Introduction
Prion diseases, also known as transmissible
spongiform encephalopathies (TSEs), are a group of fatal
neurodegenerative disorders in humans and animals, including
Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform
encephalopathies (BSEs) in cattle, and scrapie in sheep and goats.
The conversion of prion protein (PrP), coded by the PRNP
gene, from its cellular isoform PrPC to its pathogenic
isoform PrPSc through a post-translational process is
considered the etiology of these diseases. As a result of the
conversion, the portion of β-sheets within PrP is increased (from 3
to 45%) whereas that of α-helices decreases (from 42 to 30%),
therefore causing PrP to become detergent insoluble and resistant
to denaturant (isoform PrPSc) (1). The conversion also causes a series
of histopathological changes, including the depositions of
PrPSc, spongiform degenerations, neuronal loss and
astrogliosis.
According to the pathomechanisms, CJD can be
classified into sporadic CJD (sCJD), familial or genetic CJD (fCJD
or gCJD) and iatrogenic CJD (iCJD). fCJD accounts for approximately
10–15% of all CJD cases, characterized by the genetic changes of
PrP (2). To date, 56 different
mutations, including residue substitutions, insertions and
deletions, have been reported (3). For instance, proline to leucine
mutation at the 102nd position (P102L) causes
Gerstmann-Straussler-Scheinker syndrome (GSS) characterized by the
impairment of cerebellum (4).
Nevertheless, D178N leads to fatal familial insomnia (FFI)
(4), mainly harming the thalamus
region. The varied clinical manifestations indicate that different
mutants may trigger different regulatory pathways.
An efficient approach to uncovering the regulated
genes and the signaling pathways caused by PrPSc is
using global transcriptional profiling on prion-infected samples.
Studies on various prion strains in mice by Hwang et al
(5) described a network of
differentially expressed genes (DEGs) on functional pathways and
discovered that the gliosis fibril acidic protein gene (Gfap) and a
set of complement activation associated genes are highly expressed
(5). Xiang et al (6) performed their studies using human
sCJD brains and described the upregulation of immune and
stress-response factors and elements involved in cell death and
cell cycle, and the downregulation of genes encoding synaptic
proteins (6). However, there is a
lack of information on the global transcriptional profiles of gCJD
patients, since gCJD and sCJD may utilize different regulatory
mechanisms.
In our previous study, we reported a Chinese gCJD
patient with a G114V mutation in PrP (7), showing an sCJD-like
neuropathological abnormality with large amounts of
PrPSc deposit, spongiform degeneration, astrogliosis and
neuron loss in the cortex regions (8). To further investigate the molecular
mechanisms and to compare those with sCJD, the full transcriptome
pattern of the parietal cortex lobe of this G114V gCJD patient was
profiled with microarray analysis (Affymetrix Human Genome U133+
2.0 Chip) with a commercial normal human parietal cortex RNA pool
as control. This investigation revealed numerous DEGs and pathways
and our results provide information regarding gCJD that is useful
for the development of novel diagnostic and therapeutic
approaches.
Materials and methods
Brain sample of the G114V fCJD
patient
Brain tissue of the parietal cortex obtained from a
patient definitely diagnosed with G114V gCJD was enrolled in this
study. The patient was a 47-year-old (at onset) Han-Chinese woman,
whose clinical and genetic characteristics have previously been
described (7). Neuropathological
assays of 10 different brain regions revealed typical sCJD-like
abnormality and PrPSc deposits (8). A commercial normal human parietal
cortex total RNA (cat. #636571; Clontech) pooled from four
males/females aged 35–89 years was utilized as a control. Usage of
the stored human brain samples in this study was approved by the
Ethics Committee of the National Institute for Viral Disease
Prevention and Control, China CDC. We obtained written informed
consent from all participants in our study.
Microarray analysis
Total RNA of the parietal cortex of the patient with
G114V gCJD was extracted with an RNeasy Mini kit (cat. #74104;
Qiagen), according to the manufacturer’s instructions. The quality
and quantity of extracted RNAs were verified by 1.2% formaldehyde
agarose electrophoresis and ultraviolet spectrophotometry
(NanoDrop, ND-1000). The processes of labeling, hybridization and
scanning were performed at a platform of CapitalBio Corporation.
Briefly, 200 ng of each total RNA preparation was taken for
synthesis and amplification of first strand cDNAs, double-stranded
cDNAs and biotin-labeled antisense RNAs, using a MessageAmp™
Premier RNA Amplification kit (cat. #AM1792; Ambion) on a PCR
apparatus (MJ, PTC-225). After measuring the concentrations of the
labeled RNAs by ultraviolet spectrophotometry, 15 μg of each
preparation was fragmented and verified using 1.2% formaldehyde
denatured agarose electrophoresis.
The biotinylated cRNAs were hybridized to a
commercial gene chip, GeneChip® Human Genome U133+ 2.0
(Affymetrix Inc., Santa Clara, CA, USA) containing 47,000
transcripts, at 45°C for 16 h with constant rotation at a speed of
60 rpm. After washing and staining automatically on an Affymetrix
fluidics station 450 with a GeneChip Hybridization, Wash and Stain
kit (cat. #900720; Affymetrix), the chips were then scanned on
Affymetrix scanners 3000 7G.
After scanning the gene chips, the CEL images were
processed using the Affymetrix GCOS 1.4 software. The generated
documents were analyzed according to the ‘Affymetrix Statistical
Algorithms Description Document’. Briefly, the raw data were
subjected to processes including masking unusable data, background
subtraction, probe values calculation, scaling, single chip
analysis, and calculation of P-value. A probe-set was considered as
expressed if the corresponding detection P-value was <0.04.
Genes were considered to be differentially expressed if their ratio
(patient/control) was higher or lower than 2-fold.
The gene functions were formatted for both gene
ontologies (GO) and molecular function (9). The GO and molecular functions of the
genes with 6-fold differential expression compared to controls were
further analyzed by CapitalBio® Molecule Annotation
System V3.0 (MAS3.0) (http://bioinfo.capitalbio.com/mas3/). The P-value was
calculated according to a probability formula of hypergeometric
distribution, reflecting the importance of the selected pathway or
GO. The smaller the P-value, the higher its significance. The
pathways were ranked in the order of P-value and the 10 most
important ones were chosen for further analyses. Meanwhile, a
Q-value corresponding to the P-value was calculated to evaluate the
false discovery rate (FDR) of significant pathway and GO through
screening by using a single P-value as cut-off. The smaller the
Q-value, the lower the FDR.
All data are MIAME compliant and the raw data have
been deposited in the GEO database (10) with the accession number of that,
platform of GPL570, samples of GSM759883 and GSM759884, as well as
series of GSE30643.
Quantitative RT-PCR (qRT-PCR)
Prior to qRT-PCR, the RNA extracts were treated with
a commercial RQ1 RNase-Free DNase (cat. #M6101; Promega) for 1 h at
37°C according to the manufacturer’s instructions. For cDNA
synthesis, 2 μg of treated RNAs were mixed with the reagents
in Reverse Transcription System (cat. #A3500; Promega). The
real-time PCR was carried out on an ABI Prism 7900 sequencing
detector, at the conditions of denaturing at 95°C for 15 sec,
annealing at 50°C for 2 min and extension at 62°C for 1 min, 40
cycles in total. β-actin gene was used as an internal control to
normalize the expression levels of target mRNAs. The primers for
each gene are shown in Table
I.
 | Table IPrimers used for the real-time RT-PCR
target genes. |
Table I
Primers used for the real-time RT-PCR
target genes.
| Gene ID | Sense primer | Antisense primer | Product length
(bp) |
|---|
| Decreased genes | PHLDA2 |
ACAGCCTCTTCCAGCTATGG |
GGTGGTGACGATGGTGAA | 173 |
| HBB |
ACGTGGATGAAGTTGGTGGT |
CTCACTCAGTGTGGCAAAGGT | 215 |
| NR4A2 |
ACCACTCTTCGGGAGAATACAG |
ACAGGGGCATTTGGTACAAG | 180 |
| CBLN4 |
CTGGGCACAGAACGACAC |
AAGGCGACCTTGGAGTTG | 144 |
| Increased
genes | ZNF396 |
TGGAAGAGGAAGAGCAGACC |
CCTCAGCCAGAGATGACAAAG | 167 |
| ZNF292 |
GAGCAGGAGAGGTTGAGTTG |
AGATAAGGTCGGGCTTTAACA | 257 |
| EIF5B |
GACAGCACCAAGGATGACATT |
GTTTTCTGTTGGCTTCACTGC | 228 |
| UBE3A |
GAGCAGCTGCAAAGCATCTA |
CTTTCTTGGAGGGATGAGGAT | 195 |
Results
Global transcriptional profiling of the
G114V gCJD patient
Following our previous studies on the G114V gCJD
patient (7,8), we further investigated all the
transcriptional patterns of the brain sample and compared them to
those of normal brain RNA pool (control). To better understand the
expression level of screened genes, we ranked them with the
relative difference ratio using the following strategy: after
single chip normalization, each probe set was marked as present (P)
or absent (A) according to the comparison to background noise.
Probe sets marked with A in both the experimental sample and
control were discarded. Thus, we found the differentially expressed
genes based upon two criteria, i) present in sample but absent in
another sample, or ii) with the gene expression level altered over
2-fold. The genes that met both conditions were considered DEGs.
After purging the redundant probe sets reflecting same genes, a
total of 8,774 genes were determined to be differentially expressed
in G114V gCJD brains. Among them, nearly one-third (2,769) was
upregulated and two-thirds (6,005) were downregulated. Due to the
lack of sufficient sample/data from other gCJD patients, we were
unable to make parallel experiments/comparisons to minimize the
gene set of gCJD specific genes. Furthermore, we did not use data
from sCJD (6) or prion-infected
mice (5), as the different
molecular background may have induced unpredictable bias.
According to the annotations of Affy-Chip, 8,494 of
the 8,774 genes were either assigned biological functions or were
similar to genes with known functions, and 280 genes remain
annotated as encoding hypothetical proteins. GO assignment
determined these DEGs to be involved in 1,552 biological processes,
with 819 molecular functions, and to be located in 368 cell
components. In view of the significantly altered biological
processes, 87 contained >10 DEGs. The predominantly altered
processes covered the major basic cellular functions, including
regulation of transcription, signal transduction, development and
transport, oxidation reduction and apoptosis (Table II). Regarding the molecular
functions, most DEGs were related to molecular (protein,
nucleotide, ion) binding/interactions. The cellular component
assignment showed that most functional genes were located in the
membrane system (plasma membrane, mitochondria, endoplasmic
reticulum). Since the sample was collected from the gCJD patient
postmortem, the transcriptional profile represents the terminal
stage of the infected brain.
| Table IISignificantly altered biological
processes of gene ontology. |
Table II
Significantly altered biological
processes of gene ontology.
| GO term | Count | P-value | Q-value | 5 most up and
downregulated genes, respectivelya |
|---|
| GO:0006355
regulation of transcription, DNA-dependent | 114 | 3.62E-72 | 2.60E-70 | ATRX, CHD9, ZNF292,
ZNF396, JMJD1C; ZNF200, MEF2C, TBC1D9, ZMYM2, NR4A2 |
| GO:0007165 signal
transduction | 110 | 1.20E-49 | 6.34E-48 | SYCP2, CPLX2,
SLC5A3, GABRG1, BPTF; SLC8A1, GRIN2A, ATP6V1B2, GRIN2A,
GRIA4 |
| GO:0006350
transcription | 106 | 3.72E-60 | 2.18E-58 | PHF3, CHD9, ZNF292,
ZNF396, JMJD1C; ZNF200, BRWD1, MEF2C, ZMYM2, NR4A2 |
| GO:0007275
development | 71 | 2.16E-31 | 6.11E-30 | CSPP1, COL27A1,
APC, TRAPPC2L, CD47; PCDHA1, PCDH8, CD164, UCHL1, NELL1 |
| GO:0006810
transport | 71 | 7.25E-29 | 1.94E-27 | RNF130, DTNA,
IL1RL1, PKN2, IL6ST; RTN1, UCHL1, PENK, OR2L13, NR4A2 |
| GO:0006811 ion
transport | 68 | 3.08E-62 | 2.03E-60 | TIMM8A, VEGFA,
NAV1, MPPED2, FOSL2; ITM2B, FGF13, UCHL1, NELL1, GAP43 |
| GO:0007155 cell
adhesion | 67 | 3.29E-59 | 1.86E-57 | ASPH, JMJD1C,
SYCP2, CPLX2, PLOD2; PCYOX1, VAT1L, SQLE, KCNAB1,
SRD5A1 |
| GO:0007399 nervous
system development | 57 | 5.56E-45 | 2.66E-43 | ASPH, JMJD1C,
SYCP2, CPLX2, PLOD2; PCYOX1, VAT1L, SQLE, KCNAB1,
SRD5A1 |
| GO:0015031 protein
transport | 53 | 5.92E-42 | 2.68E-40 | TIMM8A, CEP290,
RASEF, RASEF, HSP90B1; ARF4, NSF, CADPS, CADPS, CADPS |
| GO:0008152
metabolism | 51 | 0.06 | 0.04 | SFRS8, THOC2, PPIG,
PRPF40A, ZRANB2; MAGOH, LSM8, THOC4, SRPK2, RBM9 |
| GO:0008150
biological_process | 50 | 1.00 | 0.58 | GABRG1, SYN2,
CTNNB1, MAPK12, MAPK11; TAC1, SYT1, SYN2, SNAP25,
SLC1A6 |
Identification of the most DEGs
According to the microarray results, the most
upregulated genes included UBE3A and RBBP6, which are involved in
the ubiquitin protein degradation system, suggesting that the
ubiquitin-dependent catabolic processes are essential in the gCJD
patient. ASPH was the second most highly expressed gene that plays
an important role in calcium homeostasis. It is well known that
destruction of calcium homeostasis is common in neurodegenerative
diseases (11). On the contrary,
the most downregulated genes included genes involved in iron ion
binding (HBB), transcriptional regulation (NR4A2), signal
transduction (OR2L13) and cell skeleton formation (NEFL) (Table III).
| Table IIIMost differentially expressed genes
of the microarray data. |
Table III
Most differentially expressed genes
of the microarray data.
| | | Gene Ontology
|
|---|
| Gene | Description | Ratio | Biological
process | Cellular
component | Molecular
function |
|---|
| UBE3A | Ubiquitin protein
ligase E3A | 75.71 | Proteinmodification
process; proteolysis; ubiquitin-dependent protein catabolic
process; brain development; modification-dependent protein
catabolic process; interspecies interaction between organisms | Intracellular;
nucleus; cytosol; protein complex | Ubiquitin-protein
ligase activity; protein binding; acid-amino acid ligase
activity |
| ASPH | Aspartate
β-hydroxylase | 72.30 | Muscle contraction;
peptidyl-amino acid modification; oxidation reduction | Integral to
membrane; integral to endoplasmic reticulum membrane | Peptide-aspartate
β-dioxygenase activity; structural molecule activity; iron ion
binding; calcium ion binding; structural constituent of muscle;
electron carrier activity; oxidoreductase activity |
| CCDC88A | Coiled-coil domain
containing 88A | 47.85 | Regulation of
protein amino acid phosphorylation; regulation of DNA replication;
membrane organization; cell migration; lamellipodium assembly;
activation of protein kinase B activity; regulation of actin
cytoskeleton organization; regulation of cell proliferation | Cytoplasm;
endoplasmic reticulum; Golgi apparatus; cytosol; plasma membrane;
membrane; lamellipodium; cytoplasmic vesicle; cell projection | Actin binding;
microtubule binding; phosphoinositide binding; protein
homodimerization activity; protein kinase B binding |
| RBBP6 | Retinoblastoma
binding protein 6 | 36.45 | Protein
ubiquitination | Ubiquitin ligase
complex; nucleus | Nucleic acid
binding; ubiquitin-protein ligase activity; zinc ion binding |
| LOC643187 | Hypothetical
LOC643187 | 30.62 | N/A | N/A | N/A |
| ANKRD12 | Ankyrin repeat
domain 12 | 30.27 | N/A | Nucleus;
ribosome | N/A |
| LOC554203 | Alanyl-tRNA
synthetase domain containing 1 pseudogene | 29.83 | N/A | N/A | N/A |
| ANKRD36 | Ankyrin repeat
domain 36 | 25.71 | N/A | N/A | N/A |
| PHF3 | PHD finger protein
3 | 25.30 | Transcription;
multicellular organismal development | Nucleus | Protein binding;
zinc ion binding |
| EIF5B | Eukaryotic
translation initiation factor 5B | 24.24 | Translation;
regulation of translational initiation | Cytoplasm | Nucleotide binding;
translation initiation factor activity; GTPase activity; protein
binding; GTP binding |
| HBB | Hemoglobin, β | 0.004 | Regulation of blood
pressure; oxygen transport; positive regulation of nitric oxide
biosynthetic process; regulation of blood vessel size | Hemoglobin
complex | Oxygen transporter
activity; iron ion binding; oxygen binding; hemoglobin binding |
| TSPYL1 | TSPY-like 1 | 0.006 | Nucleosome
assembly | Intracellular;
nucleus | N/A |
| GAP43 | Growth associated
protein 43 | 0.009 | Activation of
protein kinase C activity by GPCR protein signaling pathway;
nervous system development; response to wounding; glial cell
differentiation; axon choice point recognition; regulation of
growth; tissue regeneration; cell fate commitment | Plasma membrane;
membrane; cell junction; axon; cell projection; synapse | Calmodulin
binding |
| NR4A2 | Nuclear receptor,
subfamily 4, group A, member 2 | 0.009 | DNA-dependent
regulation of transcription; signal transduction; cellular response
to extra- cellular stimulus; respose to protein stimulus | Nucleus | Transcription
factor activity; steroid hormone receptor activity; zinc ion
binding; sequence-specific DNA binding |
| PHLDA2 | Pleckstrin
homology-like domain, family A, member 2 | 0.009 | Apoptosis; organ
morphogenesis | Cytoplasm;
membrane | N/A |
| OR2L13 | Olfactory receptor,
family 2, subfamily L, member 13 | 0.01 | Signal
transduction; GPCR protein signaling pathway; sensory perception of
smell; response to stimulus | Plasma membrane;
membrane; integral to membrane | Signal transducer
activity; olfactory receptor activity |
| NEFL | Neurofilament,
light polypeptide | 0.01 | Anterograde axon
cargo transport; retrograde axon cargo transport; axon regeneration
in the peripheral nervous system; axon transport of mitochondrion;
regulation of axon diameter; neurofilament bundle assembly;
locomotion; negative regulation of neuron apoptosis; intermediate
filament bundle assembly; neuron projection morphogenesis; positive
regulation of axonogenesis; neuromuscular process controlling
balance; neurofilament cytoskeleton organization | Intermediate
filament; neurofilament; axon; TSC1-TSC2 complex | Structural molecule
activity; structural constituent of cytoskeleton; protein
C-terminus binding; identical protein binding |
| C11orf87 | Chromosome 11 open
reading frame 87 | 0.018 | N/A | Integral to
membrane | N/A |
| CBLN4 | Cerebellin 4
precursor | 0.019 | N/A | Extracellular
region; cell junction; synapse | N/A |
| CD200 | CD200 molecule | 0.019 | N/A | Integral to plasma
membrane; integral to membrane | Protein
binding |
To evaluate the microarray results, we performed
real-time PCR, targeting some specific genes from the brain of the
G114V gCJD patient and the control RNA pool from the normal brain.
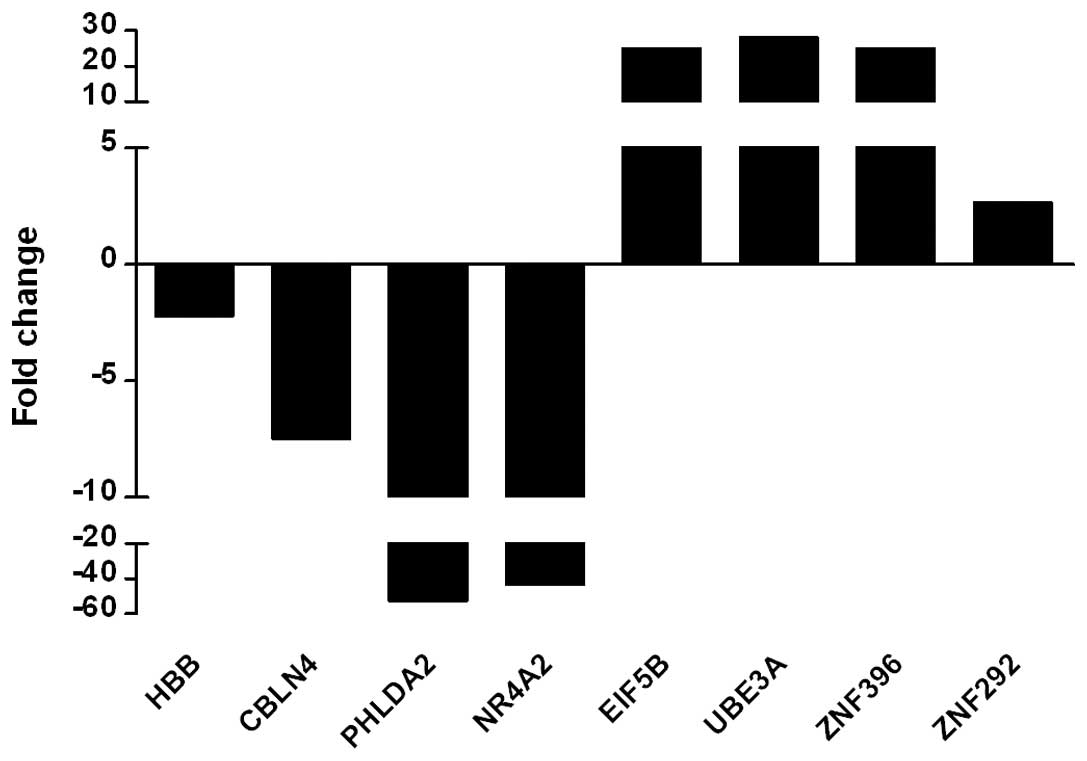
Real-time PCR showed that the transcriptional levels of the
downregulated genes HBB, CBLN4, PHLDA2 and NR4A2 from the
microarray were markedly lower than those of the normal control,
particularly PHLDA2 and NR4A2, which were >40-fold decreased and
consistent with the micro-array result. The upregulated genes
EIF5B, UBE3A, ZNF396 and ZNF292 in the microarray were also
significantly higher than the control in real-time PCR assay,
particularly EIF5B, UBE3A and ZNF396 showing a >30-fold increase
(Fig. 1). These findings indicate
that the results of the microarray are reliable.
Involvement of significant pathways in
G114V gCJD
To examine the changes of cell signaling pathways in
the brain cortex of the G114V gCJD patient, the genes expressed at
least 6-fold higher or lower than that of normal control were
subsequently analyzed with the CapitalBio® Molecule
Annotation System V3.0 (MAS 3.0) using KEGG pathways (12). In total, 169 different pathways
with significant difference in expression (P<0.05) were
identified. Briefly, 82 pathways were involved in metabolism, 28 in
human diseases, 19 in organismal systems, 16 in environmental
information processing, 14 in genetic information processing and 10
in cellular processes, according to the KEGG classification of
functional pathways.
The 10 most altered pathways ranked in the order of
P-value are summarized in Table
IV. Most of the genes in these path-ways were downregulated
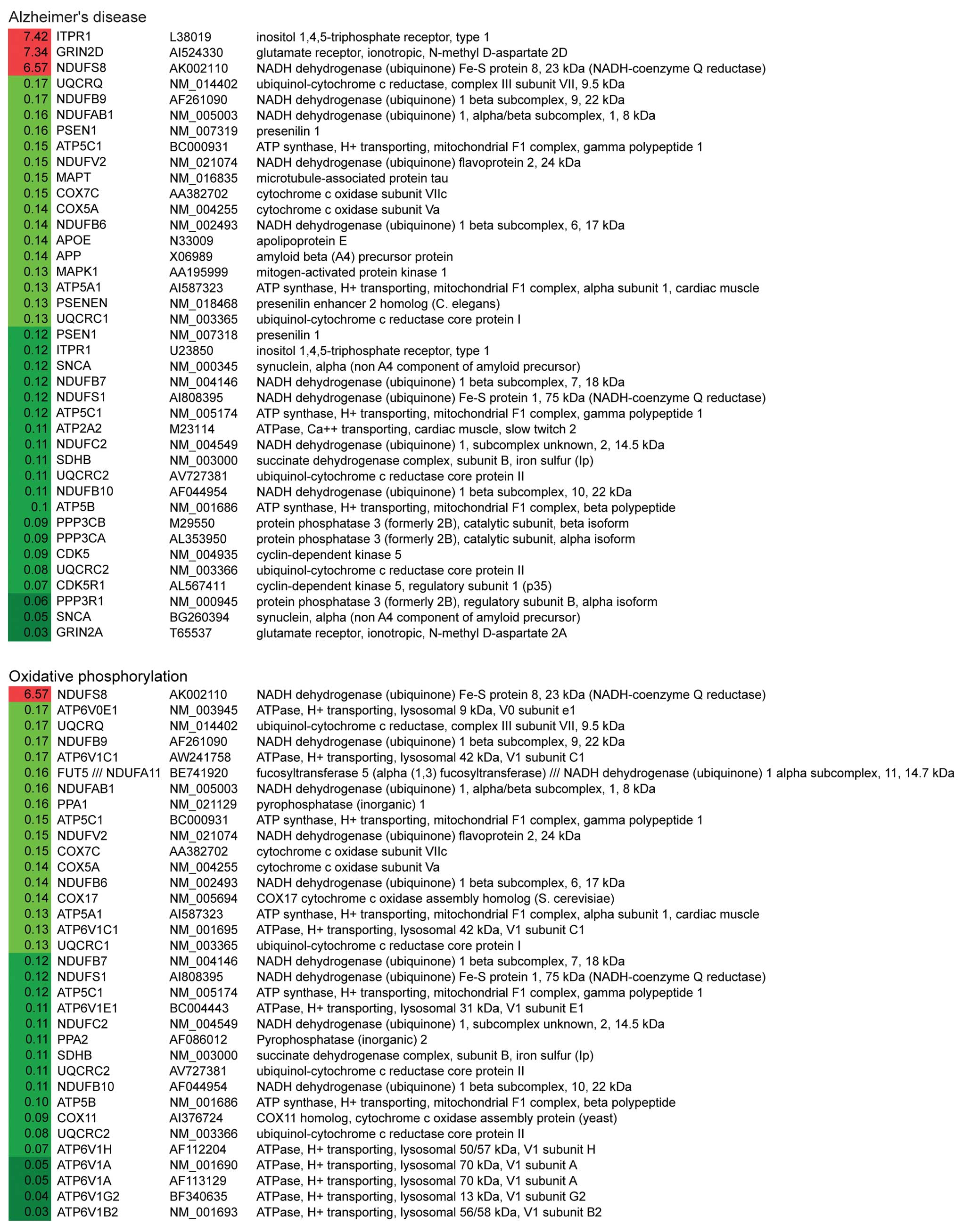
(Fig. 2). Notably, the first and
the third most altered pathways were Alzheimer’s disease (AD) and
Parkinson’s disease (PD). Within these two pathways, 31/34 genes
and 27/28 genes were significantly downregulated in AD and PD
pathways, respectively (Table
IV). The down-regulated genes related to these two pathways are
involved in mitochondrial dysfunction, ER stress and apoptosis.
These data suggest that G114V gCJD may share similar mechanisms to
AD and PD.
| Table IVThe 10 most significantly regulated
pathways deduced from the microarray data. |
Table IV
The 10 most significantly regulated
pathways deduced from the microarray data.
| Pathway | Count | P-value | Q-value |
|---|
| Alzheimer’s
disease | 34 | 1.26E-17 | 1.79E-16 |
| Oxidative
phosphorylation | 30 | 3.42E-17 | 4.60E-16 |
| Parkinson’s
disease | 28 | 1.40E-15 | 1.47E-14 |
| Regulation of actin
cytoskeleton | 32 | 2.97E-13 | 1.84E-12 |
| Pathogenic
Escherichia coli infection | 17 | 4.20E-13 | 2.45E-12 |
| MAPK signaling
pathway | 35 | 1.67E-12 | 8.40E-12 |
| Axon guidance | 24 | 1.79E-12 | 8.83E-12 |
| Gap junction | 20 | 6.58E-12 | 3.01E-11 |
| Proteasome | 15 | 8.43E-12 | 3.78E-11 |
| Purine
metabolism | 24 | 5.64E-11 | 2.20E-10 |
Genes related to oxidative phosphorylation and
purine metabolism were also significantly altered. Out of 30
differentially altered genes of involved in oxidative
phosphorylation, 29 were downregulated, including NADH
dehydrogenase, succinate dehydrogenase, cytochrome c
reductase, cytochrome c oxidase and F-type ATPase (Fig. 2). Regarding the purine metabolism
pathway, all 24 altered genes were downregulated, including
adenylosuccinate lyase, RNA polymerase II and some
phosphodiesterases (Fig. 2).
These observations indicate that there is a severe failure in the
mitochondria and a severe dysfunction of cell metabolism at the
terminal stage.
Two pathways related to the cytoskeleton were also
markedly downregulated; 29/32 altered genes involved in the
regulation of actin cytoskeleton were significantly downregulated.
All 17 affected genes related to pathogenic Escherichia coli
infection were downregulated; among them some gene products were
cell structure proteins, i.e. CDC42 and tubulin. Additionally,
other possible severely disrupted pathways included those related
to gap junctions (17/20 DEGs downregulated), which are involved in
direct communication between the cytosolic compartments of adjacent
cells and the pathway of axon guidance (24/24 DEGs downregulated),
which helps axons extend to their correct targets. The
downregulation of these two pathways suggests that there is damage
to cell communication and neuronal development in the G114V gCJD
brain.
Among the 10 most affected pathways, the
mitogen-activated protein kinase (MAPK) signaling pathway was the
only one related to environmental signaling processing. The
processes seemed to be markedly repressed as well, with 33/35
altered genes being downregulated, including kinases (MAPK1, MAPK1,
MAP2K1, MAP2K4, PRKCB, PAK1 and STK4), phosphatases (DUSP4, DUSP5,
DUSP6, PPP3CA and PPP3CB) and some regulatory factors (FGF13,
FGF14, MEF2C and RASGRF2) (Fig.
2). These genes were distributed in the three sub-pathways,
classical MAP kinase pathway, JNK and p38 MAP kinase pathway, and
extracellular-regulated kinase 5 (ERK5) pathway. This indicates
that the local information processing in the brain of the gCJD
patient was severely impaired.
The transcriptional pattern of important
prion disease associated genes
Our previous study demonstrated large amounts of
PrPSc deposits and severe gliosis in the cortex regions
of this G114V gCJD patient, while the transcription levels of
PRNP did not vary as much as in the PrPSc deposit
among 10 different regions (8).
Furthermore, microarray data showed no difference in PRNP
transcription between the patient’s brain and normal control, even
slightly downregulated in the patient’s brain with several
PRNP probes, possibly indicating deposits of
PrPSc in brains do not lead to enhancing the PRNP
transcription. The transcriptional level of the GFAP gene in the
patient was ∼2-fold increased relative to that of the control,
highlighting an active gliosis. Nevertheless, a spectrum of
neuronal biomarkers was downregulated in the patient’s brain, such
as NSE (7.25-fold), tubulin-β III (3.88-fold), MAP2 (8.13-fold),
NF-M (38.46-fold), NF-H (12.35-fold) and NF subunit NF-L
(27.78-fold), demonstrating severe neuron loss. These data are
consistent with the pathological characteristics of G114V gCJD.
Discussion
In the present study, we analyzed the global
expression patterns in the parietal cortex of a G114V gCJD patient
with a commercial gene chip containing 47,000 transcripts. This is
the highest-capacity approach to gene expression analysis used in
human prion disease thus far. After purging the redundant
transcripts, we identified 2,769 upregulated and 6,005
down-regulated genes. Further qRT-PCRs for several differentially
expressed genes confirmed the results of the microarray. Notably,
more downregulated genes in the brain of G114V gCJD are consistent
with the results of a previous study on sporadic CJD with a
relatively lower throughput microarray (18,000 transcripts), in
which 275 genes out of 287 differentially expressed genes were
downregulated (6).
In line with the observations of the pathological
abnormalities in the G114V gCJD patient (8,13),
the transcriptional level of the GFAP gene associated with gliosis
is increased and a series of genes associated with neurons are
decreased. Although the brain tissues are severely damaged
pathologically, the expression levels of prion protein gene
PRNP do not differ distinctly compared with those of normal
control, which is in accordance not only with the data of
PRNP transcription in this patient with qRT-PCR, but also
with the previous microarray findings in the sCJD patients
(6), mice infected with scrapie
or CJD agents (14) and cattle
infected with the BSE agent (15). Maintenance of active transcription
of the PRNP gene in CNS tissues at the terminal stage of
human and animal prion diseases may indicate a special environment
that facilitates the replication of prion agents locally by
supplying enough PrPC as the substrates for
PrPSc replication.
The most differentially expressed genes in G114V
gCJD seem to be involved in multiple cell processes, such as
regulation of transcription, ion transport, cell adhesion, signal
transduction, nervous system development, oxidation reduction,
protein transport, RNA splicing and synaptic transmission. In the
brains of naturally-occurring or experimental animal and human
TSEs, as well as in some prion infected cell lines (5,6,15–17), abnormal alterations in ion
transportation, transcription, cell adhesion, signal transduction
and synaptic transmission have been repeatedly observed. Numerous
differentially expressed genes involved in different cell processes
or networks in brain tissues of this G114V gCJD patient reflect an
extensive brain dysfunction at the final period of the disease.
Based on the classification of the KEGG database,
169 different pathways were significantly altered in the brain of
the patient with G114V gCJD. Most of the differentially expressed
genes in the 10 most significantly altered pathways were
down-regulated, revealing a deeply suppressed expression status of
the relevant functions. Two metabolic pathways, oxidative
phosphorylation and purine metabolism, were markedly repressed in
our study. In the oxidative phosphorylation pathway, the expression
of several key elements, such as NADH dehydrogenase, succinate
dehydrogenase, cytochrome c reductase, cytochrome c
oxidase and F-type ATPase were decreased. This result is in
accordance with previous studies by both microarray (5) and proteomics (18), reflecting a complete failure of
mitochondria. Purine metabolism includes the biological synthesis,
degradation and salvation of purines, an essential component of
nucleotides (19). Abnormality in
this pathway has not previously been observed in the prion-infected
cells, or human and animal TSEs. Besides, the pathway of cellular
proteasome in the brain of this gCJD case is significantly
involved, in which various proteasome subunits are downregulated.
The dysfunction of the cellular proteasome system is often noticed
in several neurodegenerative disorders (18), including prion disease (20). The disability of protein
degradation, especially in clearance of misfolded protein, may
contribute to the accumulation of PrPSc in brain
tissues.
It has been repeatedly observed that the
cytoskeleton and microtubule are severely destroyed in the brain of
prion disease (21–23). In our study, the pathway of
regulation of actin cytoskeleton and the pathway involved in the
expressions of cell structure proteins, such as CDC42 and tubulin,
were clearly suppressed. Reduction of expression levels of those
genes results in rearrangement of the cytoskeleton, disruption of
barrier function and an increase in monolayer permeability.
The MAPK cascade is a highly conserved pathway
involved in various cellular functions, including cell
proliferation, differentiation and migration. Our microarray
experiment illustrated the increased expression of p38 MAP kinase
and nuclear factor-κB (NF-κB) in the brain of gCJD. The increase of
those two factors has been reported in cells treated with the
peptide PrP106–126. PrP106–126 has been demonstrated to activate
p38 MAP kinase in human microglia accompanied by upregulation of
NF-κB (24), and to induce a p38
MAP kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells
independently from the amyloid fibril formation (25). The decrease of ERK in our
microarray is also in line with the observation that PrP fragment
(aa 90–231) activates p38 MAP kinase by inhibiting the activation
of extracellular-regulated kinases 1/2 (ERK1/2), followed by the
caspase-3-dependent cell apoptosis in SH-SY5Y cells (26).
Genes involved in axon guidance and gap junctions,
which are critical for cell communication and cell development, are
rarely investigated in prion diseases. Axon guidance is a subfield
of neural development concerning the process by which neurons send
out axons to reach the correct targets. Its role in prion disease
is rarely described, until recently a group performed systematical
analyses on the gene changes in the brains of eight mouse
adapted-prion strains throughout the progression of the diseases.
Axon guidance disturbance was found in the mice with shorter
incubation times (5). Gap
junctions are involved in direct communication between the
cytosolic compartments of adjacent cells. Apart from the changes of
MAPK associated genes, some receptors of monoamines and other
biogenic amine neurotransmitters, such as β-1 adrenergic receptor
(ADRB1), dopamine receptor D2 (DRD2), 5-hydroxytryptamine
(serotonin) receptor 2 (HTR2) and mGluR are suppressed, resulting
in abnormal regulation of the expressions of the genes downstream
and subsequently inducing the dysfunction of calcium signaling
pathway and transportation of other biological masses.
The most significantly altered pathways in human
diseases in the brain of the G114V gCJD patient are those of AD and
PD, strongly indicating that G114V gCJD shares the similar gene
expression profiles as these two neurodegenerative diseases. Among
these two pathways, cell death induced by changes of oxidative
phosphorylation in the mitochondria is a critical factor for neuron
loss in AD and PD (18,27). In G114V gCJD, distinct impediment
of oxidation phosphorylation is also observed. This includes
abnormal phosphorylation, ATP depletion, collapse of mitochondrial
membrane potential, increase of reactive oxygen species (ROS).
Moreover, reduction of the expression of the relevant genes in this
gCJD case highlight the presence of the similar ER stress-induced
cell death observed in AD and PD, and Fas-induced cell apoptosis
observed in AD, which have also been described in BSE (28).
Aside from numerous genes downregulated in this gCJD
case, there are several genes showing upregulation. Among the 12
upregulated genes in sCJD described previously (6), eight genes were increased in G114V
gCJD, including RAB13 (RAB13, member RAS oncogene family), inositol
1,4,5-trisphosphate 3-kinase B (ITPKB) and transcriptional
coactivator with PDZ-binding motif (TAZ) that are more than 2-fold
increased, and GFAP, cysteine and glycine-rich protein 1 (CSRP1),
tropomyosin 2 (TPM2), promoting factor 1 (PTN) and RNA binding
motif, single stranded interacting protein 3 (RBMS3) that are more
than 1.5-fold increased. The proteins of the Rab family regulate
specific tethering/docking of incoming vesicles to the correct
target organelle (29), and they
are involved in various biological processes, protein transport,
small GTPase mediated signal transduction, vesicle-mediated
transport, modification-dependent protein catabolism, ER to Golgi
vesicle-mediated transport, endocytosis and some related regulation
processes (12). Several Rab
family members have been found to be associated with the
PrPSc propagation and accumulation in the prion-infected
cells (30), as well as with the
clinical manifestations (31). In
line with the observation in sCJD, Rab genes are unregulated in the
cerebral cortex of G114V gCJD. These phenomena illustrate the
similarity of gene expression profiles between G114V gCJD and sCJD.
More up and downregulated genes in G114V gCJD rely heavily on the
usage of a relatively larger capacity gene chip in this study. In
addition, a series of genes that appeared dozens of times increased
both in microarray and qRT-PCR will provide useful insights to
further explore potential biomarkers for the diagnosis of CJD.
Acknowledgements
We thank Dr Christopher J. Vavricka
from the Institute of Microbiology, Chinese Academy of Sciences,
Beijing, China, for kindly checking the manuscript. This study was
supported by the Chinese National Natural Science Foundation Grants
30800975, 81101302 and 30800640, the National Basic Research
Program of China (973 Program) (2007CB310505), the China
Mega-Project for Infectious Disease (2009ZX10004-101 and
2008ZX10004-008), and the SKLID Development Grant
(2008SKLID102).
References
|
1
|
Prusiner SB: Prions. Proc Natl Acad Sci
USA. 95:13363–13383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Colby DW and Prusiner SB: Prions. Cold
Spring Harb Perspect Biol. 3:a0068332011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pastore M, Chin SS, Bell KL, et al:
Creutzfeldt-Jakob disease (CJD) with a mutation at codon 148 of
prion protein gene: relationship with sporadic CJD. Am J Pathol.
167:1729–1738. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Collins S, McLean CA and Masters CL:
Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia,
and kuru: a review of these less common human transmissible
spongiform encephalopathies. J Clin Neurosci. 8:387–397. 2001.
View Article : Google Scholar
|
|
5
|
Hwang D, Lee IY, Yoo H, et al: A systems
approach to prion disease. Mol Syst Biol. 5:2522009. View Article : Google Scholar
|
|
6
|
Xiang W, Windl O, Westner IM, et al:
Cerebral gene expression profiles in sporadic Creutzfeldt-Jakob
disease. Ann Neurol. 58:242–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye J, Han J, Shi Q, et al: Human prion
disease with a G114V mutation and epidemiological studies in a
Chinese family: a case series. J Med Case Rep. 2:3312008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi Q, Zhang BY, Gao C, et al: The
diversities of PrP(Sc) distributions and pathologic changes in
various brain regions from a Chinese patient with G114V genetic
CJD. Neuropathology. 32:51–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Camon E, Magrane M, Barrell D, et al: The
Gene Ontology Annotation (GOA) project: implementation of GO in
SWISS-PROT, TrEMBL, and InterPro. Genome Res. 13:662–672. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
National Center for Biotechnology
Information, US National Library of Medicine; Bethesda, MD:
http://www.ncbi.nlm.nih.gov/geo.
|
|
11
|
Mukherjee A and Soto C: Role of
calcineurin in neurodegeneration produced by misfolded proteins and
endoplasmic reticulum stress. Curr Opin Cell Biol. 23:223–230.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
KEGG: Kyoto Encyclopedia of Genes and
Genomes. http://www.genome.jp/kegg/.
|
|
13
|
Rodriguez MM, Peoc’h K, Haik S, et al: A
novel mutation (G114V) in the prion protein gene in a family with
inherited prion disease. Neurology. 64:1455–1457. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kordek R, Liberski PP, Yanagihara R,
Isaacson S and Gajdusek DC: Molecular analysis of prion protein
(PrP) and glial fibrillary acidic protein (GFAP) transcripts in
experimental Creutzfeldt-Jakob disease in mice. Acta Neurobiol Exp.
57:85–90. 1997.
|
|
15
|
Tang Y, Xiang W, Hawkins SA, Kretzschmar
HA and Windl O: Transcriptional changes in the brains of cattle
orally infected with the bovine spongiform encephalopathy agent
precede detection of infectivity. J Virol. 83:9464–9473. 2009.
View Article : Google Scholar
|
|
16
|
Martinez T and Pascual A: Identification
of genes differentially expressed in SH-SY5Y neuroblastoma cells
exposed to the prion peptide 106–126. Eur J Neurosci. 26:51–59.
2007.PubMed/NCBI
|
|
17
|
Xiang W, Windl O, Wunsch G, et al:
Identification of differentially expressed genes in
scrapie-infected mouse brains by using global gene expression
technology. J Virol. 78:11051–11060. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zabel C, Nguyen HP, Hin SC, Hartl D, Mao L
and Klose J: Proteasome and oxidative phoshorylation changes may
explain why aging is a risk factor for neurodegenerative disorders.
J Proteomics. 73:2230–2238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wikipedia website: http://en.wikipedia.org/wiki/Purine_metabolism.
|
|
20
|
Deriziotis P and Tabrizi SJ: Prions and
the proteasome. Biochim Biophys Acta. 1782:713–722. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li XL, Wang GR, Jing YY, et al: Cytosolic
PrP induces apoptosis of cell by disrupting microtubule assembly. J
Mol Neurosci. 43:316–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nieznanski K, Podlubnaya ZA and Nieznanska
H: Prion protein inhibits microtubule assembly by inducing tubulin
oligomerization. Biochem Biophys Res Commun. 349:391–399. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osiecka KM, Nieznanska H, Skowronek KJ,
Karolczak J, Schneider G and Nieznanski K: Prion protein region
23–32 interacts with tubulin and inhibits microtubule assembly.
Proteins. 77:279–296. 2009.
|
|
24
|
Fabrizi C, Silei V, Menegazzi M, et al:
The stimulation of inducible nitric-oxide synthase by the prion
protein fragment 106–126 in human microglia is tumor necrosis
factor-alpha-dependent and involves p38 mitogen-activated protein
kinase. J Biol Chem. 276:25692–25696. 2001.PubMed/NCBI
|
|
25
|
Corsaro A, Thellung S, Villa V, et al:
Prion protein fragment 106–126 induces a p38 MAP kinase-dependent
apoptosis in SH-SY5Y neuroblastoma cells independently from the
amyloid fibril formation. Ann NY Acad Sci. 1010:610–622. 2003.
|
|
26
|
Corsaro A, Thellung S, Chiovitti K, et al:
Dual modulation of ERK1/2 and p38 MAP kinase activities induced by
minocycline reverses the neurotoxic effects of the prion protein
fragment 90–231. Neurotox Res. 15:138–154. 2009.PubMed/NCBI
|
|
27
|
Higgins GC, Beart PM, Shin YS, Chen MJ,
Cheung NS and Nagley P: Oxidative stress: emerging mitochondrial
and cellular themes and variations in neuronal injury. J Alzheimers
Dis. 20(Suppl 2): S453–S473. 2010.PubMed/NCBI
|
|
28
|
Tang Y, Xiang W, Terry L, Kretzschmar HA
and Windl O: Transcriptional analysis implicates endoplasmic
reticulum stress in bovine spongiform encephalopathy. PLoS One.
5:e142072010. View Article : Google Scholar
|
|
29
|
Zerial M and McBride H: Rab proteins as
membrane organizers. Nat Rev Mol Cell Biol. 2:107–117. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gilch S, Bach C, Lutzny G, Vorberg I and
Schatzl HM: Inhibition of cholesterol recycling impairs cellular
PrP(Sc) propagation. Cell Mol Life Sci. 66:3979–3991. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ermolayev V, Cathomen T, Merk J, et al:
Impaired axonal transport in motor neurons correlates with clinical
prion disease. PLoS Pathog. 5:e10005582009. View Article : Google Scholar : PubMed/NCBI
|