Introduction
Cisplatin, a platinum-containing drug, has the
ability to bind to DNA, disrupt cell division and ultimately, cause
apoptosis. These properties have resulted in the extensive use of
cisplatin as an antineoplastic agent. However, its therapeutic
usefulness may be limited by the potential risk of acute kidney
injury. The apoptotic death of renal proximal tubule epithelial
cells is the main characteristic of cisplatin-induced kidney injury
(1,2). The activation of pro-apoptotic
proteins, mitogen-activated protein kinases (MAPKs), Src/epidermal
growth factor receptor (EGFR) and the generation of reactive oxygen
species (ROS) have been recognized as the upstream molecular
mechanisms responsible for cisplatin-induced apoptosis of renal
proximal tubule epithelial cells (3–6).
In addition, inflammation, mediated by the activation of nuclear
factor-κB (NF-κB), also plays an important role in the pathogenesis
of cisplatin-induced apoptosis (7–9).
G-protein-coupled receptor 40 (GPR40) is a member of
a subfamily of G-protein-coupled receptors and is abundantly
expressed in the pancreatic β-cells, where it regulates insulin
secretion by functioning as a receptor for long-chain free fatty
acids (10,11). To date, the expression and
function of GPR40 in the kidneys have not been established.
Recently, diverse pharmacological effects of GPR40 agonists have
been observed (12–16). GPR40 has been emerged as a novel
therapeutic target for glycemic control in type 2 diabetes mellitus
(12). The anti-diabetic effects
of GPR40 agonists have been associated with the improvement of the
preservation of pancreatic β-cells through the inhibition of
apoptosis (13,14). In addition, treatment with GPR40
agonists has been shown to suppress cutaneous immune inflammation
and prevent NF-κB activation and nuclear translocation in bone
(15,16).
In the present study, we investigated the changes
occurring in the expression of GPR40 in the kidneys during
cisplatin-induced kidney injury. In addition, we investigated the
effects of the GPR40 agonist, GW9508, on the cisplatin-induced
apoptosis of the human renal proximal tubule epithelial cell line,
HK-2.
Materials and methods
Animal model of cisplatin-induced kidney
injury
This study was approved by the Ethics Committee of
Chonnam National University Medical School, Gwangju, Korea and the
experimental procedure conformed to the Institutional Guidelines
for Experimental Animal Care and Use. Male Sprague-Dawley rats
weighing 180–200 g were injected intraperitoneally with cisplatin
(8 mg/kg; Boryung Co., Ltd., Ansan, Korea). Control rats were
treated with saline. After 2 or 4 days, the rats were anesthetized
with ketamine and blood samples were collected from the inferior
vena cava. These samples were then analyzed for blood urea nitrogen
(BUN) and creatinine levels. The kidneys were rapidly removed from
the animals and processed for semi-quantitative immunoblot
analysis, as previously described (17,18).
Cell culture and reagents
The HK-2 cells (ATCC; Manassas, VA, USA) were
cultured in 100-mm dishes containing Dulbecco’s modified Eagle’s
medium-F-12 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with
10% fetal bovine serum (FBS), 100 U/ml of penicillin and 100 μg/ml
of streptomycin (Sigma-Aldrich). The cells were grown at 37°C in a
5% CO2 humidified incubator for 24 h and subcultured to
70–80% confluence. The cells were treated with cisplatin (50 μM)
for 24 h in the presence or absence of the GPR40 agonist, GW9508
(10 μM; Cayman Chemical Co., Ann Arbor, MI, USA), for 1 h prior to
the addition of cisplatin and then harvested for further analysis.
Control cells were treated with the vehicle (dimethyl
sulfoxide).
Cell viability
The HK-2 cells were plated at 5×103
cells/well in a 96-well plate and incubated for 24 h. In order to
examine the effects of GW9508, the cells were treated with
cisplatin for 24 h in the presence or absence of GW9508 for 1 h
prior to exposure to cisplatin. Cell viability was determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. Following incubation, 50 μl of 5 mg/ml MTT (Sigma-Aldrich)
were added to each well of the 96-well plates and followed by
incubation for 3 h at 37°C. The supernatant was removed by
aspiration and then dimethyl sulfoxide was added to dissolve the
precipitated dye. The absorbance at 570 nm was detected using a
96-well ELISA reader (BioTek Instruments Inc., Winooski, VT, USA).
Cell viability was expressed as the fraction of surviving cells
relative to the vehicle-treated cells.
Detection of apoptosis
Apoptotic nuclei were detected by staining the cells
with the DNA-specific fluorescent dye,
4′-6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA,
USA). Following exposure to cisplatin in the presence or absence of
GW9508, the cells were fixed with 3% paraformaldehyde for 30 min at
room temperature and then washed twice with phosphate-buffered
saline (PBS). DAPI was added to the fixed cells for 5 min, followed
by examination under a fluorescence microscope (Nikon, Tokyo,
Japan) in order to assess chromatin condensation and the
fragmentation of nuclei. The degree of nuclear fragmentation was
evaluated by counting the percentage of apoptotic cells at ×400
magnification in 5 randomly selected fields from 3 independent
cultures. Furthermore, apoptosis was also assessed by TUNEL assay
(Chemicon, Temecula, CA, USA) according to the manufacturer’s
instructions.
Preparation of nuclear extracts
To prepare the nuclear extracts, the cells were
lysed using NE-PER nuclear extraction reagent (NER; Pierce
Biotechnology, Rockford, IL, USA) according to the manufacturer’s
instructions. Briefly, the HK-2 cells incubated with cisplatin in
the presence or absence of GW9508 were harvested by scraping the
cells into cold PBS, pH 7.2, followed by centrifugation at 14,000 ×
g for 2 min. After removing the supernatant, 100 μl of ice-cold
cytoplasmic extraction reagent (CER) I were added to the dried cell
pellets followed by incubation on ice for 10 min. Ice-cold CER II
was then added to the tube and centrifuged at 16,000 × g for 5 min.
The pellet fraction was suspended in 50 μl of ice-cold NER,
followed by centrifugation at 16,000 × g for 10 min. Finally, the
supernatant (nuclear extract) fraction was transferred to a new
tube and the protein concentrations were measured, as previously
described (19).
Semi-quantitative immunoblot
analysis
The HK-2 cells were harvested, washed twice with
cold PBS, resuspended in lysis buffer (20 mM Tris-HCl, pH 7.4, 0.01
mM EDTA, 150 mM NaCl, 1 mM PMSF, 1 μg/ml leupeptin, 1 mM
Na3VO4) and sonicated briefly. Following
centrifugation, the supernatant was prepared using the Pierce BCA
protein assay kit (Pierce Biotechnology) and the protein
concentrations were then measured. Equal amounts of protein were
separated on 9 or 12% sodium dodecyl sulfate polyacrylamide gels
(SDS-PAGE). The protein contained in the gels was then
electrophoretically transferred onto nitrocellulose membranes using
a Bio-Rad Mini Protean II apparatus (Bio-Rad Laboratories,
Hercules, CA, USA). The blots were subsequently blocked with 5%
milk in a mixture of tris-buffered saline and Tween-20 (TBST; 20 mM
Tris-HCl, 140 mM NaCl, 0.1% Tween-20, pH 8.0) for 1 h and then
incubated overnight at 4°C with primary antibodies. The membranes
were then incubated with secondary anti-rabbit or anti-mouse
horseradish peroxidase-conjugated antibodies and visualized with an
enhanced chemiluminescence system.
The anti-GPR40 and anti-caspase 3 antibodies were
purchased from Epitomics (Burlingame, CA, USA) and Santa Cruz
Biotechnology (Santa Cruz, CA, USA), respectively. The anti-cleaved
caspase-3, anti-Bax, anti-Bcl-2, anti-IκB-α, anti-NF-κB p65,
anti-histone H3, anti-Src, anti-phospho-Src, anti-EGFR,
anti-phospho-EGFR, anti-extracellular signal-regulated kinase (ERK)
and anti-phospho-ERK antibodies were obtained from Cell Signaling
Technology (Danvers, MA, USA). All antibodies were diluted in
blocking buffer and incubated overnight at 4°C. Anti-β-actin
(Sigma-Aldrich) antibody was used as a control.
Determination of ROS generation
Intracellular ROS generation was measured using a
2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA)
fluoroprobe (Molecular Probes, Eugene, OR, USA). The cells were
incubated with 5 μM H2DCF-DA for 30 min at 37°C. The
cells were then washed, collected by centrifugation and resuspended
in PBS. The fluorescence intensity was measured using a
FACSCalibur™ flow cytometer (Becton-Dickinson, San Jose, CA,
USA).
Statistical analysis
The results are expressed as the means ± standard
error of the mean (SEM) of 3 individual experiments. An unpaired
t-test was used to determine the differences between 2 groups.
Multiple comparisons among the groups were made using one-way ANOVA
and the post hoc Tukey HSD test. Differences with values of
P<0.05 were considered statistically significant.
Results
Expression of GPR40 in kidneys
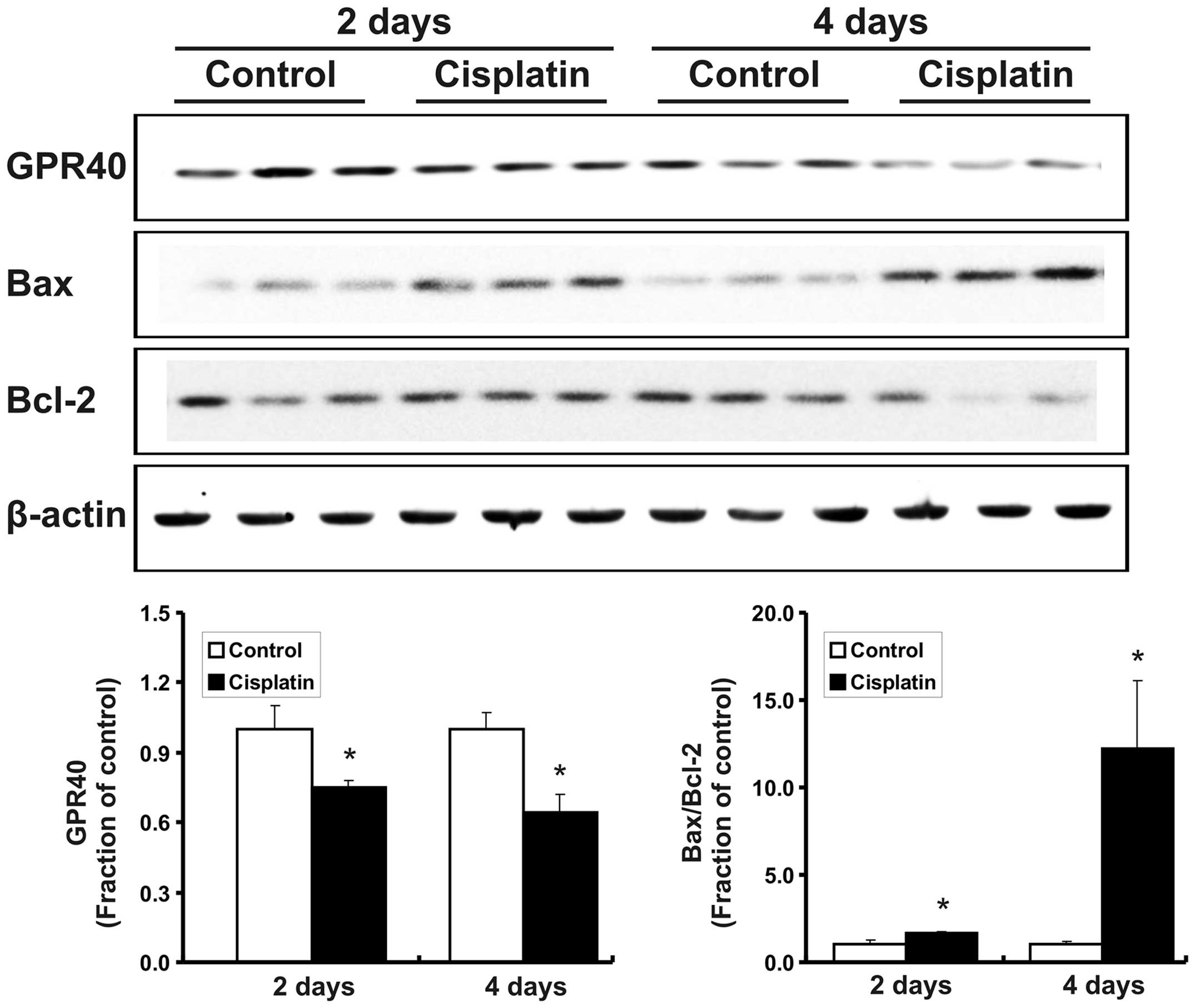
Treatment with cisplatin increased serum creatinine
levels in the rats compared with the control (after 2 days,
0.22±0.02 vs. 0.43±0.06 mg/dl, P<0.05; after 4 days, 0.25±0.02
vs. 3.89±0.26 mg/dl, P<0.05). BUN levels were also increased in
the cisplatin-treated rats (after 2 days, 16.45±0.57 vs. 29.04±2.19
mg/dl, P<0.05; after 4 days, 15.60±1.24 vs. 204.24±10.46 mg/dl,
P<0.05). Following cisplatin treatment, the protein expression
of GPR40 was decreased in the kidneys of the rats, while the
Bax/Bcl-2 expression ratio was increased (Fig. 1).
Cell viability and apoptosis
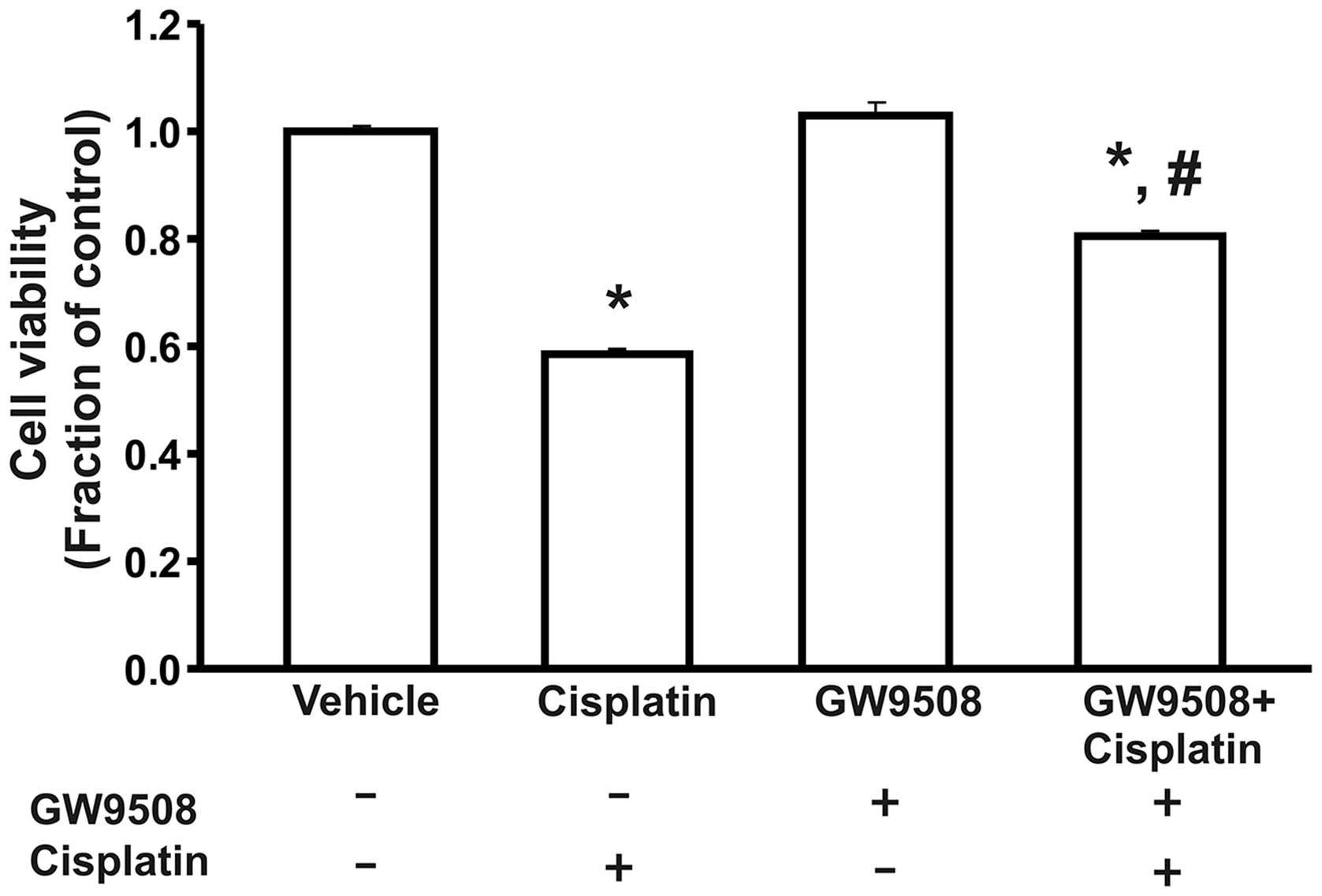
We performed an MTT assay to determine the viability
of the HK-2 cells. Our data revealed that treatment with cisplatin
decreased the viability of the cells compared with the control.
Furthermore, pre-treatment with GW9508 attenuated the
cisplatin-induced cell death (Fig.
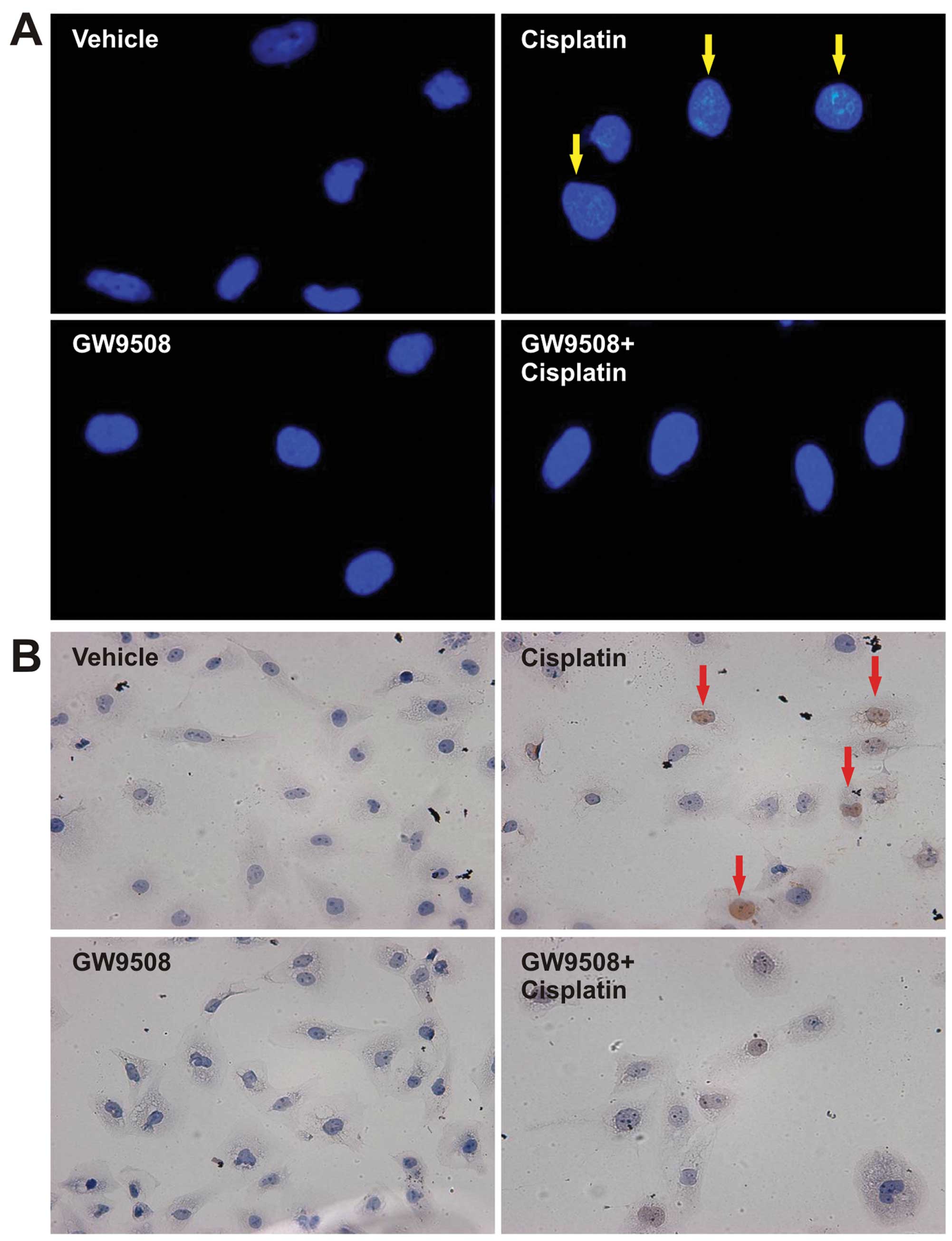
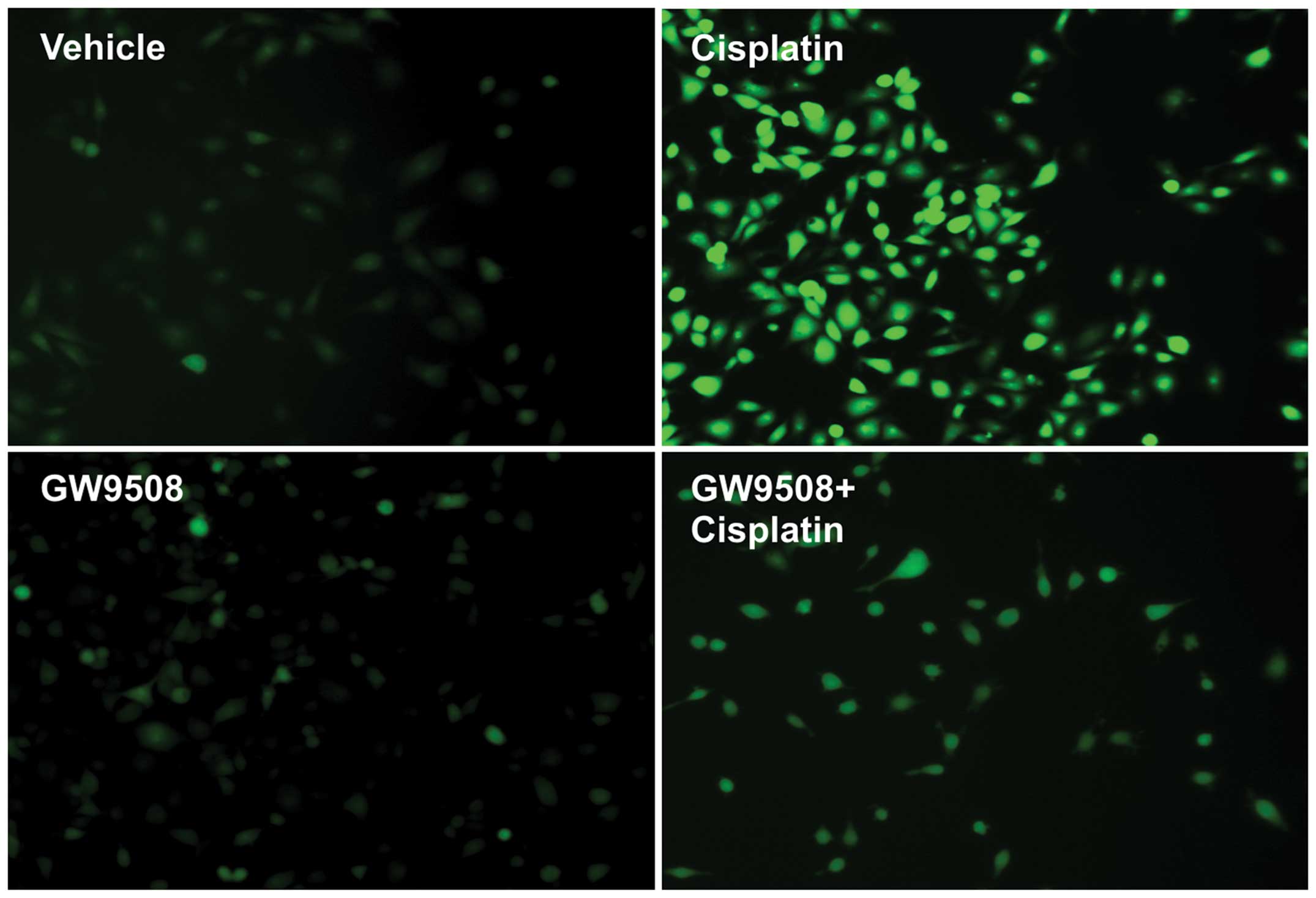
2). DAPI staining indicated that treatment with cisplatin
increased the number of cells with condensed nuclei and TUNEL
staining also demonstrated that treatment with cisplatin resulted
in increased levels of apoptosis. However, pre-treatment with
GW9508 prevented the cisplatin-induced apoptosis of HK-2 cells
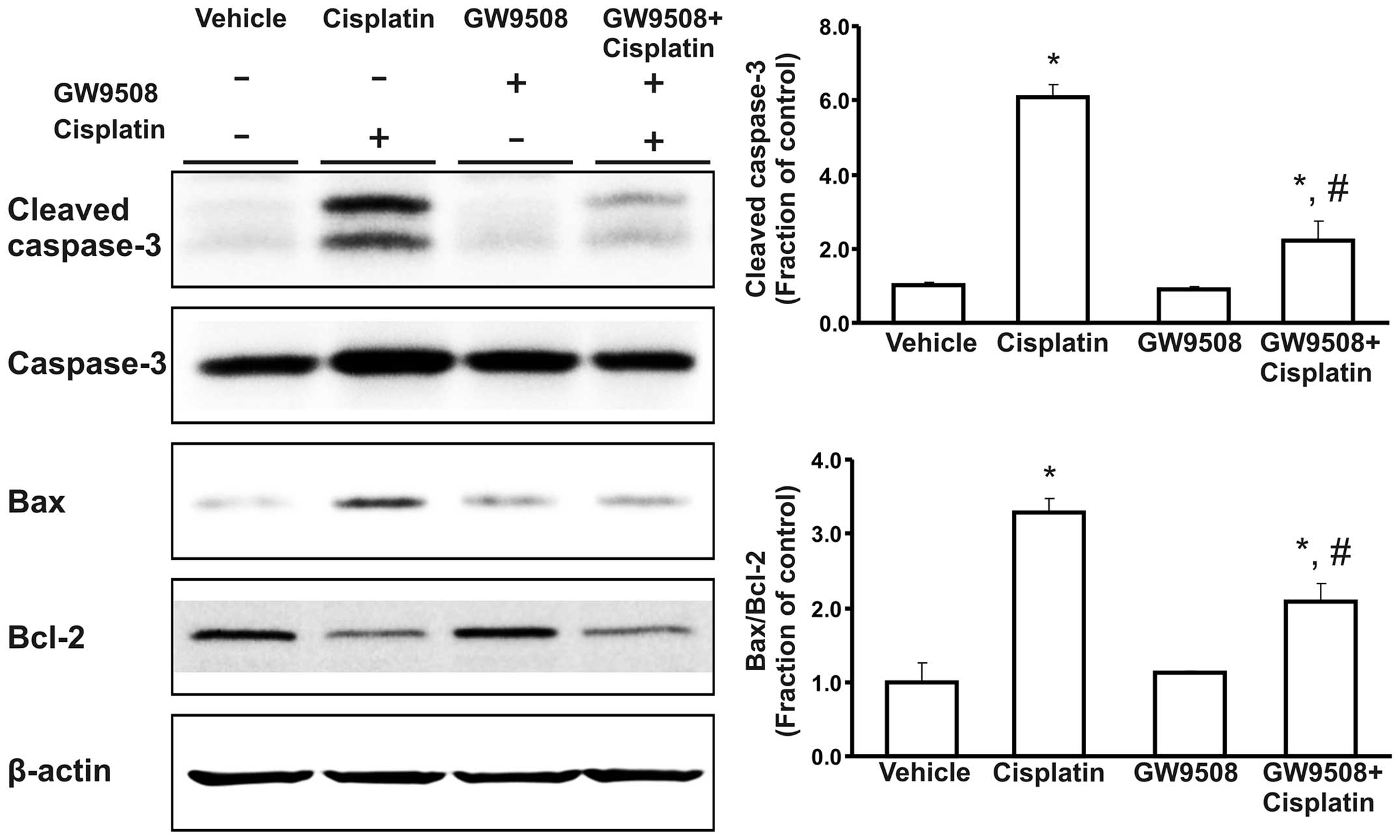
(Fig. 3). Treatment with
cisplatin also increased the expression of cleaved caspase-3 and
the Bax/Bcl-2 expression ratio in the HK-2 cells compared with the
control. These changes were attenuated by pre-treatment with GW9508
(Fig. 4).
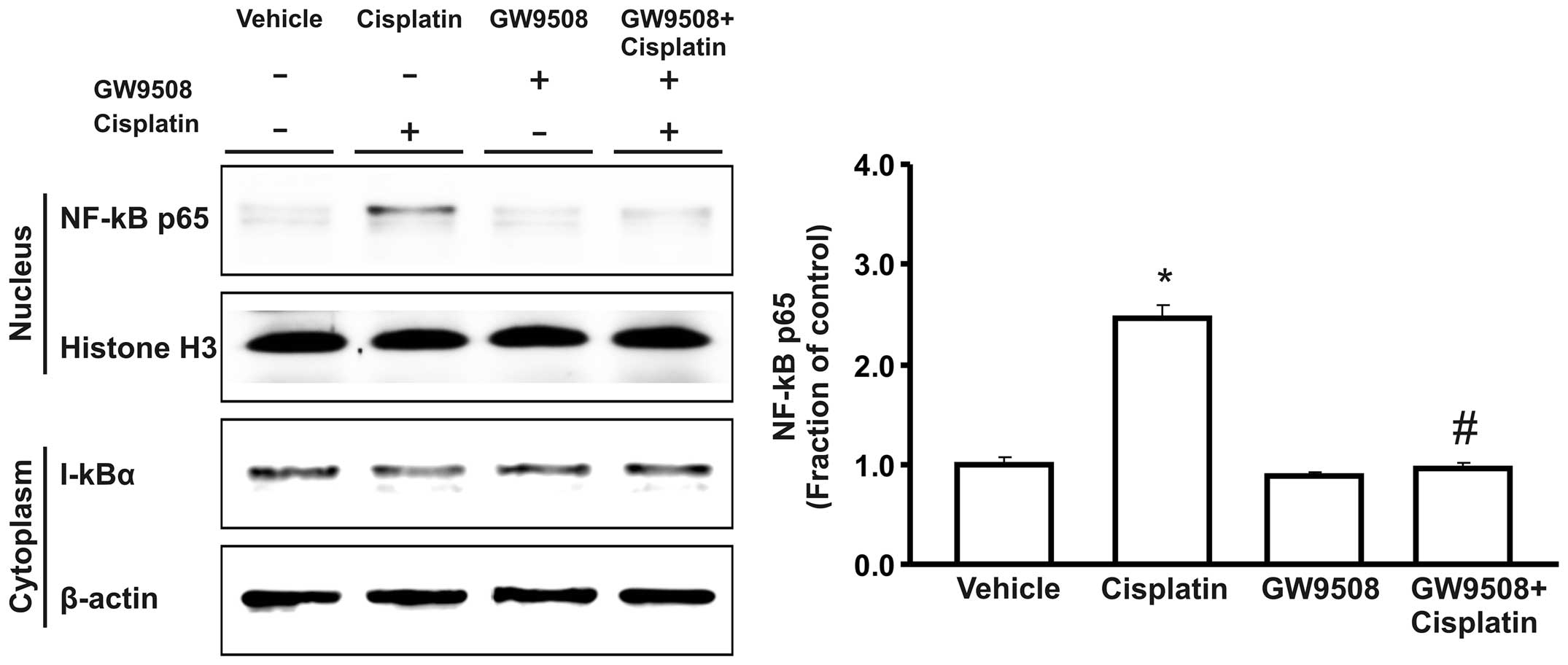
NF-κB expression
During activation, NF-κB is released from the
inhibitory subunit IκB-α, and translocates to the nucleus, where it
promotes the transcriptional activation of a number of target
genes. Importantly, the protein expression of the p65 subunit of
nuclear NF-κB was markedly increased in the cisplatin-treated HK-2
cells compared with the control, while the expression of cytosolic
IκB-α was decreased. Furthermore, the apparent increase in NF-κB
nuclear translocation induced by treatment with cisplatin was
counteracted by pre-treatment with GW9508 (Fig. 5).
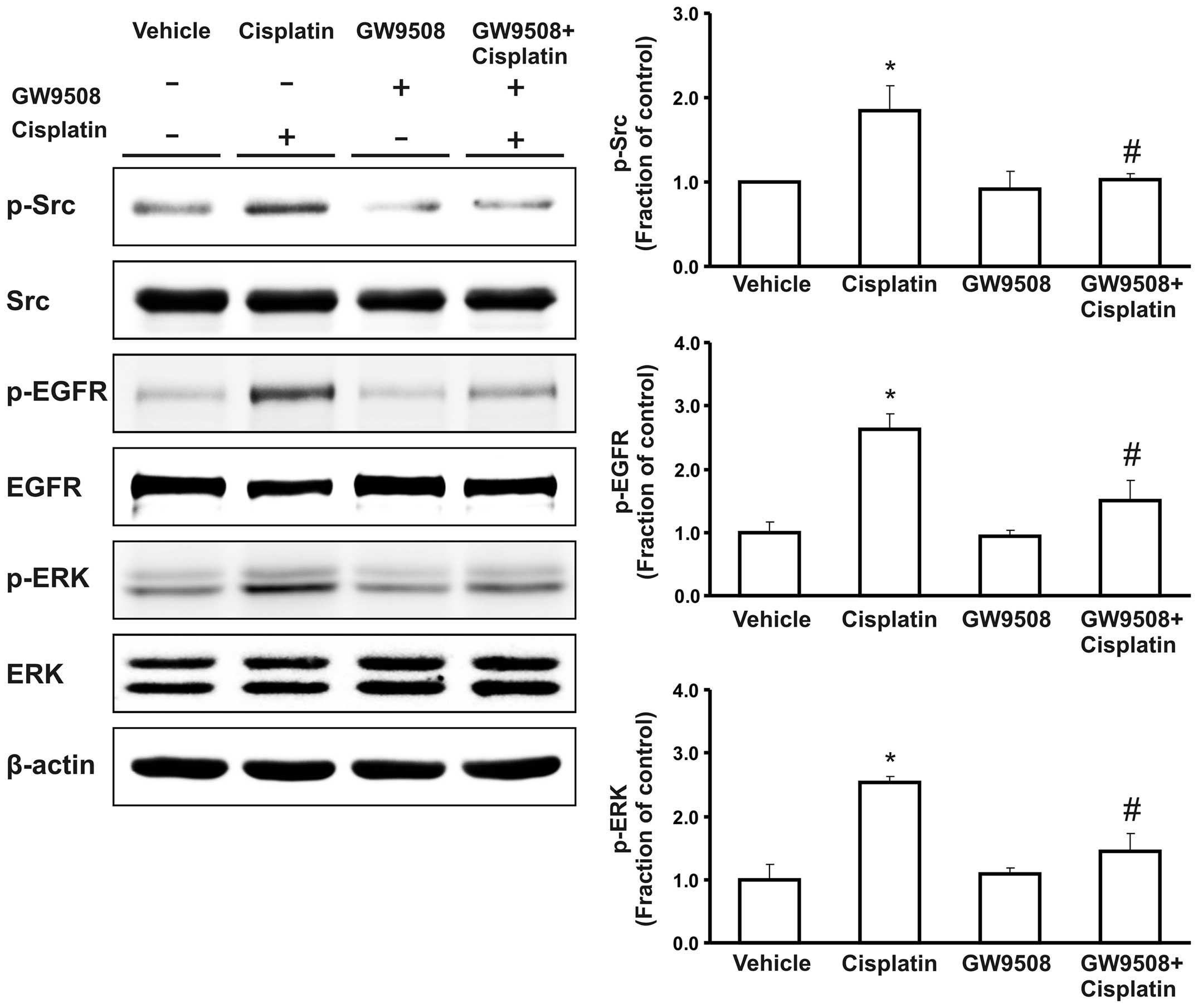
ROS generation and the Src/EGFR/ERK
signaling pathway
We measured the intracellular ROS generation using
H2DCF-DA fluoroprobe following treatment with cisplatin
in the absence or presence of pre-treatment with GW9508. We
observed that treatment with cisplatin promoted the generation of
ROS in the HK-2 cells. However, this increase was diminished by
pre-treatment of the cells with GW9508 (Fig. 6). Furthermore, following treatment
with cisplatin, the expression of the phosphorylated proteins of
Src, EGFR and ERK was markedly increased compared with the control.
Pre-treatment with GW9508 counteracted this increase in the
phosphorylation of the Src/EGFR/ERK signaling pathway (Fig. 7).
Discussion
Previously, we, as well as others have demonstrated
that treatment with cisplatin induces the apoptosis of renal
proximal tubule epithelial cells, and that the activation of
pro-apoptotic proteins is causally related to the pathogenesis of
cisplatin-induced kidney injury (17,20,21). Our findings in the present study
also confirmed that treatment with cisplatin impaired renal
functional parameters and increased the Bax/Bcl-2 expression ratio
in the kidneys.
It has been demonstrated that GPR40 is primarily
expressed in pancreatic β-cells, where it regulates the
physiological effects of fatty acids as a cell-surface receptor
(11,22). However, its cellular localization
and physiological function in the kidneys have not yet been
established. In the present study, we demonstrated that GPR40 was
expressed in rat kidneys and that treatment with cisplatin resulted
in a decrease in the protein expression of GPR40. This decrease was
also associated with an increase in serum creatinine and BUN levels
and an increase in the Bax/Bcl-2 expression ratio. Taken together,
these data suggest that the decreased expression of GPR40 in the
kidneys may be related to the pathogenesis of cisplatin-induced
kidney injury.
To the best of our knowledge, the anti-apoptotic
effects of GPR40 agonists on renal tubular epithelial cells have
not been demonstrated to date, although recent studies have
revealed that GPR40 agonists inhibit the apoptosis of pancreatic
β-cells (13,14). In the present study, treatment
with cisplatin decreased the viability of HK-2 cells and increased
apoptosis, which was associated with the increased expression of
pro-apoptotic proteins. These changes were attenuated by
pre-treatment with the GPR40 agonist, GW9508. The present study
suggests that GPR40 agonists play a protective role in the
cisplatin-induced apoptosis of renal tubular epithelial cells
through the inhibition of pro-apoptotic proteins. Furthermore, it
has been established that the activation of NF-κB plays a critical
role in the pathogenesis of cisplatin-induced kidney injury and the
inhibition of NF-κB activity has been shown to attenuate the
apoptosis of renal tubular epithelial cells (7–9).
The present study also demonstrated that treatment with cisplatin
increased the nuclear translocation of NF-κB in HK-2 cells. In
addition, our data indicate that the GPR40 agonist, GW9508,
counteracted the cisplatin-induced activation of NF-κB and
prevented cisplatin-induced apoptosis.
Increased levels of ROS also play a crucial role in
the development of cisplatin-induced kidney injury. Cisplatin leads
to the accumulation of endogenous ROS in renal tubular epithelial
cells through the depletion of glutathione and the induction of
mitochondrial dysfunction (1,2).
The inhibition of cisplatin-induced generation of ROS diminishes
cisplatin-induced apoptosis (6,9,23,24). In the present study, we also
demonstrated that treatment with cisplatin increased the generation
of ROS in HK-2 cells, which was suppressed by pre-treatment with
GW9508, a GPR40 agonist. This finding suggests that the protective
role of the GPR40 agonist (GW9508) against cisplatin-induced
apoptosis may be attributed to the inhibition of ROS
generation.
The activation of the Src/EGFR/ERK signaling pathway
is known to affect a number of processes, including cellular
proliferation, differentiation and apoptosis. Recent studies have
suggested that prolonged generation of ROS triggers the activation
of the Src/EGFR/ERK signaling pathway (25,26). The phosphorylation of Src and EGFR
has been shown to result in the activation of ERK, which plays a
pro-apoptotic role as an upstream mechanism of pro-apoptotic
proteins during cisplatin-induced kidney injury (3–5).
In the present study, we demonstrted that pre-treatment with the
GPR40 agonist, GW9508, counteracted the cisplatin-induced increase
in the phosphorylation of Src/EGFR/ERK, which may be associated
with the anti-apoptotic effects of the GPR40 agonist.
In conclusion, the data from our study demonstrate
that GPR40 expression in the kidneys is decreased in rats with
cisplatin-induced kidney injury. In HK-2 cells, the activation of
GPR40 attenuates cisplatin-induced apoptosis by inhibiting ROS
generation, the activation of the Src/EGFR/ERK signaling pathway
and the nuclear activation of NF-κB and pro-apoptotic factors.
Acknowledgements
The present study was supported by a research grant
from the Research Institute of Medical Sciences, Chonnam National
University (2012-CURIMS-DR002), Chonnam National University
(2013–2575) and the Chonnam National University Hospital Biomedical
Research Institute (CRI14012-1).
References
|
1
|
Yao X, Panichpisal K, Kurtzman N and
Nugent K: Cisplatin nephrotoxicity: a review. Am J Med Sci.
334:115–124. 2007. View Article : Google Scholar
|
|
2
|
Pabla N and Dong Z: Cisplatin
nephrotoxicity: mechanisms and renoprotective strategies. Kidney
Int. 73:994–1007. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arany I, Megyesi JK, Kaneto H, Price PM
and Safirstein RL: Cisplatin-induced cell death is EGFR/src/ERK
signaling dependent in mouse proximal tubule cells. Am J Physiol
Renal Physiol. 287:F543–F549. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jo SK, Cho WY, Sung SA, Kim HK and Won NH:
MEK inhibitor, U0126, attenuates cisplatin-induced renal injury by
decreasing inflammation and apoptosis. Kidney Int. 67:458–466.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim YK, Kim HJ, Kwon CH, Kim JH, Woo JS,
Jung JS and Kim JM: Role of ERK activation in cisplatin-induced
apoptosis in OK renal epithelial cells. J Appl Toxicol. 25:374–382.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mishima K, Baba A, Matsuo M, Itoh Y and
Oishi R: Protective effect of cyclic AMP against cisplatin-induced
nephrotoxicity. Free Radic Biol Med. 40:1564–1577. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li S, Gokden N, Okusa MD, Bhatt R and
Portilla D: Anti-inflammatory effect of fibrate protects from
cisplatin-induced ARF. Am J Physiol Renal Physiol. 289:F469–F480.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee S, Kim W, Moon SO, et al:
Rosiglitazone ameliorates cisplatin-induced renal injury in mice.
Nephrol Dial Transplant. 21:2096–2105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sung MJ, Kim DH, Jung YJ, et al: Genistein
protects the kidney from cisplatin-induced injury. Kidney Int.
74:1538–1547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sum CS, Tikhonova IG, Neumann S, Engel S,
Raaka BM, Costanzi S and Gershengorn MC: Identification of residues
important for agonist recognition and activation in GPR40. J Biol
Chem. 282:29248–29255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Itoh Y, Kawamata Y, Harada M, et al: Free
fatty acids regulate insulin secretion from pancreatic beta cells
through GPR40. Nature. 422:173–176. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bharate SB, Nemmani KV and Vishwakarma RA:
Progress in the discovery and development of small-molecule
modulators of G-protein-coupled receptor 40 (GPR40/FFA1/FFAR1): an
emerging target for type 2 diabetes. Expert Opin Ther Pat.
19:237–264. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gowda N, Dandu A, Singh J, et al:
Treatment with CNX-011-67, a novel GPR40 agonist, delays onset and
progression of diabetes and improves beta cell preservation and
function in male ZDF rats. BMC Pharmacol Toxicol. 14:282013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wagner R, Kaiser G, Gerst F, et al:
Reevaluation of fatty acid receptor 1 as a drug target for the
stimulation of insulin secretion in humans. Diabetes. 62:2106–2111.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fujita T, Matsuoka T, Honda T, Kabashima
K, Hirata T and Narumiya S: A GPR40 agonist GW9508 suppresses CCL5,
CCL17, and CXCL10 induction in keratinocytes and attenuates
cutaneous immune inflammation. J Invest Dermatol. 131:1660–1667.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wauquier F, Philippe C, Léotoing L, et al:
The free fatty acid receptor G protein-coupled receptor 40 (GPR40)
protects from bone loss through inhibition of osteoclast
differentiation. J Biol Chem. 288:6542–6551. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim SW, Lee JU, Nah MY, Kang DG, Ahn KY,
Lee HS and Choi KC: Cisplatin decreases the abundance of aquaporin
water channels in rat kidney. J Am Soc Nephrol. 12:875–882.
2001.PubMed/NCBI
|
|
18
|
Ma SK, Choi JS, Joo SY, et al: Activation
of the renal PI3K/Akt/mTOR signaling pathway in a DOCA-salt model
of hypertension. Chonnam Med J. 48:150–154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rosenau C, Emery D, Kaboord B and
Qoronfleh MW: Development of a high-throughput plate-based
chemiluminescent transcription factor assay. J Biomol Screen.
9:334–342. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bae EH, Lee J, Ma SK, et al: alpha-Lipoic
acid prevents cisplatin-induced acute kidney injury in rats.
Nephrol Dial Transplant. 24:2692–2700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park JW, Cho JW, Joo SY, et al:
Paricalcitol prevents cisplatin-induced renal injury by suppressing
apoptosis and proliferation. Eur J Pharmacol. 683:301–309. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Briscoe CP, Tadayyon M, Andrews JL, et al:
The orphan G protein-coupled receptor GPR40 is activated by medium
and long chain fatty acids. J Biol Chem. 278:11303–11311. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee S, Moon SO, Kim W, et al: Protective
role of L-2-oxothiazolidine-4-carboxylic acid in cisplatin-induced
renal injury. Nephrol Dial Transplant. 21:2085–2095. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mukhopadhyay P, Horváth B, Zsengellér Z,
et al: Mitochondrial-targeted antioxidants represent a promising
approach for prevention of cisplatin-induced nephropathy. Free
Radic Biol Med. 52:497–506. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J, Chen JK, Nagai K, et al: EGFR
signaling promotes TGFβ-dependent renal fibrosis. J Am Soc Nephrol.
23:215–224. 2012.
|
|
26
|
Chen J, Chen JK and Harris RC: Angiotensin
II induces epithelial-to-mesenchymal transition in renal epithelial
cells through reactive oxygen species/Src/caveolin-mediated
activation of an epidermal growth factor receptor-extracellular
signal-regulated kinase signaling pathway. Mol Cell Biol.
32:981–991. 2012. View Article : Google Scholar
|