Introduction
The hepatitis C virus (HCV) is an enveloped virus
with a single stranded, positive sense, nonsegmented RNA genome of
approximately 9,500 nucleotides that encodes a polyprotein of
approximately 3,000 amino acids (1,2).
Analysis of the HCV genome has demonstrated extremely high
heterogeneity in both structural and nonstructural coding regions
and has identified at least six different genotypes (1 to 6) that
have generally been divided into several subtypes (designated a, b,
c) (3–5). These genotypes have distinct
geographical distributions. Although HCV genotypes 1, 2 and 3
appear to have a worldwide distribution, their relative prevalence
varies from one geographic area to another. HCV genotype 4 was
found in the Middle East and North Africa, and genotypes 5 and 6 in
South Africa and Asia, respectively (6–11).
Genotype identification is clinically important for
prediction of responses to, and in determining the duration of,
antiviral therapy (12). This is
illustrated by the fact that genotypes 1 and 4 were more resistant
to treatment with pegylated interferon-α and ribavirin than
genotypes 2, 3 and 6 (13–15).
Patients with chronic HCV genotype 1b infection showed more severe
liver disease than patients infected with other genotypes (16). At present, most treatment
protocols require genotype information for patients infected with
HCV.
A variety of technologies have been developed for
HCV genotype determination. The majority of these assays rely on
the amplification of short HCV RNA regions from clinical specimens,
followed by a type-specific assay, such as restriction fragment
length polymorphism analysis (17), line probe reverse hybridization
(18,19), or sequence analysis (20,21). Almost all available commercial
assays target the 5′-untranslated region (5′-UTR), as the highly
conserved sequences of this region are most suitable for reverse
transcription-PCR (RT-PCR) amplification.
The Versant HCV genotype assay (LiPA) is one of the
most widely used methods for HCV genotyping. In this assay, the
5′-UTR of HCV is amplified with biotinylated primers, after which
the PCR product is hybridized to a membrane impregnated with
genotype-specific probes and detected with streptavidin linked to a
colorimetric detector (22).
Despite the high conservation of the 5′-UTR, genotype determination
of HCV based on the 5′-UTR is accurate for most genotypes (21,23,24). However, it has been noted that
methods that are based on the use of the 5′-UTR falsely identify
genotypes 6c to 6l from SEA as genotype 1, which is also the case
in the Versant HCV genotype assay (22,25). Moreover, this assay is unable to
distinguish genotype 1a from 1b in 5–10% of the cases (24,26). To improve the accuracy of
distinguishing between genotypes 1a, 1b, and 6c to 6l, a new
generation of the line probe assay (Versant HCV genotype 2.0 assay,
LiPA 2.0) that uses core sequence information in addition to the
5′-UTR was developed.
Sequencing and phylogenetic analysis of the core/E1
or NS5B region were considered to be the gold standard for HCV
genotyping since they accurately identified the subtype and were
used to establish an epidemiological picture of circulating virus
strains (27). Although such
assay involves complicated and time-consuming experimental
procedures of RNA extraction, reverse transcription, nested PCR,
DNA sequencing and phylogram construction, genotyping HCV by
sequence analysis has become increasingly more convenient than
before with the continuous improvement of biochemical reagents and
experimental techniques.
In the present study, three methods, i.e., core,
NS5B sequence analysis and line probe assay (LiPA 2.0), were
evaluated for their effectiveness in identifying HCV
genotypes/subtypes in China.
Materials and methods
Serum samples
One hundred and ten patients from Guangdong, China
were diagnosed with chronic hepatitis C in the Third Affiliated
Hospital of Sun Yet-sen University. Prior to antiviral treatment, 5
ml serum samples were collected from each patient. All patients had
signed the informed consent.
HCV viral load tests
The viral loads were determined with Amplicor HCV
monitor version 2.0 (Roche, Meylan, France). According to the
supplier’s instructions, serum samples were not prediluted except
as otherwise stated. RNA extraction, reverse transcription,
amplification, detection, and calculation of the number of HCV
copies per ml were performed according to the manufacturer’s
protocol. Briefly, 100 μl of serum was added to 400
μl of lysis buffer containing the internal quantitative
standard (IQS). After incubation for 10 min at 60°C and
precipitation, the pellet was diluted in 1 ml of specimen diluent.
For PCR, 50 μl of diluted extract was transferred into
reaction tubes containing 50 μl of the PCR master mixture.
Reverse transcription was performed at 60°C for 30 min, and
amplification was performed for 2 cycles at 95°C for 15 sec and
60°C for 20 sec and then 33 cycles at 90°C for 15 sec and 60°C for
20 sec in GeneAmp® PCR System 9700 (ABI, Alabaster, AL,
USA). Following amplification, 100 μl of denaturing reagent
was transferred to each reaction tube, and the tubes were incubated
for at least 10 min. Detection was performed on microtiter plates
coated with capture oligonucleotides specific for HCV sequences and
containing 100 μl of hybridization buffer/well by pipetting
25 μl of the amplification product into the wells of the
first row and then performing 1/5, 1/25, 1/125 and 1/625 dilutions
in the following rows. Similarly, a 1/1, 1/5 and 1/25 dilution
series was performed in wells coated with capture oligonucleotides
specific for the IQS. Following incubation, washing, and the color
reaction, the OD of each well was measured in an ELISA reader
RT-6000 (Rayto, Shenzhen, China) at A450. The HCV concentration was
calculated by multiplying the OD value of HCV and IQS of the
highest dilution that gave an OD of between 0.5 and 2.0 by the
respective dilution factor and dividing the value for HCV by the
value for the IQS. The result was then multiplied by 100 for the
IQS copies and a sample dilution factor of 200, resulting in the
number of HCV copies/ml of serum. For each sample, the OD for the
IQS was above 0.5 in at least one of the wells and the ODs of HCV
and IQS were subtracted by the background value according to the
supplier’s instructions.
RNA extraction and reverse
transcription
RNA was isolated from the first RNA-positive serum
sample obtained from each patient using 500 μl serum and a
RNAiso™ Plus extraction kit (Takara, Dalian, China). HCV RNA was
eluted in 10 μl of Tris-EDTA (TE) buffer and was
subsequently transcribed into cDNA using the ReverTra Ace-α-reverse
transcription kit (Toyobo, Shanghai, China). This cDNA was used as
the input for two separate PCR assays targeting the HCV core and
NS5B regions.
Amplifying and sequencing core and NS5B
fragments
The core and NS5B regions were amplified using a
nested polymerase chain reaction (nPCR). Primers used for
amplifying the core region were the same as previously reported
Lole et al (28). The core
outer primers were: forward, 5′-ACTGCCTG ATAGGGTGCTTGC-3′ and
reverse, 5′-ATGTACCCCAT GAGGTCGGC-3′; the inner primers were:
forward, 5′-AGG TCTCGTAGACCGTGCA-3′ and reverse, 5′-CATGTGAG
GGTATCGATGAC-3′. The primers used for amplifying the NS5B region
derived from Laperche et al (29). The NS5B outer primers were:
forward, 5′-CNTAYGGITTCCA RTACTCICC-3′ and reverse, 5′-GAG
GARCAIGATGTT ATIARCTC-3′; the inner primers were: forward, 5′-TATGA
YACCCGCTGYTTTGACTC-3′ and reverse, 5′-GCNGAR
TAYCTVGTCATAGCCTC-3′.
Outer PCR (30 μl): 10X PCR buffer 3 μl
2.5 mM dNTP 2 μl, dH2O 17.6 μl, primers
(10 pmol/μl) 1.5 μl, Taq enzyme (2.5 U/μl) 0.4
μl, template cDNA 4 μl. Inner PCR system (30
μl): 10X PCR buffer 3 μl, 2.5 mM dNTP 2 μl,
dH2O 19.6 μl, primers (10 pmol/μl) 1.5
μl of each, Taq enzyme (2.5 U/μl) 0.4 μl,
template cDNA 2 μl. PCR conditions were: 94°C for 5 min;
94°C for 30 sec, 55°C for 1 min, 72°C for 40 sec, 30 cycles; 72°C
for 10 min.
Following verification by agarose gel
electrophoresis, the PCR products were sent to the Beijing Genomics
Institute at Guangzhou for sequencing.
Sequence genotyping
Basic local alignment search tool (BLAST) and
phylogenetic analysis were used to identify HCV genotypes. First,
the nucleotide sequences from the core and NS5B regions were
analyzed by HCV BLAST in the Los Alamos HCV sequence
database (http://hcv.lanl.gov/content/index). Then, using the
ClustalW 1.8 software package, the sequences of HCV strains were
then aligned with a reference panel of sequences representative of
each subtype provided by the Los Alamos National Laboratory
(30). Pairwise distances were
generated using the Jukes-Cantor corrected distance algorithm of
the program MEGA 4.0 (31).
Phylogenetic analysis was performed using the neighbor-joining
method for tree drawing. The reliability of phylogenetic
classification was evaluated by a 1,000-cycle bootstrap test.
LiPA 2.0
The LiPA 2.0 was a reverse hybridization line probe
assay in which biotinylated DNA PCR products were hybridized to
immobilized oligonucleotide probes that were specific for the
5′-UTRs and core regions of the six HCV genotypes. The probes were
bound to a nitrocellulose strip by a poly (T) tail. After
hybridization of the biotinylated targets to the probes,
unhybridized PCR products were washed from the strips, and alkaline
phosphatase-labeled streptavidin (conjugate) was bound to the
biotinylated hybrid. After washing the strips,
5-bromo-4-chloro-3-indolylphosphate (BCIP)-nitroblue tetrazolium
chromogen (substrate) reacted with the conjugate, forming a
purple/brown precipitate, which resulted in a visible line pattern
on the strip that was specific for each genotype. Each strip had
three control lines and 22 parallel DNA probe lines containing
sequences specific for HCV genotypes 1–6. The conjugate control at
line 1 monitored the color development reaction and gave a positive
result if the strip was processed correctly. The amplification
control at line 2 (AMPL CTRL 1) contained universal probes that
hybridized to PCR products from the 5′-UTR. AMPL CTRL 2 was located
at line 23 and contained universal probes that hybridize to PCR
products from the core region. HCV genotypes were determined by
aligning the strips with a reading card and comparing the line
patterns from the strip with the patterns on the interpretation
chart.
Statistical analysis
Statistical analysis was performed with the SPSS
17.0 software package. Statistical significance was defined as a
2-sided P-value of ≤0.05. The differences in the distribution of
categorical variables were assessed by Pearson’s Chi-squared
test.
Results
Determination of HCV genotype by sequence
analysis
Of the 110 HCV samples, 8 failed to be amplified in
any region, 57 were amplified in both regions, 40 were amplified in
the core region only and 5 were amplified in the NS5B region only
(Tables I and II). The amplification rate of the core
region (92.7%) was significantly higher (P<0.001) than that of
the NS5B region (56.4%). Correlation analysis revealed that
amplification rate of both regions was correlated with viral load
(Table I). When the viral load
was ≥1.E+03 IU/ml, the amplification rate of the core region was
satisfactorily high (87.5–100%). However, it decreased markedly
(P<0.001) to 41.7% when the viral load was <1.E+03 IU/ml. A
similar phenomenon was observed in amplifying the NS5B region. When
the viral load was ≥1.E+04 IU/ml, the amplification rate ranged
from 62.1 to 73.9%, whereas when the viral load was <1.E+04
IU/ml, it decreased significantly (P<0.001) to only 26.7% at
1.E+03–1.E+04 IU/ml and 16.7% at <1.E+03 IU/ml.
 | Table I.The amplification rate of core/NS5B
regions with different viral loads. |
Table I.
The amplification rate of core/NS5B
regions with different viral loads.
| | No. of amplified
fragments
|
|---|
| RNA quantity
(IU/ml) | Samples (n) | Core, n (%) | NS5B, n (%) |
|---|
| 1.E+07–1.E+08 | 15 | 14 (93.3) | 10 (66.7) |
| 1.E+06–1.E+07 | 37 | 34 (91.9) | 23 (62.1) |
| 1.E+05–1.E+06 | 23 | 23 (100) | 17 (73.9) |
| 1.E+04–1.E+05 | 8 | 7 (87.5) | 6 (75) |
| 1.E+03–1.E+04 | 15 | 14 (93.3) | 4 (26.7) |
| <1.E+03 | 12 | 5 (41.7) | 2 (16.7) |
| Total | 110 | 97 (92.7) | 62 (56.4) |
| Table II.Genotyping HCV by core and NS5B
sequencing analysis. |
Table II.
Genotyping HCV by core and NS5B
sequencing analysis.
| No. of genotyped
samples amplified
| |
|---|
| Genotype | Core only | Core and NS5B | NS5B only | Total |
|---|
| 1b | 23 | 38 | 2 | 63 |
| 2a | 8 | 1 | 1 | 10 |
| 3a | 0 | 4 | 0 | 4 |
| 3b | 1 | 2 | 1 | 4 |
| 6a | 8 | 12 | 1 | 21 |
| Total | 40 | 57 | 5 | 102 |
Amplicons of the core and NS5B regions were
sequenced directly. Then, the sequences were submitted to GenBank
(accession no. JN572940 to JN572983) and were aligned with all
genotyped sequences in the Los Alamos HCV sequence database using
HCV BLAST (http://hcv.lanl.gov/content/sequence/BASIC_BLAST/basic_blast.html).
All the core sequences hit highly similar sequences in the database
with scores >450 bit and identities ≥97%. Similarly, all the
NS5B sequences matched sequences from the database with scores
>450 and identities ≥93%. Since the sequence similarities in the
core and NS5B regions between tested sample and strains from the
HCV database were >93 and 87.8% (which were the thresholds
between variants of the same genotypes for the core and NS5B
regions, respectively) (27,38), genotypes of sequences at the top
BLAST outputs were considered as identical to the tested samples.
Based on the BLAST result, genotypes identified by core and NS5B
sequence analysis were compared. The result showed that genotypes
assigned by sequence analysis of both regions were identical. In
summary, of the 102 HCV samples amplified in either or both
regions, 63 (61.8%) were identified as subtypes 1b, 10 (9.8%) as
2a, 4 (3.9%) as 3a, 4 (3.9%) as 4a, and 21 (20.6%) as 6a (Table II).
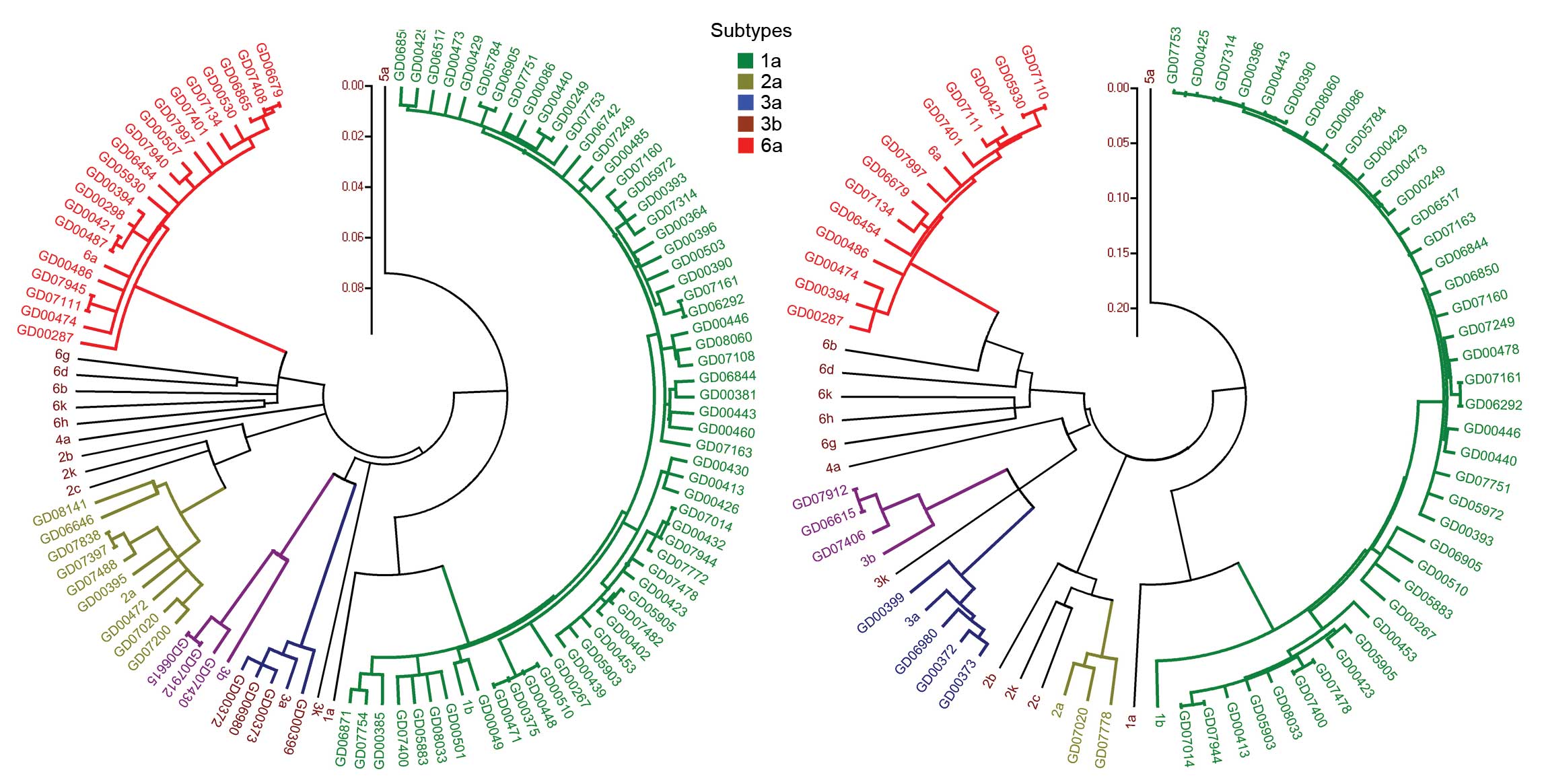
To further verify genotypes of HCV, phylograms were
constructed with the reference stains for the core and NS5B regions
(Fig. 1). The results showed that
the core sequences of 97 samples and the reference strains of
genotypes 1b, 2a, 3a, 3b and 6a were grouped into five clusters.
The bootstrap values of each cluster exceeded 80%, indicating that
the topology of core sequence was highly reliable. The genotypes
identified by phylogenetic analysis were identical to those
assigned by BLAST analysis. Similarly, the NS5B sequences were
grouped into five clusters with high bootstrap values (74–96%), and
the results of phylogenetic analysis were consistent with those of
BLAST analysis. Comparison of the phylograms of the core and NS5B
regions showed that the topology of both regions was similar, but
the Jukes-Cantor molecule genetic distance of NS5B was larger than
that of core, which was consistent with the fact that the NS5B
region is more variable than the core region.
Genotyping by LiPA 2.0
Table III shows
that except for genotype 6, the other genotypes were distinguished
correctly by LiPA 2.0 at the genotype level. However, at the
subtype level, only 1b and 3b were distinguished accurately.
| Table III.Comparison of LiPA genotyping with
core/NS5B sequence. |
Table III.
Comparison of LiPA genotyping with
core/NS5B sequence.
| Genotyping by
core/NS5B sequence
| |
|---|
| Genotyping by
INNO-LiPA 2.0 | 1b | 2a | 3a | 3b | 6a | Total |
|---|
| 1b | 63 | | | | 11 | 74 |
| 2a/c | | 8 | | | | 8 |
| 2 | | 2 | | | | 2 |
| 3a | | | 1 | | | 1 |
| 3b | | | 3 | 4 | | 7 |
| 6a/b | | | | | 10 | 10 |
| Total | 63 | 10 | 4 | 4 | 21 | 102 |
The accuracy for genotyping 1b was 100% with LiPA
2.0. Interpreting the results obtained from all genotype 1 samples
with LiPA 2.0 according to the amplified region, 44 (69.8%) were
correctly genotyped by taking into account the 5′-UTR alone,
whereas 63 (100%) were genotyped correctly using the additional
information on the core region. In this study, 8 (80%) and 2 (20%)
of the subtype 2a samples were incompletely classified into subtype
2a/2c and genotype 2, respectively. LiPA 2.0 was unable to
completely distinguish 2a from 2c, due to the lack of probes
specific to 2a and 2c. Despite all 8 of the genotype 3 samples (4
were 3a, and 4 were 3b) correctly identified at the genotype level,
there were 37.5% misidentified at the subtype level (i.e., 3
subtype 3a samples were misidentified as 3b). Compared with the
other genotype, the accuracy of identifying genotype 6 by LiPA 2.0
was low. Ten of the 21 genotype 6a samples were incompletely
classified as 6a/6b, and the others (52.4%) were misclassified into
1b. Considering the whole panel, the overall rate of concordance
(correct genotype and correct subtype) was 66.7% (68 samples) for
LiPA 2.0. The percentage of incomplete results (indistinguishable
or not identified subtype) was 19.6% (20 samples).
Misclassifications were observed for 13.7% (14) of the tested samples.
Discussion
The most accurate method for genotyping is
sequencing the entire genome of HCV. However, this is
time-consuming and difficult to apply on a large scale. Despite the
sequence diversity of HCV, all genotypes share an identical
component of co-linear genes of similar or identical size in the
large open reading frame, and the genetic inter-relationships of
HCV variants are remarkably consistent throughout the genome
(4). This has enabled many of the
recognized variants of HCV to be provisionally classified, based on
partial sequences from subgenomic regions (27). Core/E1 or NS5B are considered the
most reliable regions for genotyping HCV, as sufficient genetic
diversity is presented in these regions. Although genotyping
results in these regions were consistent, amplification rates of
these regions appear different. In the present study, the
amplification rate of the core region was higher than that of NS5B
with the same viral load, which indicated that the amplification
rate of subgenomic regions was not only associated with viral load,
but also with sequence conservation. Since the core region was more
conservative than NS5B, to ensure a satisfactory amplification rate
and high genotyping accuracy, amplification of the core region was
recommended.
The accuracy of genotyping HCV by BLAST analysis
depends on the number of genotyped sequences in the HCV database.
Recently, with genotyped sequences being continuously submitted,
genotyping HCV by BLAST analysis has become increasingly more
reliable. In the present study, all the sequences of tested samples
hit highly similar genotyped sequence in the HCV database. These
results highly support BLAST analysis. Moreover, compared with
phylogenetic analysis, BLAST analysis was simpler and more
timesaving.
The accuracy of identifying genotype 6, subtypes c
to l, was improved greatly in LiPA 2.0 using sequence motifs from
the core region in addition to the 5′-UTR (32). In addition, the accuracy of
identifying 1a and 1b was also improved greatly in LiPA 2.0 (Ross
et al) (33). In the
present study, only 69.8% of genotype 1b would have been
characterized correctly using 5′-UTR information alone, whereas
100% of them have been correctly genotyped 1b with core
information. The results confirmed the benefit of including the
core region in LiPA 2.0. However, the capacity to identify the
other genotypes/subtypes was not improved in LiPA 2.0, since only
5′-UTR information was used. Despite several studies showing that
most genotypes, excluding subtypes 1a, 1b and 6c-l, could be
distinguished correctly by 5′-UTR (21,24,34), it is not always the case. In some
countries or regions, the HCV genotypes were not well distinguished
by 5′-UTR. For example, Stuyver et al (22) used LiPA to analyze 506
HCV-infected sera from different geographical regions, representing
a multitude of subtypes, and found that only 11% of HCV samples
from Western Africa could be identified at the subtype level by
5′-UTR. The present study showed that only 66.7% of HCV samples
were genotyped accurately, 52.4% of genotype 6a samples were
misclassified in 1b. Based on this result, the suitability of LiPA
2.0 for genotyping HCV from South China should be reconsidered. A
recent study showed that subtype 1b remained the most prevalent and
widely distributed genotype in China (35). However, subtype 6a was more
prevalent in the southern provinces of China (36,37). Clearly, with the increasing
prevalence of 6a in South China, using LiPA 2.0 to identify HCV
strains will lead to biased diagnostic and research results.
Therefore, LiPA 2.0 was not suitable for identifying HCV genotypes
in South China.
In addition, the LiPA 2.0 kit is relatively
expensive. In China, the reagent cost was about 200 dollars/sample
by LiPA 2.0, but less than 5 dollars by sequencing analysis.
Genotyping HCV by sequencing analysis is therefore more
cost-effective. Moreover, since the performance of commercial
reverse transcriptase enzymes and DNA polymerases are improving
continuously, genotyping HCV by sequencing analysis is not as
difficult as previously. Currently, a reliable genotyping result
can be obtained in 48 h with direct sequencing and BLAST analysis
by a skilled technician. Therefore, direct sequencing of HCV core
fragment plus BLAST analysis represents an ideal genotyping
approach for laboratories with skilled technicians.
Abbreviations:
|
HCV
|
hepatitis C virus;
|
|
UTR
|
untranslated region;
|
|
IQS
|
internal quantitative standard;
|
|
TE
|
Tris-EDTA;
|
|
BLAST
|
basic local alignment search tool
|
Acknowledgements
This study was supported by the
National Science and Technology Key Project during the 11th
Five-Year Plan Period (no. 2008ZX10002-013). Mention of trade names
or commercial products in this publication is solely for the
purpose of providing specific information and does not imply
recommendation or endorsement by the Ministry of Health of
China.
References
|
1.
|
Choo QL, Richman KH, Han JH, Berger K, Lee
C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ, Weiner A,
Bradley DW, Kuo G and Houghton M: Genetic organization and
diversity of the hepatitis C virus. Proc Natl Acad Sci USA.
88:2451–2455. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Inchauspe G, Zebedee S, Lee DH, Sugitani
M, Nasoff M and Prince AM: Genomic structure of the human prototype
strain H of hepatitis C virus: comparison with American and
Japanese isolates. Proc Natl Acad Sci USA. 88:10292–20296. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bukh J, Purcell RH and Miller RH: Sequence
analysis of the core gene of 14 hepatitis C virus genotypes. Proc
Natl Acad Sci USA. 91:8239–8243. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Robertson B, Myers G, Howard C, Brettin T,
Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O,
Netesov S, Nishioka K, Shin-i T, Simmonds P, Smith D, Stuyver L and
Weiner A: Classification, nomenclature, and database development
for hepatitis C virus (HCV) and related viruses: proposals for
standardization. Arch Virol. 143:2493–2503. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Stuyver L, Arnhem W, Wyseur A, Hernandez
F, Delaporte E and Maertens G: Classification of hepatitis C
viruses based on phylogenetic analysis of envelope 1 and
nonstructural 5b regions and identification of five additional
subtypes. Proc Natl Acad Sci USA. 91:10134–10138. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bukh J, Purcell RH and Miller RH: At least
12 genotypes of hepatitis C virus predicted by sequence analysis of
the putative E1 gene of isolates collected worldwide. Proc Natl
Acad Sci USA. 90:8234–8238. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mellor J, Holmes EC, Jarvis LM, Yap PL and
Simmonds P: Investigation of the pattern of hepatitis C virus
sequence diversity in different geographical regions: implications
for virus classification. The International HCV Collaborative Study
Group. J Gen Virol. 76:2493–2507. 1995. View Article : Google Scholar
|
|
8.
|
Ndjomou J, Pybus OG and Matz B:
Phylogenetic analysis of hepatitis C virus isolates indicates a
unique pattern of endemic infection in Cameroon. J Gen Virol.
84:2333–2341. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Simmonds P: Genetic diversity and
evolution of hepatitis C virus - 15 years on. J Gen Virol.
85:3173–3188. 2004.PubMed/NCBI
|
|
10.
|
Tokita H, Okamoto H, Luengrojanakul P,
Vareesangthip K, Chainuvati T, Iizuka H, Tsuda F, Miyakawa Y and
Mayumi M: Hepatitis C virus variants from Thailand classifiable
into five novel genotypes in the sixth (6b), seventh (7c, 7d) and
ninth (9b, 9c) major genetic groups. J Gen Virol. 76:2329–2335.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tokita H, Okamoto H, Tsuda F, Song P,
Nakata S, Chosa T, Iizuka H, Mishiro S, Miyakawa Y and Mayumi M:
Hepatitis C virus variants from Vietnam are classifiable into the
seventh, eighth, and ninth major genetic groups. Proc Natl Acad Sci
USA. 91:11022–11026. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Zein NN, Rakela J, Krawitt EL, Reddy KR,
Tominaga T and Persing DH: Hepatitis C virus genotypes in the
United States: epidemiology, pathogenicity, and response to
interferon therapy. Collaborative Study Group. Ann Intern Med.
125:634–639. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Fung J, Lai CL, Hung I, Young J, Cheng C,
Wong D and Yuen MF: Chronic hepatitis C virus genotype 6 infection:
response to pegylated interferon and ribavirin. J Infect Dis.
198:808–812. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
McHutchison JG, Gordon SC, Schiff ER,
Shiffman ML, Lee WM, Rustgi VK, Goodman ZD, Ling MH, Cort S and
Albrecht JK: Interferon alfa-2b alone or in combination with
ribavirin as initial treatment for chronic hepatitis C. N Engl J
Med. 339:1485–1492. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Zylberberg H, Chaix ML and Brechot C:
Infection with hepatitis C virus genotype 4 is associated with a
poor response to interferon-alpha. Ann Intern Med. 132:845–846.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Silini E, Bono F, Cividini A, Cerino A,
Bruno S, Rossi S, Belloni G, Brugnetti B, Civardi E, Salvaneschi L
and Mondelli MU: Differential distribution of hepatitis C virus
genotypes in patients with or without liver function abnormalities.
Hepatology. 21:285–290. 1995.PubMed/NCBI
|
|
17.
|
Buoro S, Pizzighella S, Boschetto R,
Pellizzari L, Cusan M, Bonaguro R, Mengoli C, Caudai C, Padula M,
Egisto-Valensin P and Palu G: Typing of hepatitis C virus by a new
method based on restriction fragment length polymorphism.
Intervirology. 42:1–8. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Stuyver L, Rossau R, Wyseur A, Duhamel M,
Vanderborght B, Van Heuverswyn H and Maertens G: Typing of
hepatitis C virus isolates and characterization of new subtypes
using a line probe assay. J Gen Virol. 74:1093–1102. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Stuyver L, Wyseur A, Arnhem van W, Lunel
F, Laurent-Puig P, Pawlotsky JM, Kleter B, Bassit L, Nkengasong J,
van Doorn LJ and Maertens G: Hepatitis C virus genotyping by means
of 5′-UR/core line probe assays and molecular analysis of
untypeable samples. Virus Res. 38:137–157. 1995.
|
|
20.
|
Simmonds P, Alberti A, Alter HJ, Bonino F,
Bradley DW, Brechot C, Brouwer JT, Chan SW, Chayama K and Chen DS:
A proposed system for the nomenclature of hepatitis C viral
genotypes. Hepatology. 19:1321–1324. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Weck K: Molecular methods of hepatitis C
genotyping. Expert Rev Mol Diagn. 5:507–520. 1994. View Article : Google Scholar
|
|
22.
|
Stuyver L, Wyseur A, Arnhem van W,
Hernandez F and Maertens G: Second-generation line probe assay for
hepatitis C virus genotyping. J Clin Microbiol. 34:2259–2266.
1996.PubMed/NCBI
|
|
23.
|
Simmonds P, Bukh J, Combet C, Deléage G,
Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens
G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon
E, Shin-I T, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ and Widell
A: Consensus proposals for a unified system of nomenclature of
hepatitis C virus genotypes. Hepatology. 42:962–973. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Smith DB, Mellor J, Jarvis LM, Davidson F,
Kolberg J, Urdea M, Yap PL and Simmonds P: Variation of the
hepatitis C virus 5′ non-coding region: implications for secondary
structure, virus detection and typing. The International HCV
Collaborative Study Group. J Gen Virol. 76:1749–1761. 1995.
|
|
25.
|
Chinchai T, Labout J, Noppornpanth S,
Theamboonlers A, Haagmans BL, Osterhaus AD and Poovorawan Y:
Comparative study of different methods to genotype hepatitis C
virus type 6 variants. J Virol Methods. 109:195–201. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Chen Z and Weck KE: Hepatitis C virus
genotyping: interrogation of the 5′ untranslated region cannot
accurately distinguish genotypes 1a and 1b. J Clin Microbiol.
40:3127–3134. 2002.PubMed/NCBI
|
|
27.
|
Simmonds P, Smith DB, McOmish F, Yap PL,
Kolberg J, Urdea MS and Holmes EC: Identification of genotypes of
hepatitis C virus by sequence comparisons in the core, E1 and NS-5
regions. J Gen Virol. 75:1053–1061. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lole KS, Jha JA, Shrotri SP, Tandon BN,
Prasad VG and Arankalle VA: Comparison of hepatitis C virus
genotyping by 5′ noncoding region- and core-based reverse
transcriptase PCR assay with sequencing and use of the assay for
determining subtype distribution in India. J Clin Microbiol.
41:5240–5244. 2003.
|
|
29.
|
Laperche S, Lunel F, Izopet J, Alain S,
Dény P, Duverlie G, Gaudy C, Pawlotsky JM, Plantier JC, Pozzetto B,
Thibault V, Tosetti F and Lefrère JJ: Comparison of hepatitis C
virus NS5b and 5′ noncoding gene sequencing methods in a
multicenter study. J Clin Microbiol. 43:733–739. 2005.
|
|
30.
|
Kuiken C, Yusim K, Boyki L and Richardson
R: The Los Alamos hepatitis C sequence database. Bioinformatics.
21:379–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tamura K, Dudley J, Nei M and Kumar S:
MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software
version 4.0. Mol Biol Evol. 24:1596–1599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Noppornpanth S, Sablon E, De Nys K, Truong
XL, Brouwer J, Van Brussel M, Smits SL, Poovorawan Y, Osterhaus AD
and Haagmans BL: Genotyping hepatitis C viruses from Southeast Asia
by a novel line probe assay that simultaneously detects core and 5′
untranslated regions. J Clin Microbiol. 44:3969–3974.
2006.PubMed/NCBI
|
|
33.
|
Ross RS, Viazov S and Roggendorf M:
Genotyping of hepatitis C virus isolates by a new line probe assay
using sequence information from both the 5′ untranslated and the
core regions. J Virol Methods. 143:153–160. 2007.
|
|
34.
|
Lee JH, Roth WK and Zeuzem S: Evaluation
and comparison of different hepatitis C virus genotyping and
serotyping assays. J Hepatol. 26:1001–1009. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Lu L, Nakano T, He Y, Fu Y, Hagedorn CH
and Robertson BH: Hepatitis C virus genotype distribution in China:
predominance of closely related subtype 1b isolates and existence
of new genotype 6 variants. J Med Virol. 75:538–549. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Fu Y, Wang Y, Xia W, Pybus OG, Qin W, Lu L
and Nelson K: New trends of HCV infection in China revealed by
genetic analysis of viral sequences determined from first-time
volunteer blood donors. J Viral Hepat. 18:42–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Zhou Y, Wang X, Mao Q, Fan Y, Zhu Y, Zhang
X, Lan L, Jiang L and Tan W: Changes in modes of hepatitis C
infection acquisition and genotypes in southwest China. J Clin
Virol. 46:230–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Simmonds P, Holmes EC, Cha TA, Chan SW,
McOmish F, Irvine B, Beall E, Yap PL, Kolberg J and Urdea MS:
Classification of hepatitis C virus into six major genotypes and a
series of subtypes by phylogenetic analysis of the NS-5 region. J
Gen Virol. 74:2391–2399. 1993. View Article : Google Scholar : PubMed/NCBI
|