the extrapolation of in vitro data
to the clinical setting should be considered with caution, these
results may have implications for the rational development of
chemotherapeutic regimens, including erlotinib and pemetrexed, for
the treatment of pancreatic cancer. Although our results explain
the synergistic effects of a sequential administration of
pemetrexed followed by erlotinib, we are unable to explain the
synergistic effects of a concomitant exposure of both agents and
erlotinib treatment followed by pemetrexed. This raises the
possibility of the involvement of another cellular signaling
mechanism, which will be the focus of our next stage of
research.
References
|
1.
|
Michaud DS: Epidemiology of pancreatic
cancer. Minerva Chir. 59:99–111. 2004.PubMed/NCBI
|
|
2.
|
Saidi RF, Remine SG and Jacobs MJ:
Interferon receptor alpha/beta is associated with improved survival
after adjuvant therapy in resected pancreatic cancer. HPB (Oxford).
9:289–294. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Burris HA III, Moore MJ, Andersen J, et
al: Improvements in survival and clinical benefit with gemcitabine
as first-line therapy for patients with advanced pancreas cancer: a
randomized trial. J Clin Oncol. 15:2403–2413. 1997.PubMed/NCBI
|
|
4.
|
Moore MJ, Goldstein D, Hamm J, et al:
erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: a phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar
|
|
5.
|
Taylor EC, Kuhnt D, Shih C, et al: A
dideazatetrahydrofolate analog lacking a chiral center at C-6:
N-[4-[2-(2-amino-3,4-dihydro-4-oxo-7H-pyrrolo
[2,3-d]pyrimidin-5yl)ethyl [benzoyl]-L-glutamic acid is an
inhibitor of thymidylate synthase. J Med Chem. 35:4450–4454.
1992.PubMed/NCBI
|
|
6.
|
Chattopadhyay S, Moran RG and Goldman ID:
pemetrexed: biochemical and cellular pharmacology, mechanisms, and
clinical applications. Mol Cancer Ther. 6:404–417. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Miller KD, Picus J, Blanke C, John W,
Clark J, Shulman LN, Thornton D, Rowinsky E and Loehrer PJ Sr:
Phase II study of the multitargeted antifolate LY231514 (ALIMTA,
MTA, pemetrexed disodium) in patients with advanced pancreatic
cancer. Ann Oncol. 11:101–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Giovannetti E, Mey V, Danesi R, Mosca I
and Del Tacca M: Synergistic cytotoxicity and pharmacogenetics of
gemcitabine and pemetrexed combination in pancreatic cancer cell
lines. Clin Cancer Res. 10:2936–2943. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Li T, Ling YH, Goldman ID and Perez-Soler
R: Schedule-dependent cytotoxic synergism of pemetrexed and
erlotinib in human non-small cell lung cancer cells. Clin Cancer
Res. 13:3413–3422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Solomon B and Bunn PA Jr: Clinical
activity of pemetrexed: a multitargeted antifolate anticancer
agent. Future Oncol. 1:733–746. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Lu YY, Jing DD, Xu M, Wu K and Wang XP:
Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer
cell line. World J Gastroenterol. 14:5403–5411. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Giovannetti E, Lemos C, Tekle C, Smid K,
Nannizzi S, Rodriguez JA, Ricciardi S, Danesi R, Giaccone G and
Peters GJ: Mechanisms underlying the synergistic interaction of
erlotinib, an epidermal growth factor receptor tyrosine kinase
inhibitor, with the multitargeted antifolate pemetrexed in
non-small-cell lung cancer cells. Mol Pharmacol. 73:1290–1300.
2008. View Article : Google Scholar
|
|
13.
|
Davies AM, Ho C, Lara PN Jr, Mack P,
Gumerlock PH and Gandara DR: Pharmacodynamic separation of
epidermal growth factor receptor tyrosine kinase inhibitors and
chemotherapy in non-small-cell lung cancer. Clin Lung Cancer.
7:385–388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Mahaffey CM, Davies AM, Lara PN Jr, Pryde
B, Holland W, Mack PC, Gumerlock PH and Gandara DR:
Schedule-dependent apoptosis in K-ras mutant non-small-cell lung
cancer cell lines treated with docetaxel and erlotinib: rationale
for pharmacodynamic separation. Clin Lung Cancer. 8:548–553. 2007.
View Article : Google Scholar
|
|
15.
|
Giovannetti E, Lemos C, Tekle C, et al:
Molecular mechanisms underlying the synergistic interaction of
erlotinib, an epidermal growth factor receptor tyrosine kinase
inhibitor, with the multitargeted antifolate pemetrexed in
non-small-cell lung cancer cells. Mol Pharmacol. 73:1290–1300.
2008. View Article : Google Scholar
|
|
16.
|
Li J, Li Y, Feng ZQ and Chen XG:
Anti-tumor activity of a novel EGFR tyrosine kinase inhibitor
against human NSCLC in vitro and in vivo. Cancer Lett. 279:213–220.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lu YY, Jing DD, Xu M, Wu K and Wang XP:
Anti-tumor activity of erlotinib in the BxPC-3 pancreatic cancer
cell line. World J Gastroenterol. 14:5403–5411. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue
P, Fu H and Khuri FR: Activation of Akt and eIF4E survival pathways
by rapamycin-mediated mammalian target of rapamycin inhibition.
Cancer Res. 65:7052–7058. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Roskoski R Jr: The ErbB/HER receptor
protein-tyrosine kinases and cancer. Biochem Biophys Res Commun.
319:1–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Tobita K, Kijima H, Dowaki S, et al:
Epidermal growth factor receptor expression in human pancreatic
cancer: Significance for liver metastasis. Int J Mol Med.
11:305–309. 2003.PubMed/NCBI
|
|
21.
|
Frolov A, Schuller K, Tzeng CW, Cannon EE,
Ku BC, Howard JH, Vickers SM, Heslin MJ, Buchsbaum DJ and Arnoletti
JP: ErbB3 expression and dimerization with EGFR influence
pancreatic cancer cell sensitivity to erlotinib. Cancer Biol Ther.
6:548–554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Giovannetti E, Mey V, Nannizzi S,
Pasqualetti G, Del Tacca M and Danesi R: Pharmacogenetics of
anticancer drug sensitivity in pancreatic cancer. Mol Cancer Ther.
5:1387–1395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Liles JS, Arnoletti JP, Tzeng CW, Howard
JH, Kossenkov AV, Kulesza P, Heslin MJ and Frolov A: ErbB3
expression promotes tumorigenesis in pancreatic adenocarcinoma.
Cancer Biol Ther. 10:555–563. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Yonezawa M, Wada K, Tatsuguchi A, Akamatsu
T, Gudis K, Seo T, Mitsui K, Nagata K, Tanaka S, Fujimori S and
Sakamoto C: Heregulin-induced VEGF expression via the ErbB3
signaling pathway in colon cancer. Digestion. 80:215–225. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Miller TW, Pérez-Torres M, Narasanna A, et
al: Loss of phosphatase and tensin homologue deleted on chromosome
10 engages ErbB3 and IGF-IR signaling to promote antiestrogen
resistance in breast cancer. Cancer Res. 69:4192–4201. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Chen K, Iribarren P, Gong W and Wang JM:
The essential role of phosphoinositide 3-kinases (PI3Ks) in
regulating pro-inflammatory responses and the progression of
cancer. Cell Mol Immunol. 2:241–252. 2005.PubMed/NCBI
|
|
27.
|
Buck E, Eyzaguirre A, Haley JD, Gibson NW,
Cagnoni P and Iwata KK: Inactivation of Akt by the epidermal growth
factor receptor inhibitor erlotinib is mediated by HER-3 in
pancreatic and colorectal tumor cell lines and contributes to
erlotinib sensitivity. Mol Cancer Ther. 5:2051–2059. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Engelman JA, Jänne PA, Mermel C, Pearlberg
J, Mukohara T, Fleet C, Cichowski K, Johnson BE and Cantley LC:
ErbB-3 mediates phosphoinositide 3-kinase activity in
gefitinib-sensitive non-small cell lung cancer cell lines. Proc
Natl Acad Sci USA. 102:3788–3793. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Engelman JA, Zejnullahu K, Mitsudomi T, et
al: MET amplification leads to gefitinib resistance in lung cancer
by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Turke AB, Zejnullahu K, Wu YL, et al:
Preexistence and clonal selection of MET amplification in EGFR
mutant NSCLC. Cancer Cell. 17:77–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Arteaga CL: HER3 and mutant EGFR meet MET.
Nat Med. 13:675–677. 2007. View Article : Google Scholar : PubMed/NCBI
|