Introduction
The adult brain has been considered insensitive to
radiation due to the relatively radio-resistant nature of mature
neurons which in certain model systems show no signs of injury
following exposure up to 22 Gy of X-rays (1). Brain damage induced by prenatal
irradiation is however a major concern and an important issue in
radioprotection (2–4). One of the most important factors
apart from dosage, in determining the nature of the damage to the
embryo from exposure to ionising radiation, is the developmental
stage. Indeed, the phenomenon of radiosensitivity is usually
recognized as the vulnerability of mitotic cells to ionising
radiation. However, brain development is characterized by the
succession of various critical periods, whose disturbance may have
severe consequences. Maturation of post-mitotic neurons and early
synaptogenesis are thus one of these critical periods. The proper
establishment and functioning of synapses is necessary for normal
brain development, thus improper synaptogenesis may disturb the
cognitive functions and lead to mental retardation and autism
(5–7). Prenatal exposure to toxic agents,
including radiation may prevent normal synaptogenesis caused by
cell loss (8), which may disturb
the cognitive functions. Active early synaptogenesis around week 18
of human gestation (9) and day 16
(E16) in embryonic rats (10) is
thus a crucial period during which neuronal cells may be highly
sensitive. During brain development, cell death is a natural
phenomenon that occurs in order to eliminate cells that did not
succeed in establishing strong contacts within the neuronal network
(11); however, an abnormal rate
of cell death during this period that dramatically reduces the
number of neuronal cells within the newly established network may
lead to neuroanatomical malformation and cognitive disabilities
(12).
High doses of ionising radiation clearly damage
immature neuronal cells (13),
but also more resistant cells such as neuroblastoma in
radiotherapy. At low doses, radiation-induced neuronal death has
also been observed through the activation of the P53 signaling
pathway, the guardian of the genome and upstream of the classical
apoptotic pathway (14).
Nevertheless, the mechanisms involved in P53-mediated apoptosis may
be more complex and may involve other factors, such as the
glutamate N-methyl D-aspartate receptor (NMDAr). Indeed, a
correlation between P53 induction and NMDAr activation and their
involvement in apoptosis has been proven (15).
NMDAr is highly expressed during brain development
and is involved in critical biological processes in brain
development such as neuronal modulation and synapse maturation
(16). The excessive activation
of NMDAr is known to be involved in excitotoxicity, a phenomenon
described in various neurode-generative pathologies such as
Alzheimer’s and Parkinson’s disease (17,18). The main element of glutamate
excitotoxicity is the downstream events of NMDAr over-activation
that are mainly related to the altered Ca2+ homeostasis
and its consequences, including neuronal death (19). Calpain, a
Ca2+-dependent protease is thus activated downstream and
plays a central role in the initiation of the cell death pathway
(20).
The block of glutamatergic neurotransmission via the
use of dizocilpine (MK-801), a non-competitive NMDAr antagonist has
been shown to confer significant protection against brain damage
caused by ionising radiation when administered subsequent to
exposure to 2.5 Gy of γ-rays and has been shown to confer a
dose-dependent protection in the dentate gyrus (21). Based on the above, we
investigated, in vivo and in vitro the
radiation-induced apoptosis in fetal cortex. We evaluated the
possible role of NMDAr and of the intracellular Ca2+
concentration in this process, by using MK-801 which blocks
Ca2+ neuronal influx, and nimodipine, an L-type
Ca2+ channel blocker.
Furthermore, in order to investigate the possible
involvement of apoptotic enzyme activation, known to occur at high
Ca2+ concentrations, we examined the protective effect
of an inhibitor of calpain on irradiated fetal brains and neurons
in cultures. Understanding the cellular and molecular mechanisms
may aid in the development of strategies to either increase the
radiation tolerance or treat central nervous system (CNS)
alterations induced by irradiation.
Materials and methods
Animals
BALB/c mice purchased from Janvier Laboratories (Le
Genest-St.-Isle, France) and Wistar R/Cnb rats obtained from Vito
(Mol, Belgium) were maintained for breeding in a conventional
animal facility under the recognition number LA 1100122 according
to the national legislation and the guidance of the Ethics
Committee of the Belgian Nuclear Research Centre (SCK-CEN) and the
Flemish Institute for Technological Research (Vito) for the care
and use of laboratory animals.
The Wistar R/Cnb rats were used for in vivo
study. The animals were mated between 06:00 and 08:30, and the day
of fertilization is referred as day 0 (E0). This short mating
duration, 150 min, was used in order to obtain very homogeneous
groups of embryos at a similar developmental stage. Mice were used
at day 17 (E17) of pregnancy and rats at day 15 (E15).
Neuronal culture
Primary cortical neuronal cultures were prepared
from BALB/cJ Rj (Janvier Laboratories) mouse fetuses, on embryonic
day 17 (E17). Pregnant females were sacrificed by cervical
dislocation and fetuses were extracted, mice were decapitated and
the heads were quickly placed into a dissection medium of cold
Hank’s buffered salt solution containing 0.5% glucose and 2.5 U/ml
penicillin/streptomycin (all from Invitrogen, Paisley, UK). The
brain cortices from each litter were dissected, pooled (between 6
and 8) and enzymatically dissociated for 20 min at 37°C in
dissection medium containing 0.1% Trypsin (Invitrogen) and 10 mg/ml
DNase I (Sigma-Aldrich, St. Louis, MO, USA). The reaction was
terminated by replacing the enzyme solution with dissection medium
containing 10% fetal bovine serum (Invitrogen). Mechanical
dissociation was carried out in dissection medium containing 5
mg/ml DNase I by trituration through the pipette tip. Dissociated
cells were pelleted by centrifugation at 1500 x g for 5 min at room
temperature. Cells were then re-suspended in the plating medium
containing minimum essential medium, 1 mM sodium pyruvate, 0.6%
glucose, 10% fetal bovine serum and 5 U/ml penicillin/streptomycin
(all from Invitrogen). The cells were plated at a density of
1.5×105 cells/well onto 13 mm diameter glass coverslips
for microscopy or 3×106 cells per flask of 25
cm2 seeding surface for cell lysis preparation.
Coverslips and flasks were pre-coated with 100 mg/ml poly-D-lysine
(Sigma-Aldrich) in 0.1 M borate buffer pH 8.5 and were incubated
for 60 min at 37°C in a humidified incubator containing 5%
CO2. The medium was then replaced with the serum-free
growth medium, consisting of neurobasal medium with 2% B27
supplement, 2.5 mM glutamine, 5 U/ml of penicillin/streptomycin
(all from Invitrogen) and 25 nM glutamate (Sigma-Aldrich). Cells
were grown for 7 days prior to treatment and irradiation. Half of
the growth medium was replaced after 3 days with the same medium
without glutamate.
Irradiation
Animals and cell cultures were irradiated at room
temperature with 250 kV-15 mA, 1 mm Cu-filtered X-rays (Pentak
HF420 RX machine), delivered at 5 mGy/sec. The farmer 2570-EMI
dosimeter was under the control of the Intercomparison Committee
for Dosimetry (former EULEP).
Cells were exposed to low (0.1 and 0.2 Gy) and
moderate (0.5 Gy) doses of X-rays. Sham-exposed cells were
subjected to the same conditions as the irradiated ones and were
considered as the controls. Animals used for in vivo study
(2–3 pregnant rats/group) were whole-body-irradiated with 0.6 Gy of
X-rays.
Treatments
Animal treatment
Rats were divided into 4 groups. At 20 min following
irradiation, one of those groups was injected intraperitoneally
with a 10 mg/ml saline solution to a dose of 3 mg/kg body weight of
dizocilpine (MK-801; Sigma-Aldrich), an NMDAr antagonist. The
second group was injected with 32 mg/kg of PD 150606, a calpain
inhibitor (Calbiochem, Darmstadt, Germany). The third group was
injected with 10 mg/kg body weight of nimodipine (Sigma-Aldrich),
an L-type Ca2+ channel blocker. The fourth irradiated
group was injected with the vehicle (saline solution) and used as
the positive control for DNA laddering.
Sham-exposed animals untreated or injected with the
vehicle, MK-801, calpain inhibitor or nimodipine were used as the
negative controls. An average of 9 embryos was collected per
female. The brain cortices of the embryos were examined
individually by DNA ladder electrophoresis 3 h following
injection.
Cell culture treatment
For each treatment, 3 replicates from 3 different
mouse litters were used. Cells were treated 2 h prior to
irradiation as follows: one group of cell cultures was treated with
10 μM of dizocilpine (MK-801; Sigma-Aldrich), the second group was
treated with 30 μM of calpeptin, a calpain inhibitor (Calbiochem),
the third group was not treated and used as the positive control of
the irradiation effect and the fourth group was sham-exposed and
used as the negative control.
Following irradiation, cell cultures were placed
back for 1 h in the incubator. The cells were then washed and fresh
medium was added. Cells were placed back into the incubator and
grown for 24 h until further manipulation.
γ-H2AX detection by immunofluorescence
microscopy
Irradiated and non-irradiated neurons plated on
coverslips were fixed with 4% paraformaldehyde 20 min after
irradiation. They were permeabilized using 0.1% Triton X-100
(Sigma-Aldrich) then blocked for 30 min with 3% bovine serum
albumin (Sigma-Aldrich) and incubated overnight at 4°C with a
primary mouse monoclonal antibody against the phosphorylated form
of the histone, H2AX (γ-H2AX) (Abcam, Cambridge, UK) diluted 1:300,
followed by incubation with a FITC-linked secondary polyclonal goat
anti-mouse antibody diluted 1:300 for 1 h at room temperature. The
nuclei were counterstained by incubating the coverslips for 5 min
with 0.5 μg/ml Hoechst. Coverslips were mounted on glass slides
using the vectashield mounting medium (Vector Laboratories,
Peterborough, UK). The images were captured using Nikon Eclipse Ti
(an automated inverted wide field epifluorescence microscope)
equipped with a ×40 oil immersion objective and a Nikon DS-Qi1Mc
camera. Images were taken as 16 different frames/coverslip, 19
plains of depth of 0.6 µm thickness. Images were then analyzed
using ImageJA freeware version 1.45 b and the number of nuclei and
γ-H2AX spots were counted as previously described (22) using an algorithm (provided by Dr
Winnok Devos, Ghent University, Ghent, Belgium). In total,
approximately 1,200 nuclei from 3 different coverslips were scored
and the number of foci/nucleus was reported. The algorithm also
determined the average spot occupancy, the area of the nucleus
occupied by one focus. The mean number of foci/nucleus and spot
occupancy was calculated for each irradiation dose using GraphPad
Prism version 5.00 for Windows (GraphPad Software, San Diego, CA,
USA).
DNA ladder electrophoresis
DNA was extracted from the cortices of the embryos.
Mice were sacrificed at 3 h and 20 min after irradiation, and fetal
and mother brains were collected. The cortex areas were dissected
and genomic DNA was extracted using the Wizard Genomic DNA kit
(Promega, Madison, USA). DNA concentration was determined by
spectrophotometry, by measuring the absorbance at 260 and 280 nm.
DNA (2 μg) was loaded on a 1% agarose gel. The DNA Molecular Weight
Marker XIII (Roche Applied Science, Vilvoorde, Belgium) was used as
a reference.
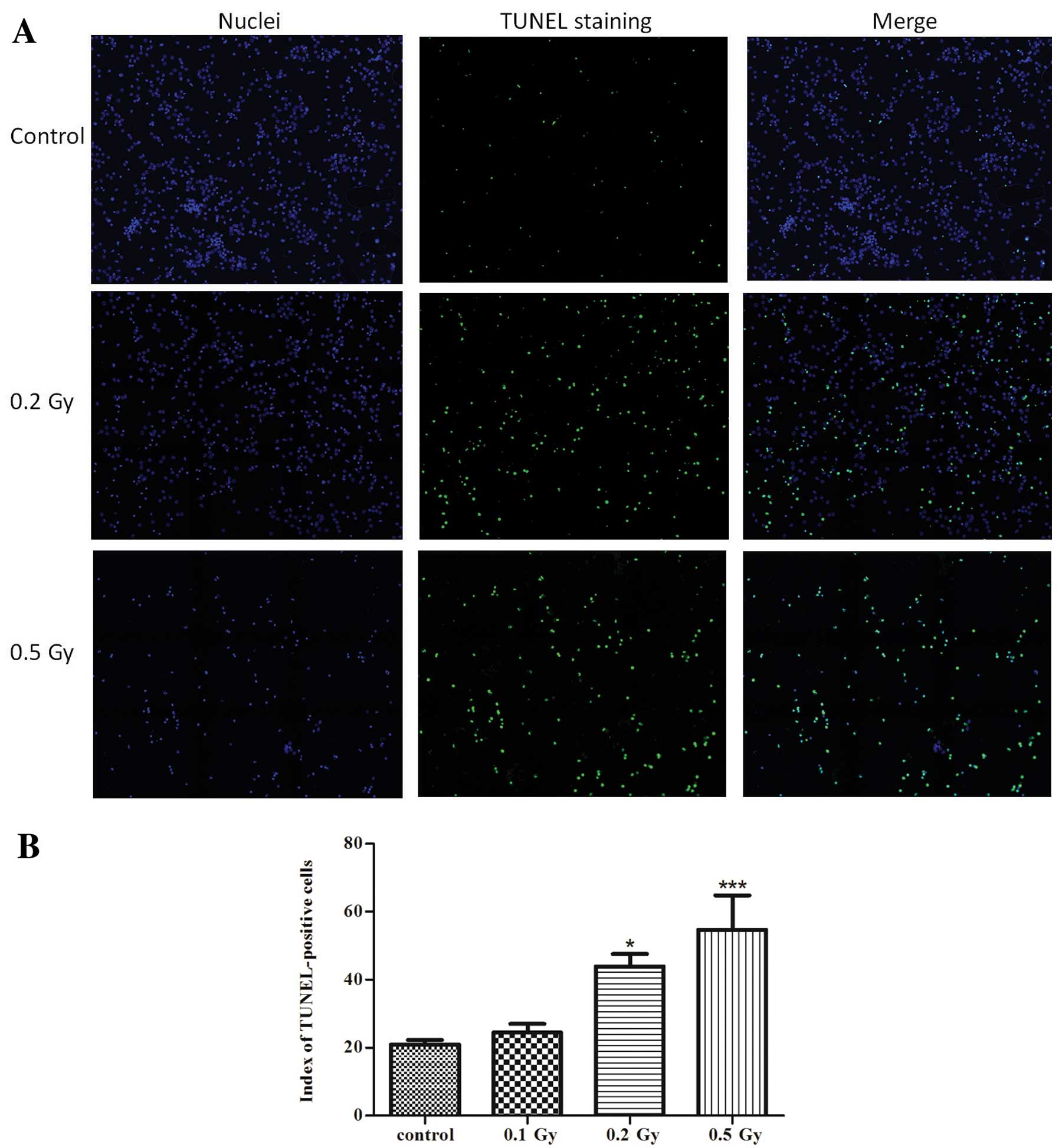
Cell death analysis
Treated and non-treated neuronal cultures mounted on
coverslips were fixed with 4% paraformaldehyde. The cells were then
permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in 0.1% sodium
citrate. Apoptotic cells were identified by TUNEL staining using an
In Situ Cell Death Detection kit (Fluorescein; Roche Applied
Science) according to the manufacturer’s instructions. In brief,
cells were incubated with the TUNEL reaction mixture containing
terminal deoxynucleotidyl transferase (TdT) enzyme and
fluoresceindUTP for 1 h at 37°C in a humidified chamber, followed
by washing with PBS 3 times. Nuclei were counterstained with
Hoechst 0.5 μg/ml for 5 min for total nuclei number counting.
Coverslips were mounted on glass slides using the vectashield
mounting medium (Vector Laboratories). TUNEL-positive nuclei were
counted in 4 randomly selected large fields in each coverslip. One
field consisted of a mosaic of 3×3 stitched images acquired by
fluorescence microscopy using Nikon Eclipse Ti (an automated
inverted wide field epifluorescence microscope) equipped with a ×20
plan dry objective and Nikon DS-Qi1Mc camera). Images were
processed using NIS-element Nikon software. A minimum of 10,000
cells was counted in each condition. TUNEL-positive nuclei (green
fluorescence) and total nuclei (Hoechst-positive, blue
fluorescence) were analyzed with ImageJA freeware version 1.45 b,
using an algorithm (provided by Dr Winnok Devos) that automatically
counts the number of nuclei detected in the two fluorescence
channels. TUNEL-positive but Hoechst-negative cells were excluded.
Apoptotic index was calculated as the percentage of TUNEL-positive
cells (positive cells/total cells ×100%).
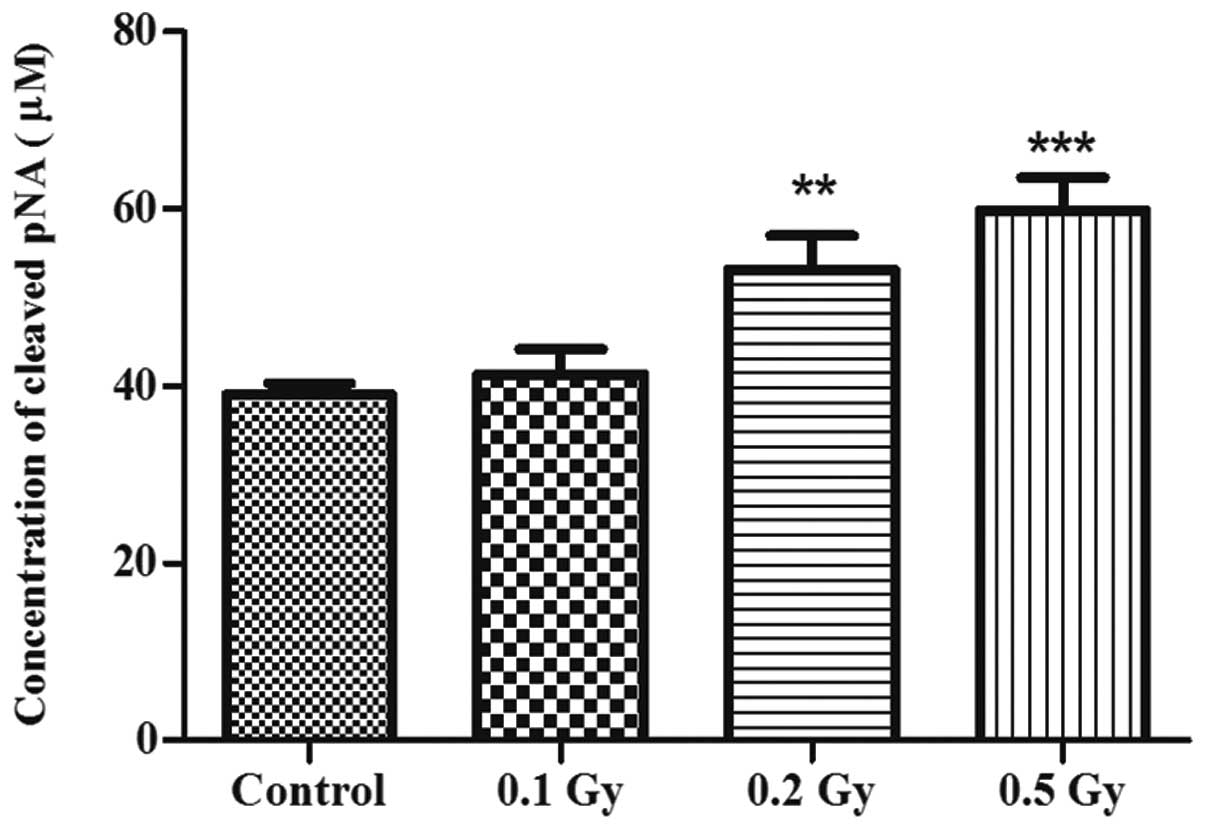
Caspase-3 activity test
Caspase-3 activity was examined in the different
conditions using a colorimetric activity assay kit (Millipore,
Darmstadt, Germany) according to the manufacturer’s instructions.
In brief, cells were directly lysed in the flasks using the lysis
buffer provided in the kit and scraped, and then the cell lysate
was centrifuged to keep only the cytosolic extract. Protein
concentration was assessed using the Quick Start Bradford Protein
Assay kit 3 (Bio-Rad, Hercules, CA, USA) and the same total protein
concentration in all the samples was used for further manipulation.
The samples were then incubated with a mixture provided in the kit,
containing Ac-DEVD-pNA, the substrate of caspase-3. The optical
density (coloration) resulting from the cleavage of the substrate
and the release of pNA, was detected and quantified with a
microtiter plate reader (Multiscan Ascent; Thermo Labsystems) at
405 nm. A standard curve was also generated using a series of
diluted pNA with known concentrations. The standard samples were
processed in the same plate and treated as the other samples. The
concentration in μM of the released pNA was calculated by
projecting the optical densities on the standard curve.
Statistical analysis
Analyses of γ-H2AX, TUNEL and caspase-3 activity
were carried out in 2 independent experiments using 3 biological
replicates.
Data from the cultures exposed only to radiation
were processed using the analysis of variance (one-way ANOVA),
followed by Tukey’s test. Statistical significance was achieved at
P<0.05. Data from cultures exposed to 2 treatments (radiation +
inhibitor of calpain or radiation + blocker of NMDAr) were
processed using the two-way ANOVA method followed by the Bonferroni
multicomparison test. Statistical significance was achieved at
P<0.05.
Results
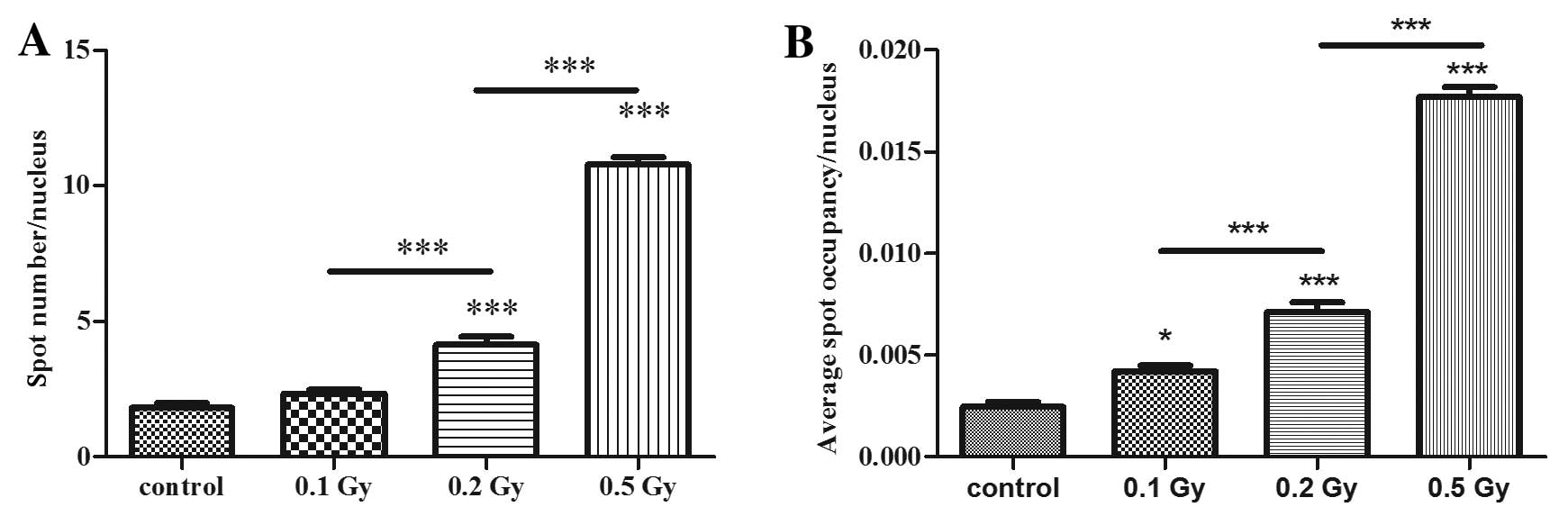
Low and moderate doses of ionising
radiation induce DNA damage in maturing neuronal cells
The ability of radiation to induce DNA damage was
further assessed with immunofluorescence of γ-H2AX foci assay 20
min following irradiation. Cells treated with 0.2 and 0.5 Gy showed
a significantly higher number of γ-H2AX foci than the control cells
in a dose-dependent manner. The doses of 0.2 and 0.5 Gy caused 2-
and 5-fold more DNA double-strand breaks, respectively than those
naturally occurring observed in the control cultures. The dose of
0.1 Gy did not show any significant effect (Fig. 1A).
The spot occupancy that indicates the size of the
foci (percentage of nucleus area occupied by one focus) was also
calculated in order to overcome the issue of foci clustering that
may be counted as only one focus due to spot segmentation issues.
This parameter confirmed the result of the first one with a
dose-dependent induction of DNA double-strand breaks; however, this
parameter showed that 0.1 Gy also induced a significant effect on
DNA damage (Fig. 1B).
Ionising radiation causes a
dose-dependent decrease in cell viability
First we investigated whether low to moderate doses
of ionising radiation induce cell death in neurons. DNA
fragmentation was then assessed as one of the principal hallmarks
of apoptosis using the TUNEL method (Fig. 2). Ionising radiation significantly
increased the rate of TUNEL-positive cells after 24 h by 2-fold
(P<0.05) compared to the control in the cultures irradiated with
0.2 Gy and by 2.6-fold (P<0.001) in the cultures irradiated with
0.5 Gy, which indicates a dose-dependent induction of apoptosis by
ionising radiation. The low dose of 0.1 Gy did not induce any
significant increase in apoptosis.
In order to corroborate these results and to
investigate whether this observed apoptosis was caspase-dependent,
the activity of caspase-3, a central factor in apoptosis
regulation, was examined by colorimetry, indicating the
concentration of the released pNA resulting from the cleavage of
Ac-DEVD-pNA by caspase-3 (Fig.
3). The activity of caspase-3 consistently increased by
1.3-fold following exposure to 0.2 Gy (P<0.01) and by 1.5
following exposure to 0.5 Gy (P<0.001) of ionising radiation in
comparison with the control, but not following exposure to the
lowest dose of 0.1 Gy.
These results clearly indicate the induction of cell
death by moderate but not low doses of ionising radiation.
Nevertheless, the fold change observed in cell death by TUNEL assay
following irradiation was higher than the one observed in caspase-3
activity, suggesting that the radiation-induced cell death assessed
in this study was partly caspase-dependent.
Radiation-induced apoptosis is
mediated by NMDA receptor activation in vivo
Glutamate mediated-excitotoxicity is the most common
cause of neuronal death due to a massive entry of calcium into the
cell leading to the activation of apoptotic or necrotic
pathways.
In an effort to examine whether this mechanism is
involved in radiation-induced cell death, the effects of NMDAr were
examined in vivo by administering a specific NMDAr blocker,
MK-801, to a group of pregnant rats and apoptosis induction in E15
fetal cortices before and after treatment was evidenced using the
DNA ladder technique.
At 3 h and 20 min after exposure, a 0.6 Gy of X-ray
dose elicited a clear apoptotic response (Fig. 4A, lane 5 and Fig. 4B, lane 2). This radiation-induced
apoptosis could be prevented by injection, 20 min after exposure,
of MK-801 (Fig. 4A, lane 6).
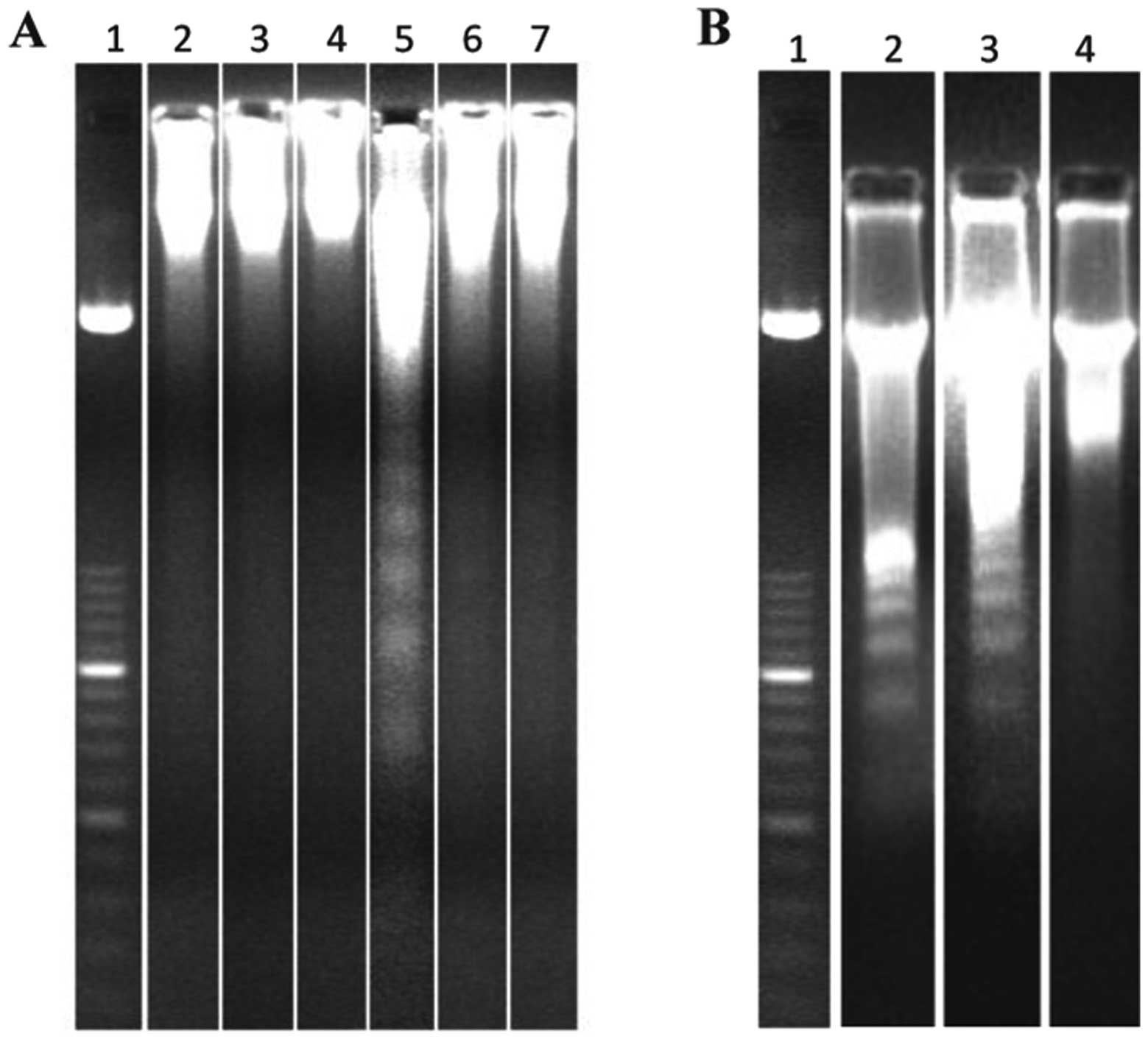 | Figure 4Gel electrophoresis for the detection
of DNA fragmentation (DNA ladder) in cortical cells. (A) Animals
were treated in vivo with 3 mg/kg MK-801 or 32 mg/kg calpain
inhibitor 20 min after irradiation with 0.6 Gy of X-rays (lane 1,
molecular weight marker; lane 2, sham-exposed; lane 3, MK-801; lane
4, calpain inhibitor; lane 5, 0.6 Gy of X-rays; lane 6, 0.6 Gy of
X-rays + MK-801; lane 7, 0.6 Gy of X-rays + calpain inhibitor). (B)
Animals were treated in vivo with 10 mg/kg of nimodipine 20
min after irradiation with 0.6 Gy of X-rays (lane 1, molecular
weight marker; lane 2, 0.6 Gy of X-rays; lane 3, 0.6 Gy of X-rays +
nimodipine; lane 4, sham-exposed). |
The downstream response of NMDAr-mediated
cytotoxicity suggests the activation of calpain, a
calcium-dependent enzyme. NMDAr was also investigated for its
involvement in radiation-induced neuronal death by administering a
calpain inhibitor to another group of pregnant rats irradiated with
0.6 Gy of X-rays. Electrophoresis of genomic DNA from cortical
cells of the fetuses showed no DNA laddering (Fig. 4A, lane 7) indicating that the
calpain inhibitor effectively prevented the fetal cortex from
radiation-induced cell death.
The fetal cortices of non-irradiated rats, whether
treated or not with MK-801 or calpain inhibitor, did not elicit
laddering in DNA electrophoresis (Fig. 4A, lanes 2–4).
In order to dismiss the possible implication of
other types of calcium channels in the radiation-induced
cytotoxicity, the blockade of L-type Ca2+ channels (high
threshold Ca2+ channels) was performed using the
nimodipine blocker. This treatment did not prevent DNA laddering
(Fig. 4B, lane 3) indicating that
these channels are not involved.
The adult brains (from the mother rats) from all the
groups (irradiated, non-irradiated, treated or not with MK-801,
nimodipine or calpain inhibitor) did not show any DNA laddering
(data not shown). This demonstrates the radiation-resistance of the
adult brain. Thus, apoptosis in such adult animals, if any, may
exist only at a low level not detectable at least by the method
used.
Radiation-induced apoptosis is
mediated by NMDA receptor activation in vitro
As regards the high variety of cells that constitute
the brain and knowing that NMDA receptors may also be present in
glial cells (23), it is very
hard to specifically assign the radiation response to one cell type
in vivo. Thus, in vitro study was performed to
investigate the specific neuronal role in this response and to
avoid any interaction with other cell types. From this perspective
the same treatment as for the in vivo study was applied to
7-day primary cultures of cortical neurons. Neurons in these
cultures were able to establish a network in vitro and
therefore we chose to use them as a model of neuronal maturation.
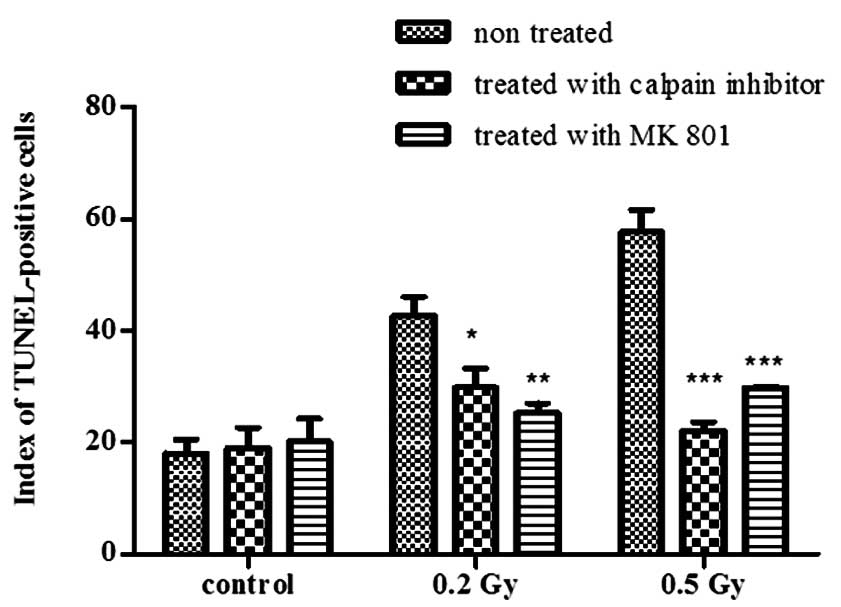
To assess radiation-induced cell death with or without treatment
with MK-801 and calpain inhibitor, TUNEL assay was performed. At 24
h following irradiation, the apoptotic index was significantly
reduced using either of the two treatments (Fig. 5) following exposure to 0.2 Gy
(P<0.05 for calpain inhibitor and P<0.01 for MK-801) and to
0.5 Gy (P<0.001 for both treatments) and was compared to the
control samples (no significant difference), indicating that both
treatments prevented radiation-induced cell death.
The non-irradiated cultures were also subjected to
the same treatments and showed no effect on the apoptotic index
(Fig. 5). These results confirm
the in vivo findings and assign the response specifically to
neurons.
In order to verify the downstream pathway used in
NMDAr-dependent excitotoxicity following irradiation, caspase-3
activity was also assayed. Consistent with the previous observation
using TUNEL assay, caspase-3 activity was also significantly
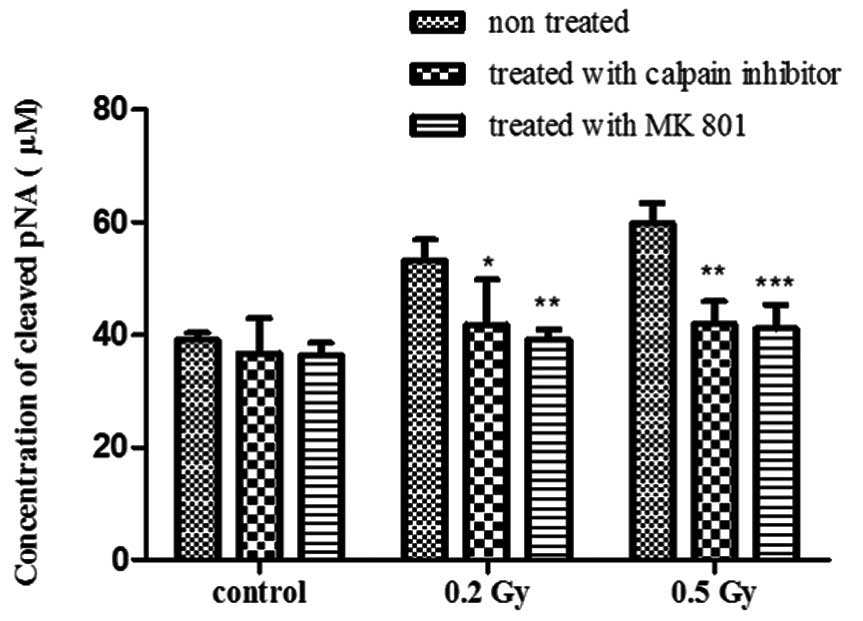
reduced following treatment with MK-801 (P<0.01 with 0.2 Gy and
P<0.001 with 0.5 Gy) or with calpain inhibitor (P<0.05 with
0.2 Gy and P<0.01 with 0.5 Gy), compared to the non-treated
cultures (Fig. 6). The
non-irradiated cultures were also subjected to the same treatments
and showed no change in the concentration of cleaved pNA (Fig. 6). This indicates that the response
to radiation implicates the activation of caspase-3-dependent
pathway.
Discussion
Cell death as a result of exposure to ionising
radiation has been extensively investigated; however, the
complexity of the various mechanisms involved in this response
remains a key topic of interest. DNA is known to be the most
critical radio-sensitive component of cells, directly targeted by
radiation or indirectly via water radiolysis that produces reactive
oxygen species. These products are responsible for the induction of
damage to DNA, including double-strand breaks (24,25), the most damaging lesion that can
lead, in the case of repair failure, to cell death, particularly
following exposure to low doses of ionising radiation (26). Double-strand breaks were revealed
in this study by detecting the phosphorylated histone, H2AX
(γ-H2AX), as one of the most effective markers of response to
radiation-induced double-strand breaks. This response triggers a
signaling cascade by the activation of an important component in
double-strand break signaling, the ATM protein kinase. ATM is
responsible for the phosphorylation of the H2AX histone and the
indirect activation of cell cycle check-points proteins required
for cell cycle arrest and DNA repair (27). ATM also regulates the P53 protein,
known as the guardian of the genome (28) for its key role in stress response
by the induction of cell cycle arrest, DNA repair and apoptosis
regulation (29). Its activation
has been widely associated with cell death induction (30,31), a phenomenon that was observed in
this study following exposure to the moderate doses of 0.2 and 0.5
Gy.
Nevertheless, in the nervous system, multiple
pathways leading to neuronal death exist depending on the nature of
the stressor, and involve key proteins, such as the Bcl-2 family
responsible for the induction of the mitochondrial pathway, leading
to the activation of caspase proteins (32) and calpains, calcium-dependent
enzymes, involved in cell death induction (33). Evidence of a crosstalk between
these pathways makes the process even more complex. A particularity
of the neuronal system is the excitability of the cells. The
over-activation of NMDA receptors by a high concentration of
glutamate, the main excitatory neurotransmitter in the mammalian
CNS, causes the cells to die from excitotoxicity, due to a massive
entry of calcium ions inside the cell (34). NMDArs are glutamate-gated ion
channels, which are selectively activated by the artificial
glutamate analog, NMDAr. These channels when open, are highly
permeable to Ca2+(35).
Attention has been paid to the pathological
significance of calcium accumulation in the CNS following insult to
the brain, including radiation damage. Excitotoxicity is linked to
chronic neurological disorders, including Alzheimer’s and
Parkinson’s disease (17,18), and acute CNS insults, including
hypoxia/ischemia (36).
Over-activation of NMDArs in the brain leads to a sustained influx
of Ca2+ through NMDA and non-NMDA Ca2+
channels. Such disturbances in calcium homeostasis may result in
the activation of several calcium-dependent cysteine proteases,
including calpain (an intracellular cysteine protease proenzyme
activated by autocatalytic cleavage in the presence of high calcium
concentrations) and caspases involved in cytotoxicity downstream
(37,38). Hence, the selective inhibition of
calcium entry by the blockade of ion gated channels to limit
neuronal damage after irradiation appears to be an attractive
method of evaluating the role of calcium homeostasis in the
radiation-induced neurodegenerative processes. We therefore
investigated the possible role of NMDAr and Ca2+ in the
induction of radiation-induced neuronal cell apoptosis.
We showed that a 0.6 Gy of X-ray exposure in
utero, led to a clear apoptotic response in E15 fetal rat
cortices. This apoptotic response was not observed in the different
fetal brains of non-irradiated animals used as the controls
(Sham-exposed and MK-801, nimodipine or calpain inhibitor-treated
animals). The same results were obtained following irradiation of
7-day primary cultures of cortical neurons with 0.2 and 0.5 Gy
using the TUNEL test, which indicated radiation-induced cell death.
Caspase-3 activity, a key factor in apoptosis induction, was also
increased following exposure to the same doses indicating that cell
death by apoptosis is caspase-dependent. However, following
irradiation, the cell death index was higher than caspase-3
activity, suggesting that other apoptotic mechanisms which are
caspase-3-independent may be responsible for this difference in
response to radiation. The number of TUNEL-stained cells and
caspase-3 activity were not significantly increased in the control
cultures (non-irradiated but treated with MK-801 or calpain
inhibitor) and the cultures irradiated with the low dose of
X-rays.
The apoptotic response including DNA fragmentation
(TUNEL) and caspase-3 activation induced in the irradiated cultures
with 0.2 and 0.5 Gy was prevented by treatment with MK-801, which
selectively blocks NMDAr and neuronal Ca2+ influx. This
indicates that radiation-induced apoptosis is mediated through
NMDAr and is affected by massive entry of Ca2+ into the
cells.
Calpain was also a good candidate in
excitotoxicity-mediated neuronal death; thus, neuronal cultures
were treated with calpain inhibitor prior to irradiation. Our
results showed that calpain inhibitor prevented the apoptotic
response in irradiated cultures, thus supporting our hypothesis of
the importance of a calpain-mediated effect in radiation-induced
apoptosis in the fetal brain.
Similar results were also observed after in
vivo treatment of pregnant rats by an injection of MK-801 or
calpain inhibitor 20 min following exposure to 0.6 Gy of X-rays.
Both treatments prevented DNA laddering, indicating that they can
protect the fetal brain from apoptotic response. The in vivo
experiment also allowed us to eliminate the implication of other
Ca2+ channels in this radiation-induced excitotoxicity,
such as the L-type, high threshold and voltage-dependent
Ca2+ channels. The blockade of these channels by
nimodipine did not prevent irradiation-induced DNA laddering;
Therefore, it cannot protect the fetal brain from radiation-induced
apoptosis, indicating that the sensitivity of the fetal brain to
Ca2+ influx through NMDA channels is specific and
indicates a particular radiosensitivity of the cell bearing these
receptors. Thus, apoptosis induced in immature neurons, by
activation of Ca2+-dependent proteolytic enzymes such as
calpain, plays a key role in the radiation-induced damage of the
developing fetal brain.
Our results showing the protective effect of either
MK-801 or calpain inhibitor on radiation-induced apoptosis in the
fetal cortex and in vitro, specifically in established
neuronal network of 7-day cultured cortical neurons, further
suggest the involvement of various pathways leading to neuronal
cell death following exposure to low and moderate doses of ionising
radiation.
Indeed, the activation of caspase-3 that was
observed following irradiation is a classical response to
Ca2+ influx, responsible for apoptosis induction by the
cleavage of several proteins involved in this process. The
inhibition of caspase-3 protects cortical neurons from
NMDAr-induced apoptosis (38).
The activation of caspase-3 has been reported to be a downstream
effector of mitochondrial disruption following the release of
cytochrome c (38,39) and is involved in the execution
phase of apoptosis.
On the other hand, calpain is involved in several
actions following the entry of calcium. Calpain is a proteolytic
enzyme directly activated by calcium entry (40) and is mainly known for its capacity
to cleave cytoskeletal proteins, such as α-spectrin, a phenomenon
that suggests its important role in various neurodegenerative
diseases (41). Attention has
been paid to the novel roles of calpain in the excitotoxicity
phenomenon. It has been found to contribute to the further
disturbance of calcium homeostasis by cleaving different substrates
involved in calcium extruding, such as the
Na+/Ca2+ exchanger and
sarcoplasmic/endoplasmic reticulum calcium ATPase (42,43) or in cytosolic calcium homeostasis,
such as the protein phosphatase calcineurin (44). When activated following the
cleavage by calpain, the latter triggers downstream effectors known
to induce apoptosis, including cytochrome c release from the
mitochondria, leading to caspase-3 activation. This has been
further proven by the overexpression of 48-kDa calcineurin A
(truncated active form), that has been shown to induce an increase
in caspase-3 activity and TUNEL-positive apoptotic cells (44). The same finding has been reported
using a Parkinson’s disease model, where caspase-3 activation was
calpain-dependent (45). A recent
study also established a link between the calcium-dependent
activation of calpain and the induction of apoptosis via
caspases-12, 9 and 3 (46). Our
results showing a decrease in caspase-3 activity and DNA
fragmentation following treatment with calpain inhibitor also
confirm these findings, which permit us to establish a link between
calpain and caspase-3 activity, a link that has not always been
clear since these two enzymes were believed to be involved in two
independent pathways ultimately leading to cell death. Other
studies had even described caspase-3 as being directly activated
following cleavage by calpain (47,48), indicating another contribution of
calpain to the apoptotic induction of caspase-dependent
apoptosis.
Our results also demonstrate a radiation-induced DNA
damage by detecting double-strand breaks. This damage was shown to
proportionally increase with the dose. Such damage is believed to
enhance the expression of P53 protein which plays a key role in
apoptosis induction (49) through
the activation of Bax, a pro-apoptotic protein (50). A P53-dependent activation of Bax
has also been shown to be involved in NMDAr-mediated neuronal death
(51). Of note, it has been found
that calpain activity may be induced following DNA damage and
furthermore leads to the activation of P53 (52,53), indicating another role of calpain
in the induction of caspase-dependent apoptosis via the activation
of P53 response following DNA damage. Furthermore, the fact that
the inhibition of calpain in our study almost completely prevented
cells from radiation induced-apoptosis, including the fraction of
cells that died independently from caspase-3 activation, leads us
to suggest an involvement of calpain in both caspase-dependent and
-independent pathways.
These studies together with our results indicate a
central role of calpain in radiation-induced excitotoxicity, but
also indicate an evident crosstalk of several cell death pathways.
These interactions and their nature (synergistic or competitive),
remain poorly understood; thus investigating these interactions is
of high interest for the elaboration of neuroprotective therapies
for neurodegenerative diseases caused by excitotoxicity and this
study opens new perspectives for radiation protection of the
developing brain.
Our results reveal a new non-conventional
radiation-induced cell death pathway, involving the excitotoxicity
principle mediated by NMDAr activation, not dependent on direct
radiation DNA damage. This pathway involves the activation of
calpain enzyme but also caspase-3 activation, suggesting the
eventual direct or indirect interaction of these two proteins and
their respective classical pathways. P53 activation by calpain
following radiation-induced DNA damage remains a hypothesis that
requires further investigation.
Acknowledgements
The authors wish to acknowledge Dr
Winnok Devos (Ghent University, Ghent, Belgium) for providing the
algorithms used in apoptosis and DNA damage quantifications and Ms.
Leysen Liselotte for her technical assistance. This study was
supported by a doctoral SCK-CEN/VUB grant and partly funded by the
FP7 Euratom Network of Excellence DoReMi (Low Dose Research Towards
Multidisciplinary Integration) (grant no. 249689) and CEREBRAD
(cognitive and cerebrovascular effects induced by low dose ionising
radiation) (GA no. 295552).
References
|
1
|
Li YQ, Jay V and Wong CS: Oligodendrocytes
in the adult rat spinal cord undergo radiation-induced apoptosis.
Cancer Res. 56:5417–5422. 1996.PubMed/NCBI
|
|
2
|
Saito S, Aoki I, Sawada K and Suhara T:
Quantitative assessment of central nervous system disorder induced
by prenatal X-ray exposure using diffusion and manganese-enhanced
MRI. NMR Biomed. 25:75–83. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thabet A, Kalva SP, Liu B, Mueller PR and
Lee SI: Interventional radiology in pregnancy complications:
indications, technique, and methods for minimizing radiation
exposure. Radiographics. 32:255–274. 2012. View Article : Google Scholar
|
|
4
|
Otake M and Schull WJ: Radiation-related
brain damage and growth retardation among the prenatally exposed
atomic bomb survivors. Int J Radiat Biol. 74:159–171. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sudhof TC: Neuroligins and neurexins link
synaptic function to cognitive disease. Nature. 455:903–911. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bassell GJ and Warren ST: Fragile X
syndrome: loss of local mRNA regulation alters synaptic development
and function. Neuron. 60:201–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garcia O, Torres M, Helguera P, Coskun P
and Busciglio J: A role for thrombospondin-1 deficits in
astrocyte-mediated spine and synaptic pathology in Down’s syndrome.
PLoS One. 5:e142002010.PubMed/NCBI
|
|
8
|
Luo J: GSK3beta in ethanol neurotoxicity.
Mol Neurobiol. 40:108–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zecevic N: Synaptogenesis in layer I of
the human cerebral cortex in the first half of gestation. Cereb
Cortex. 8:245–252. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Balslev Y, Saunders NR and Mollgard K:
Synaptogenesis in the neocortical anlage and early developing
neocortex of rat embryos. Acta Anat (Basel). 156:2–10. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oppenheim RW: Cell death during
development of the nervous system. Annu Rev Neurosci. 14:453–501.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wolvetang EJ, Wilson TJ, Sanij E, et al:
ETS2 overexpression in transgenic models and in Down syndrome
predisposes to apoptosis via the p53 pathway. Hum Mol Genet.
12:247–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okamoto M, Suzuki Y, Shirai K, et al:
Effect of radiation on the development of immature hippocampal
neurons in vitro. Radiat Res. 172:718–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bolaris S, Bozas E, Benekou A, Philippidis
H and Stylianopoulou F: In utero radiation-induced apoptosis and
p53 gene expression in the developing rat brain. Int J Radiat Biol.
77:71–81. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Poulaki V, Benekou A, Bozas E, Bolaris S
and Stylianopoulou F: p53 expression and regulation by NMDA
receptors in the developing rat brain. J Neurosci Res. 56:427–440.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Perez-Otano I and Ehlers MD: Learning from
NMDA receptor trafficking: clues to the development and maturation
of glutamatergic synapses. Neurosignals. 13:175–189. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hynd MR, Scott HL and Dodd PR:
Glutamate-mediated excitotoxicity and neurodegeneration in
Alzheimer’s disease. Neurochem Int. 45:583–595. 2004.
|
|
18
|
Blandini F: An update on the potential
role of excitotoxicity in the pathogenesis of Parkinson’s disease.
Funct Neurol. 25:65–71. 2010.PubMed/NCBI
|
|
19
|
Lipton SA and Rosenberg PA: Excitatory
amino acids as a final common pathway for neurologic disorders. N
Engl J Med. 330:613–622. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brorson JR, Marcuccilli CJ and Miller RJ:
Delayed antagonism of calpain reduces excitotoxicity in cultured
neurons. Stroke. 26:1259–1267. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Alaoui F, Pratt J, Trocherie S, Court L
and Stutzmann JM: Acute effects of irradiation on the rat brain:
protection by glutamate blockade. Eur J Pharmacol. 276:55–60. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Vos WH, Van Neste L, Dieriks B, Joss GH
and Van Oostveldt P: High content image cytometry in the context of
subnuclear organization. Cytometry A. 77:64–75. 2010.PubMed/NCBI
|
|
23
|
Verkhratsky A and Kirchhoff F: NMDA
Receptors in glia. Neuroscientist. 13:28–37. 2007. View Article : Google Scholar
|
|
24
|
Wallace SS: Enzymatic processing of
radiation-induced free radical damage in DNA. Radiat Res. 150(Suppl
5): S60–S79. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jeggo P and Lavin MF: Cellular
radiosensitivity: how much better do we understand it? Int J Radiat
Biol. 85:1061–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chistiakov DA, Voronova NV and Chistiakov
PA: Genetic variations in DNA repair genes, radiosensitivity to
cancer and susceptibility to acute tissue reactions in
radiotherapy-treated cancer patients. Acta Oncol. 47:809–824. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurz EU and Lees-Miller SP: DNA
damage-induced activation of ATM and ATM-dependent signaling
pathways. DNA Repair (Amst). 3:889–900. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Harris CC and Hollstein M: Clinical
implications of the p53 tumor-suppressor gene. N Engl J Med.
329:1318–1327. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Verheyde J, de Saint-Georges L, Leyns L
and Benotmane MA: The role of Trp53 in the transcriptional response
to ionizing radiation in the developing brain. DNA Res. 13:65–75.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lai CY, Tsai AC, Chen MC, et al:
Aciculatin induces p53-dependent apoptosis via MDM2 depletion in
human cancer cells in vitro and in vivo. PLoS One. 7:e421922012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Laptenko O and Prives C: Transcriptional
regulation by p53: one protein, many possibilities. Cell Death
Differ. 13:951–961. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koike T, Yang Y, Suzuki K and Zheng X:
Axon and dendrite degeneration: its mechanisms and protective
experimental paradigms. Neurochem Int. 52:751–760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vosler PS, Gao Y, Brennan CS, et al:
Ischemia-induced calpain activation causes eukaryotic (translation)
initiation factor 4G1 (eIF4GI) degradation, protein synthesis
inhibition, and neuronal death. Proc Natl Acad Sci USA.
108:18102–18107. 2011. View Article : Google Scholar
|
|
34
|
Arundine M and Tymianski M: Molecular
mechanisms of calcium-dependent neurodegeneration in
excitotoxicity. Cell Calcium. 34:325–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Waring P: Redox active calcium ion
channels and cell death. Arch Biochem Biophys. 434:33–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wroge CM, Hogins J, Eisenman L and
Mennerick S: Synaptic NMDA receptors mediate hypoxic excitotoxic
death. J Neurosci. 32:6732–6742. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
D’Orsi B, Bonner H, Tuffy LP, et al:
Calpains are downstream effectors of bax-dependent excitotoxic
apoptosis. J Neurosci. 32:1847–1858. 2012.PubMed/NCBI
|
|
38
|
Tenneti L and Lipton SA: Involvement of
activated caspase-3-like proteases in N-methyl-D-aspartate-induced
apoptosis in cerebrocortical neurons. J Neurochem. 74:134–142.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Siman R and Noszek JC: Excitatory amino
acids activate calpain I and induce structural protein breakdown in
vivo. Neuron. 1:279–287. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Czogalla A and Sikorski AF: Spectrin and
calpain: a ‘target’ and a ‘sniper’ in the pathology of neuronal
cells. Cell Mol Life Sci. 62:1913–1924. 2005.
|
|
42
|
Bano D, Young KW, Guerin CJ, et al:
Cleavage of the plasma membrane Na+/Ca2+
exchanger in excitotoxicity. Cell. 120:275–285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
French JP, Quindry JC, Falk DJ, et al:
Ischemia-reperfusion-induced calpain activation and SERCA2a
degradation are attenuated by exercise training and calpain
inhibition. Am J Physiol Heart Circ Physiol. 290:H128–H136. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu HY, Tomizawa K, Oda Y, et al: Critical
role of calpain-mediated cleavage of calcineurin in excitotoxic
neurodegeneration. J Biol Chem. 279:4929–4940. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saito Y, Nishio K, Ogawa Y, et al:
Molecular mechanisms of 6-hydroxydopamine-induced cytotoxicity in
PC12 cells: involvement of hydrogen peroxide-dependent and
-independent action. Free Radic Biol Med. 42:675–685. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Boehmerle W and Endres M: Salinomycin
induces calpain and cytochrome c-mediated neuronal cell death. Cell
Death Dis. 2:e1682011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Blomgren K, Zhu C, Wang X, et al:
Synergistic activation of caspase-3 by m-calpain after neonatal
hypoxia-ischemia: a mechanism of ‘pathological apoptosis’. J Biol
Chem. 276:10191–10198. 2001.PubMed/NCBI
|
|
48
|
McGinnis KM, Gnegy ME, Park YH, Mukerjee N
and Wang KK: Procaspase-3 and poly(ADP)ribose polymerase (PARP) are
calpain substrates. Biochem Biophys Res Commun. 263:94–99. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Komarova EA, Chernov MV, Franks R, et al:
Transgenic mice with p53-responsive lacZ: p53 activity varies
dramatically during normal development and determines radiation and
drug sensitivity in vivo. EMBO J. 16:1391–1400. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Morris EJ, Keramaris E, Rideout HJ, et al:
Cyclin-dependent kinases and P53 pathways are activated
independently and mediate Bax activation in neurons after DNA
damage. J Neurosci. 21:5017–5026. 2001.PubMed/NCBI
|
|
51
|
Djebaili M, Rondouin G, Baille V and
Bockaert J: p53 and Bax implication in NMDA induced-apoptosis in
mouse hippocampus. Neuroreport. 11:2973–2976. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sedarous M, Keramaris E, O’Hare M, et al:
Calpains mediate p53 activation and neuronal death evoked by DNA
damage. J Biol Chem. 278:26031–26038. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hama Y, Katsuki H, Izumi Y, Kume T and
Akaike A: Excitotoxicity-associated p53 expression in adult rat
retina is mediated by calpain activity but not by Cl−
influx. J Pharmacol Sci. 110:493–496. 2009. View Article : Google Scholar : PubMed/NCBI
|