Introduction
Airway epithelial cells protect the underlying
tissue against inhaled allergens, environmental pollutants and
respiratory viruses. Airway epithelial cells constitute a highly
regulated and impermeable barrier formed by tight junctions (TJs),
which are composed of zonula occludens (ZO)1–3, occludin and
claudin 1–5, as well as adhesion junctions (AJs), which consist of
E-cadherin, β-catenin and junctional adhesion molecule (JAM)
(1). E-cadherin is regarded as
the ‘gatekeeper’ in the airway mucosa and in allergic sensitization
due to its key role in maintaining the stability of epithelial
junctions, including TJs (2).
Studies have demonstrated that the bronchial epithelial barrier in
asthmatic patients is compromised. The expression of ZO-1 and
E-cadherin in bronchial epithelial cells obtained from asthmatic
patients has been shown to be significantly lower than that in
cells obtained from non-asthmatic subjects (3,4).
The disruption of the epithelial barrier may facilitate the
transport of allergens to allergen-presenting cells and promote the
pro-inflammatory activities of the epithelium (5). Increasing evidence suggests that the
loss of airway epithelial integrity contributes significantly to
asthma pathogenesis (2).
In recent years, lower vitamin D levels have been
found to be associated with impaired lung function, increased
airway hyperresponsiveness (AHR) and a reduced glucocorticoid
response in subjects with asthma (6). However, the detailed mechanisms
involved remain poorly understood.
Toluene diisocyanate (TDI) is currently one of the
leading causes of occupational asthma. In previous studies, we
demonstrated that TDI-human serum albumin (HSA) impaired TJ
function, induced E-cadherin redistribution and increased the
permeability of bronchial epithelial cells both in vitro and
in vivo (7,8). In this study, we aimed to determine
whether 1,25-dihydroxyvitamin D3
[1,25(OH)2D3 or 1,25D3] preserves
airway epithelial barrier integrity and whether the inhibition of
extracellular signal-regulated kinase (ERK)1/2 is involved in this
process.
Materials and methods
Animals and agents
Six-week-old male BALB/c mice were purchased from
Southern Medical University (Guangzhou, China). The mice were
housed in a specific pathogen-free (SPF) environment with a 12-h
dark/light cycle. All experiments were conducted in accordance with
the guidelines outlined by the committee of Southern Medical
University on the use and care of animals. The protocols were
approved by the Animal Subjects Committee of Nanfang Hospital. The
vehicle (AOO) used to dissolve TDI consisted of a mixture of 2
volumes of acetone and 3 volumes of olive oil for dermal
sensitization, and 1 volume of acetone and 4 volumes of olive oil
for airway challenge. The concentrations of TDI were provided as a
percentage (v/v) in AOO. TDI (toluene-2,4-diisocyanate), acetone
and 1,25D3 were obtained from Sigma-Aldrich (Shanghai,
China). Enzyme-linked immunosorbent assay (ELISA) kits for IgE,
interleukin (IL)-4 and interferon-γ (IFN-γ) were from Boshide
Biotechnology (Wuhan, China).
Animal experimental protocol
All the mice were randomly divided into the
following 3 groups as follows: i) the AOO group: vehicle
(AOO)-sensitized/AOO-challenged and phosphate-buffered saline
(PBS)-treated; ii) the TDI group: TDI-sensitized/TDI-challenged and
PBS-treated; and iii) 1,25D3 group:
TDI-sensitized/TDI-challenged and 1,25D3-treated. The
construction of the model of TDI-induced asthma was carried out as
previously described (8).
Briefly, on days 1 and 8, the animals were dermally treated with
0.3% TDI or the vehicle on the dorsum of both ears (20
µl/ear). On days 15, 18 and 21, the mice underwent an
oropharyngeal aspiration (20 µl) with 0.01% TDI or the
vehicle. 1,25D3 (100 ng/mouse) dissolved in 300
µl PBS containing 0.9% ethanol was administered to the mice
by intraperitoneal injection 1 day prior to challenge with TDI for
8 consecutive days. The control mice received 300 µl PBS
containing 0.9% ethanol by comparison. The methods for the
measurements of airway reactivity, airway inflammation, and the
expression levels of E-cadherin, ZO-1 and phosphorylated (p-)
ERK1/2 in the airway mucosa of the mice were as previously
described (8). The measurement of
airway reactivity was carried out using methacholine. AHR to
methacholine was assessed 24 h after the third challenge. Briefly,
the mice were placed in a barometric plethysmographic chamber
(Buxco Electronics, Troy, NY, USA). Aerosolized methacholine
(Sigma-Aldrich) in increasing concentrations (0–50 mg/ml) was
nebulized through an inlet of the main chamber for 3 min. Readings
were taken and averaged for 3 min after each nebulization. The
bronchopulmonary resistance was expressed as enhanced pause
(Penh).
Hematoxylin and eosin (H&E) staining was used
for the measurement of airway inflammation. Briefly, the mice were
humanely euthanized with pentobarbital (100 mg/kg body weight,
administered intraperitoneally) and the lungs were removed. The
left lungs were infused with 4% formaldehyde. The lungs were fixed
and embedded in paraffin. Sections (4-µm-thick) were cut
using a Leica microtome 2030 (Leica Microsystems Nussloch GmbH,
Nussloch, Germany). The slides were stained with H&E.
Preparation of TDI-HSA conjugates
TDI-HSA conjugates were prepared by a modification
of the method described in the study by Son et al (9) and the method used for the
calculation of the amount of TDI bound to HSA was as previously
described (7).
Epithelial cell culture and exposure to
TDI-HAS
The human bronchial epithelial cell line, 16HBE,
(Shanghai Fuxiang Biological Technology Co., Ltd., Shanghai, China)
was used in this study due to its highly characterized
intercellular adhesion properties (10). The 16HBE cells were grown in a
cell culture flask in Dulbecco’s modified Eagle’s medium (DMEM)
(Ginuo Biopharm Technology Co., Shanghai, China) with 10% fetal
calf serum (FCS; Invitrogen, Gibco, Carlsbad, CA, USA) and placed
in a humidified incubator at 37°C with 5% CO2. When
reaching 90% confluence, the cells were trypsinized and seeded into
proper culture plates at a density of 104–105
cells/cm2. When the cells had grown to complete
confluence, they were pre-treated with or without 1,25D3
(0.1 to 100 nM for 6 to 24 h) or the ERK1/2 selective inhibitor,
U0126 (25 µM, for 1 h; Cell Signaling Technology, Beverly,
MA, USA). Subsequently, 100 µg/ml TDI-HSA conjugate were
added to the culture medium followed by incubation for 24 h. The
concentration of 1,25D3 and U0126 used in this study was
in accordance with that used in previous studies (11,12). Cell viability was detected using a
MTT colorimetric assay.
Measurement of the transepithelial
electrical resistance (TER) of the epithelial monolayer
The method used for measuring TER was as described
in the study of Sekiyama et al (13). In brief, the 16HBE cells were
seeded at a density of 2×105 cells/cm2 onto
the apical chamber of Polyester Membrane Transwell-Clear Inserts
(Cat. no. 3460; Corning Inc., Corning, NY, USA) and incubated at
37°C until complete confluence was reached. Cell monolayer
integrity was evaluated by measuring TER using a Millicell ERS-2
Epithelial Volt-Ohm Meter (Millipore, Billerica, MA, USA). TER (Ω ×
cm2) was calculated by (TER sample - TER blank) ×
surface area (cm2). The percentage change in TER
following treatment was calculated as follows: TER(%) = (TER
test/TER control) ×100, where the TER sample is the initial reading
of each chamber, the TER blank is the reading of the no-cell
control chamber, the TER test is the real value of each chamber,
and the TER control is the reading of the no treatment control
chamber.
Measurement of the permeability of
fluorescein isothiocyanate-dextran (FITC-Dx) in the epithelial
monolayer
The measurement of the FITC-Dx (70 kDa) flux was
carried out using a previously described method (7). Briefly, the 16HBE cells were grown
in Transwell inserts to achieve complete confluence. The cells were
pre-treated with or without 1,25(OH)2D3, or
the ERK1/2 inhibitor, U0126, and 100 µg/ml TDI-HSA
conjugates were added to the culture medium followed by incubation
for 24 h. At the end of the exposure period, the apical and
basolateral chambers were washed twice with PBS, and FITC-Dx (0.5
mg/ml in phenol red-free DMEM) was then added to the apical chamber
at a level of 0.5 ml, and 1.0 ml phenol red-free DMEM (Gibco,
Carlsbad, CA, USA) was added to the basolateral chambers. The
plates were then incubated at 37°C for 90 min. The samples from the
apical and basolateral chambers were collected and data were read
using a TECAN Infinite M200 fluorometer (Tecan, Maennedorf,
Switzerland) with an excitation/emission wavelength of 495/520
nm.
Western blot analysis and
immunofluorescence staining
The cells were grown to confluence, and then
harvested and washed twice with ice-cold PBS. The cells were
subsequently lysed in cell lysis buffer (KeyGen Biotech, Nanjing,
China) containing protease inhibitor, calcineurin inhibitors and
PMSF, and centrifuged at 12,000 x g for 15 min at 4°C. The protein
products were normalized and boiled with standard SDS sample
buffer, then separated by 8% (ZO-1) or 10% (E-cadherin, occludin,
claudin-1/2, ERK1/2 and p-ERK1/2) sodium dodecyl
sulfate-polyacrylamide (SDS) gel electrophoresis and transferred
onto polyvinylidene fluoride membranes. The membranes were then
blocked with 5% BSA (for p-ERK1/2 only) or skim milk at room
temperature for 2 h, and incubated with anti-ZO-1 (sc-10804),
anti-E-cadherin (sc-7870), anti-occludin (sc-5562),
anti-claudin-1/2 (sc-28668) (Santa Cruz Biotechnology, Santa Cruz,
CA, USA), anti-ERK1/2 and anti-p-ERK1/2 (#9101S) antibodies (Cell
Signaling Technology) at 4°C overnight, and then incubated with
anti-rabbit IgG (#7074; Cell Signaling Technology) at room
temperature for 1 h. The immunoreactive bands were detected using
an enhanced chemiluminescence ECL system (DingGuo Biotech Co.,
Ltd., Guangzhou, China).
In a parallel experiment, the 16HBE cells were
seeded at a density of 1×105 cells/cm2 on a
cell culture dish (35×12 mm, 15 mm glass bottom; Nest Biotechnology
Co., Ltd., Shanghai, China) until confluent. The cells were then
fixed with 4% para-formaldehyde at room temperature for 15 min,
washed with ice-cold PBS for 15 min, incubated with 0.2% Triton
X-100 in PBS for 10 min, and rinsed again with PBS. The cells were
blocked with 5% skim milk for 2 h. All samples were subsequently
incubated with rabbit polyclonal anti-E-cadherin and FITC-linked
anti-rabbit IgG (ZF-0311; Zhongshan Jinqiao Biotechnology Co.,
Ltd., Beijing, China). The cell nuclei were stained with
4′,6-diamidino-2-phenylindole dihydrochloride (Sigma-Aldrich). A
laser scanning confocal microscope (Olympus, Tokyo, Japan) was
utilized to examine the distribution of junction proteins in the
16HBE cells. The images were processed with FV10-ASW1.7 Viewer and
Photoshop.
Statistical analysis
Statistical analysis was performed using SPSS
software version 13.0. Data are expressed as the means ± standard
error (SE) and comparisons among groups were analyzed by one-way
ANOVA accompanied by the LSD test for multiple comparisons. A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
1,25D3 decreases AHR, and the
levels of serum IgE, IL-4 and IFN-γ in the mouse model of
TDI-induced asthma
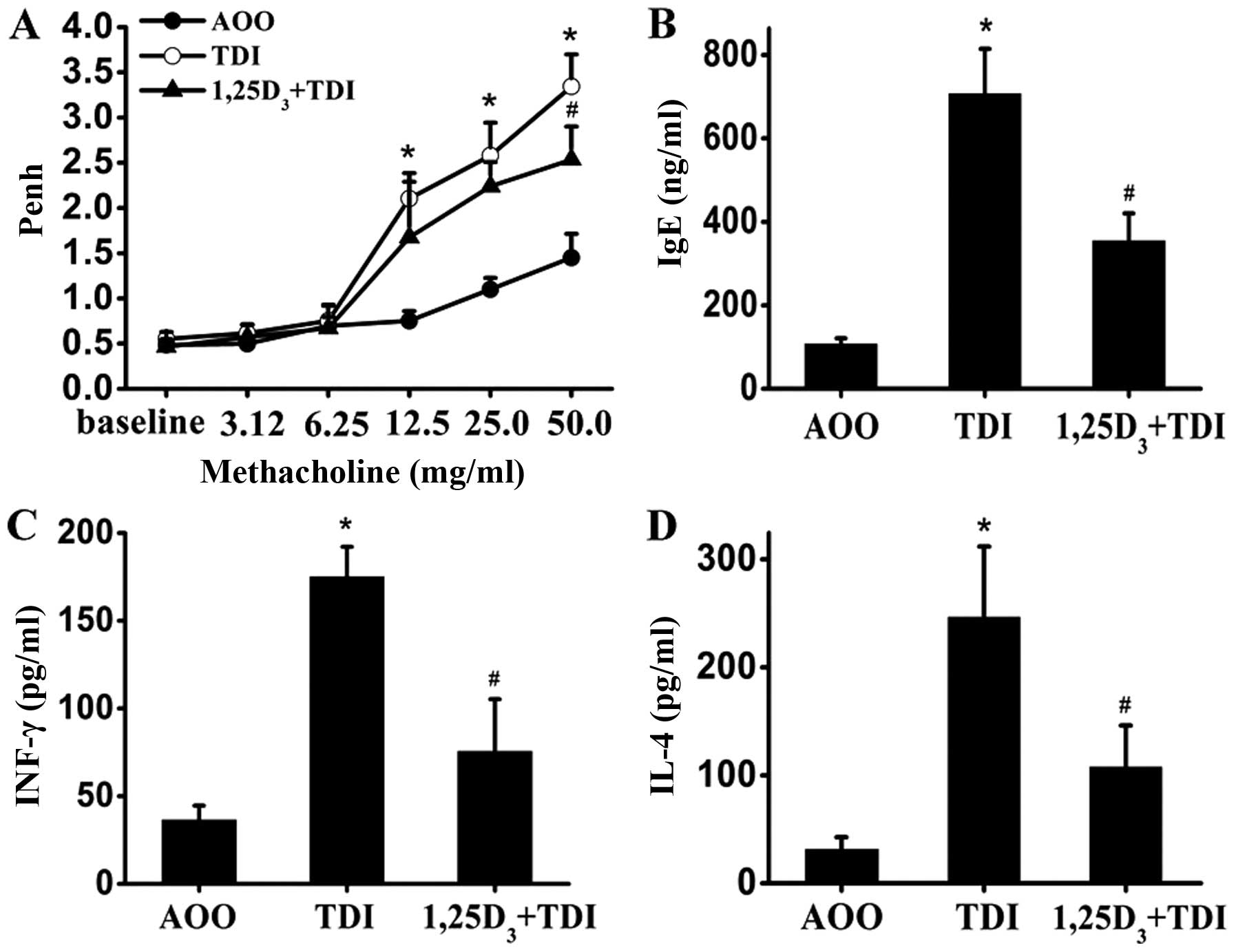
The pulmonary assessment of enhanced pause (Penh)
was used to determine airway reactivity to methacholine. As shown
in Fig. 1A, the Penh values were
significantly increased in the TDI-treated mice compared with the
controls following stimulation with methacholine (12.5, 25 and 50
mg/ml) (P<0.05); these values decreased after the administration
of 1,25D3 (methacholine, 50 mg/ml) (P<0.05).
Similarly, there was a robust elevation in the serum IgE levels
when the mice were sensitized and challenged with TDI; this effect
was inhibited by 1,25D3 (Fig. 1B). At the same time, the
TDI-induced release of IL-4 (Th2-related) and IFN-γ (Th1-related)
was also suppressed by treatment with 1,25D3 (Fig. 1C and D).
1,25D3 prevents TDI-induced
airway inflammation
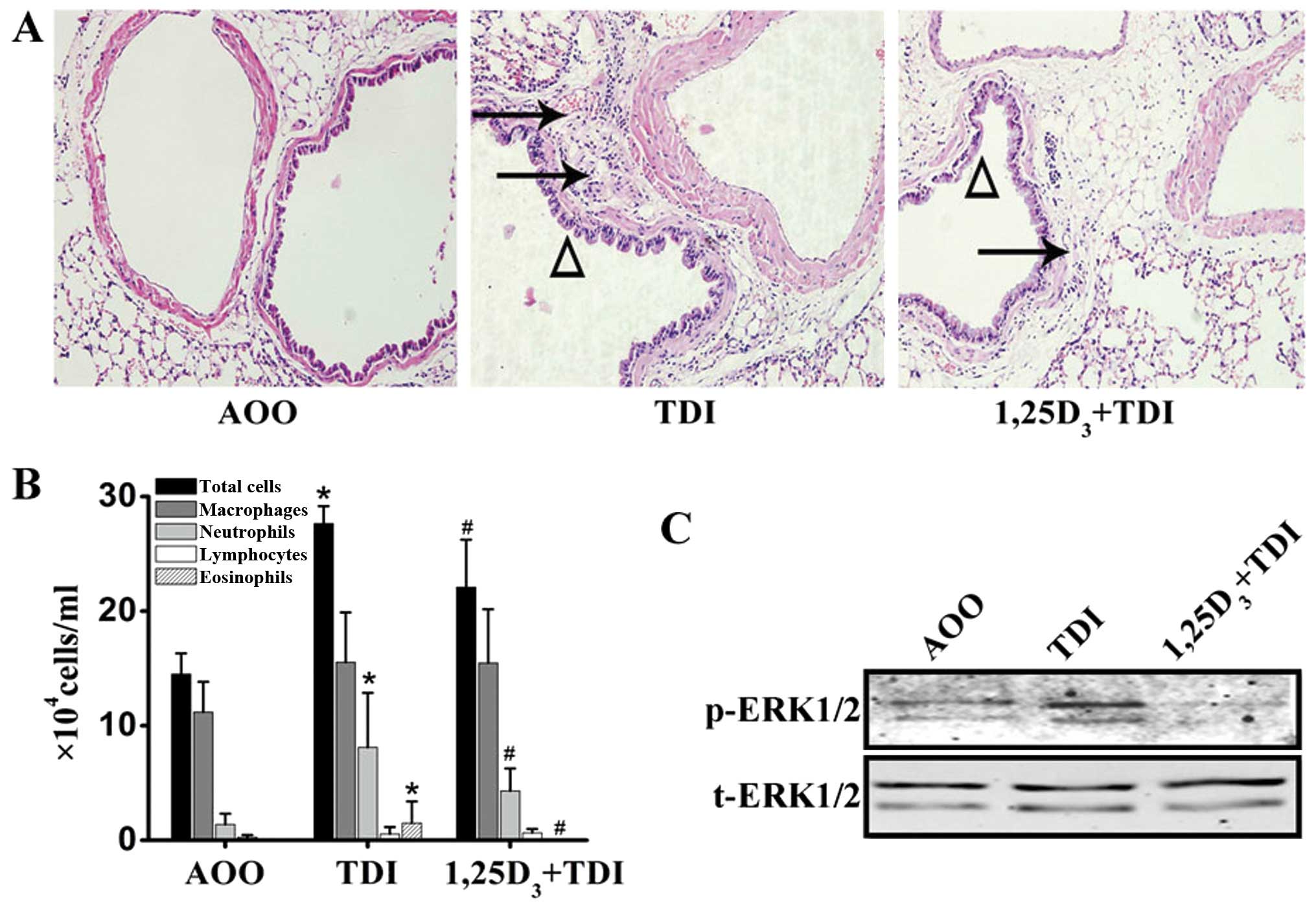
The mice sensitized and challenged with TDI
displayed marked inflammation in the peribronchial regions, as well
as epithelial hyperplasia, as indicated by H&E staining of the
sections (Fig. 2A). The analysis
of the number of total and differential cells in the
bronchoalveolar lavage (BAL) fluid revealed a significant increase
in the number of total inflammatory cells, neutrophils and
eosinophils following challenge with TDI (Fig. 2B). Treatment with
1,25D3 markedly mitigated peribronchial inflammation and
inflammatory cell accumulation into the airway lumen (Fig. 2A and B).
1,25D3 inhibits the
phosphorylation of ERK1/2 in the lungs induced by TDI
The pulmonary expression of p-ERK1/2 was determined
by western blot analysis. There was a marked increase in the
expression levels of activated ERK1/2 following airway challenge
with TDI, which was then significantly inhibited by treatment with
1,25D3 (n=3) (Fig.
2C).
1,25D3 inhibits the
TDI-induced delocalization of E-cadherin and ZO-1 at the epithelial
cell-cell contact sites of the airway mucosa
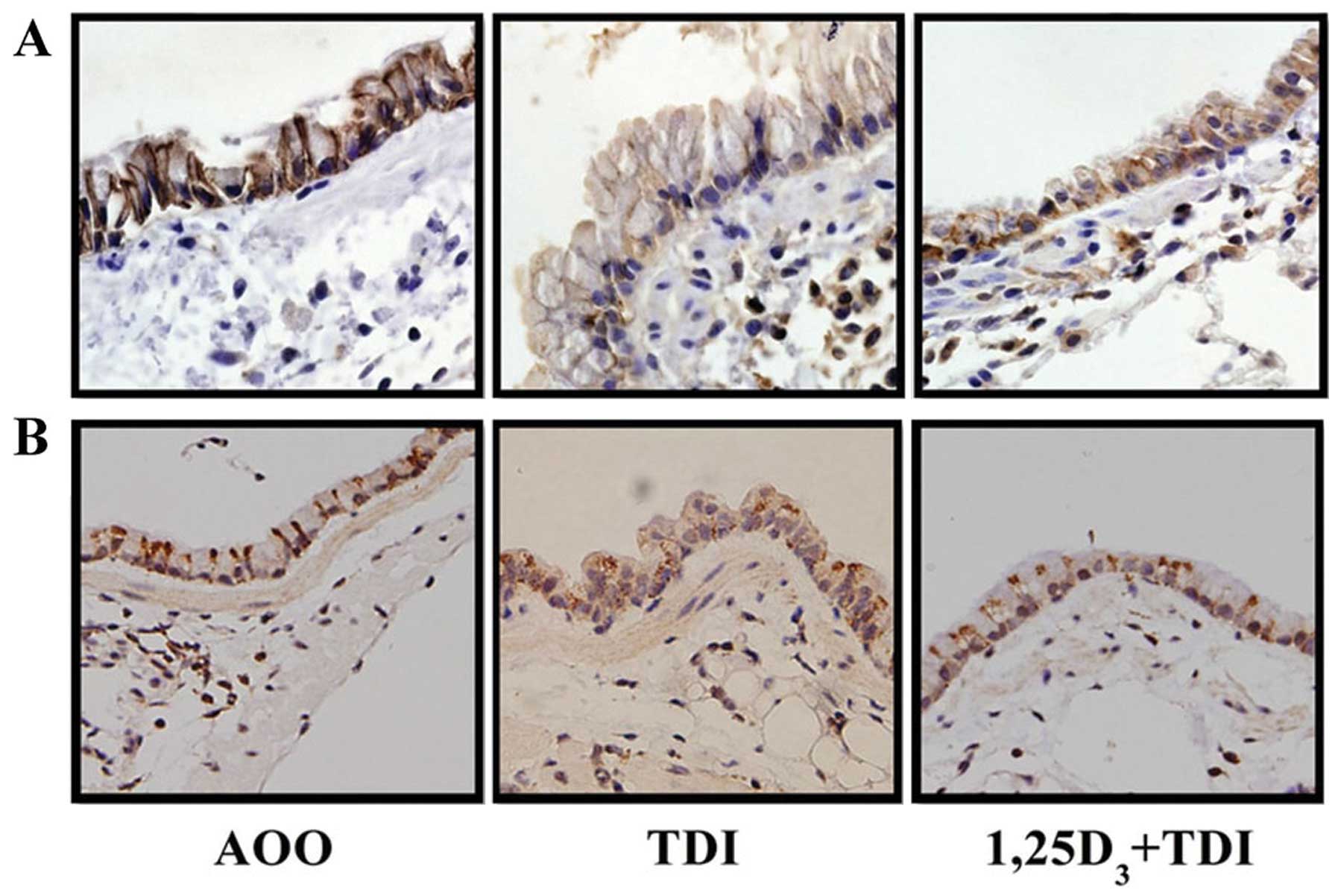
The sections subjected to immunofluorescence
staining showed a strong immunoreactivity of E-cadherin and ZO-1 at
the lateral side and apicolateral border of the airway epithelial
cells, while challenge with TDI resulted in much fainter staining
of the two junction proteins at the epithelial cell-cell contact
sites; these effects were partially reversed by treatment with
1,25D3 (Fig. 3).
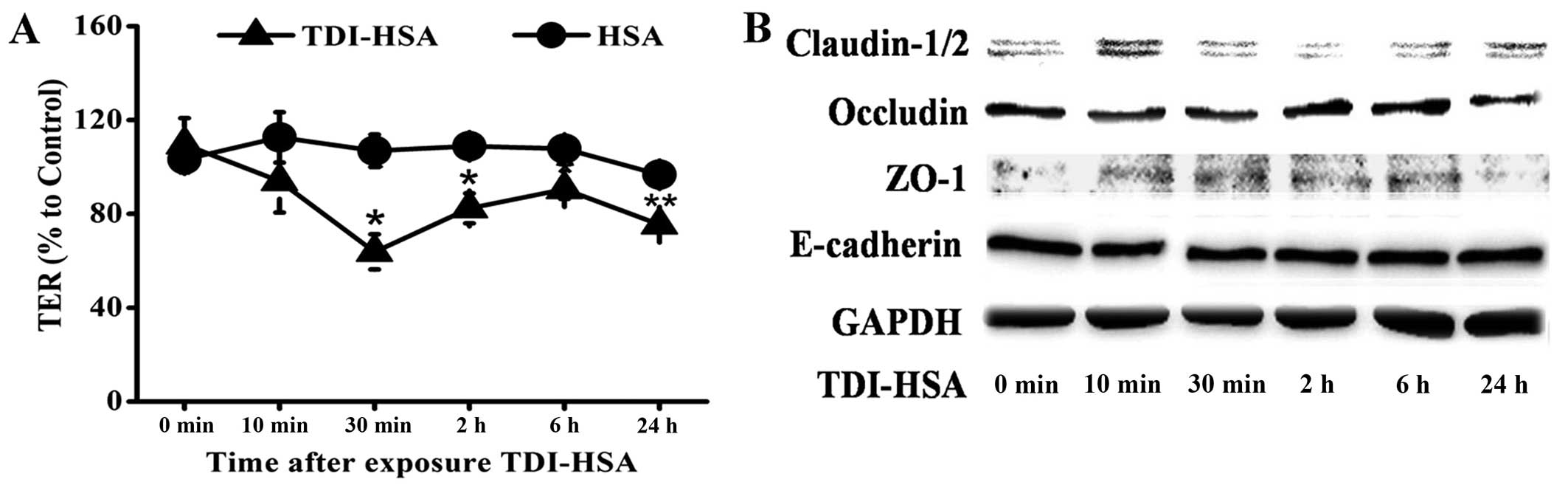
TDI-HSA impairs the barrier integrity of
16HBE cells
Each concentration of TDI-HSA we used had no
detectable effect on cell viability (data not shown). TER was
measured to determine epithelial barrier integrity. TER decreased
immediately following the addition of TDI-HSA (100 µg/ml) to
the culture plates and this decrease lasted for >24 h (Fig. 4A).
We then assessed the total cell lysates by western
blot analysis to determine whether the epxression levels of TJ and
AJ proteins were downregulated. We observed a decrease in the
expression of occludin that was in parallel with the changes
observed in TER. A transient abnormal expression of ZO-1 and
claudin-1/2 was also detected, while the total protein expression
of E-cadherin was relatively unaltered (Fig. 4B).
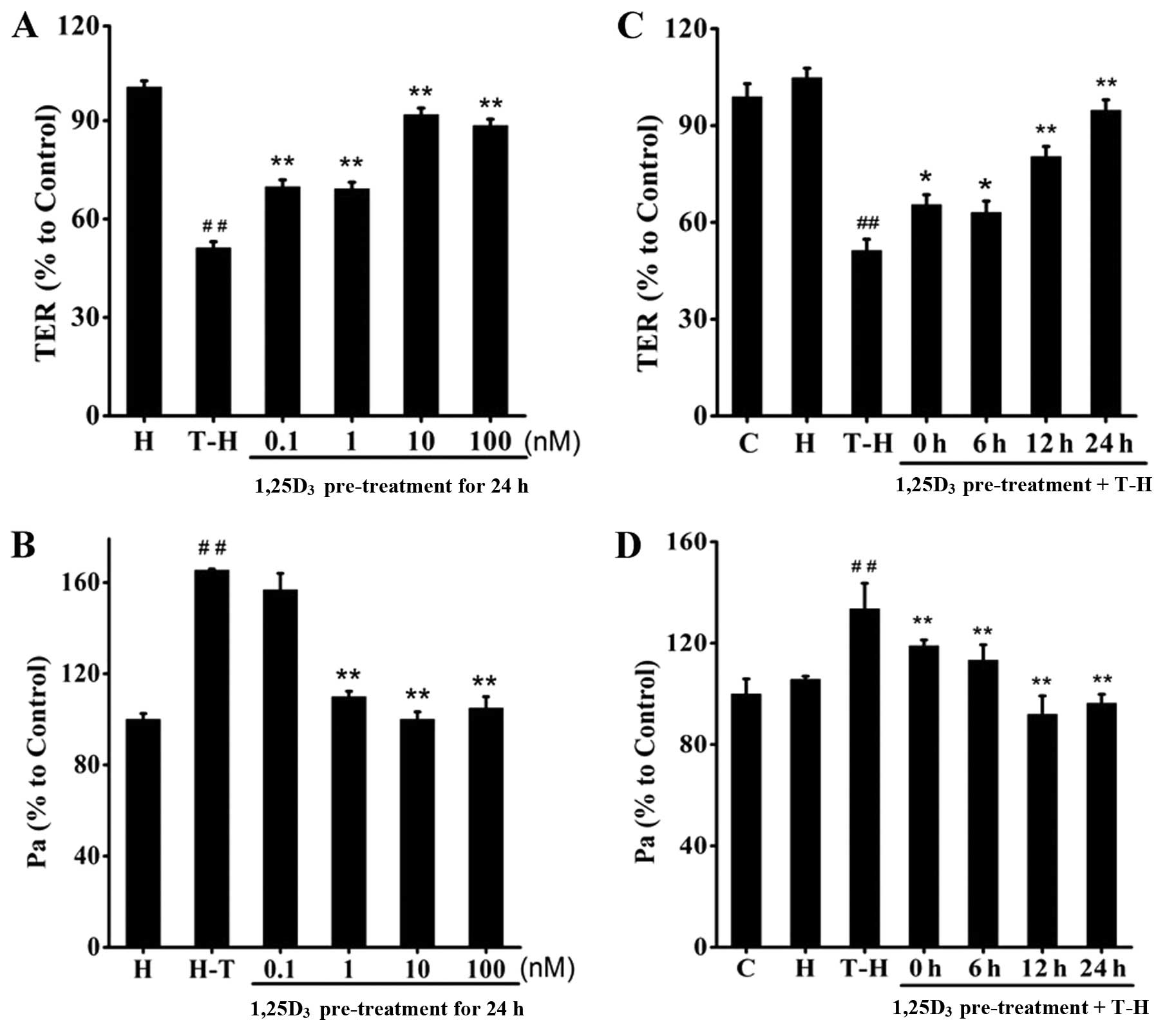
1,25D3 prevents TDI-induced
barrier disruption in 16HBE cells
The TDI-HSA-treated 16HBE cells were pre-treated
with 1,25D3 at the indicated concentrations (0.1–100 nM)
(Fig. 5A and B) for the indicated
periods of time (0, 6, 12 and 24 h) (Fig. 5C and D). We observed that
treatment with 1,25D3 significantly reversed the
decrease in TER and increased FITC-Dx permeability. Treatment with
1,25D3 at the dose of 10 nM for 24 h achieved the most
prominent protective effects.
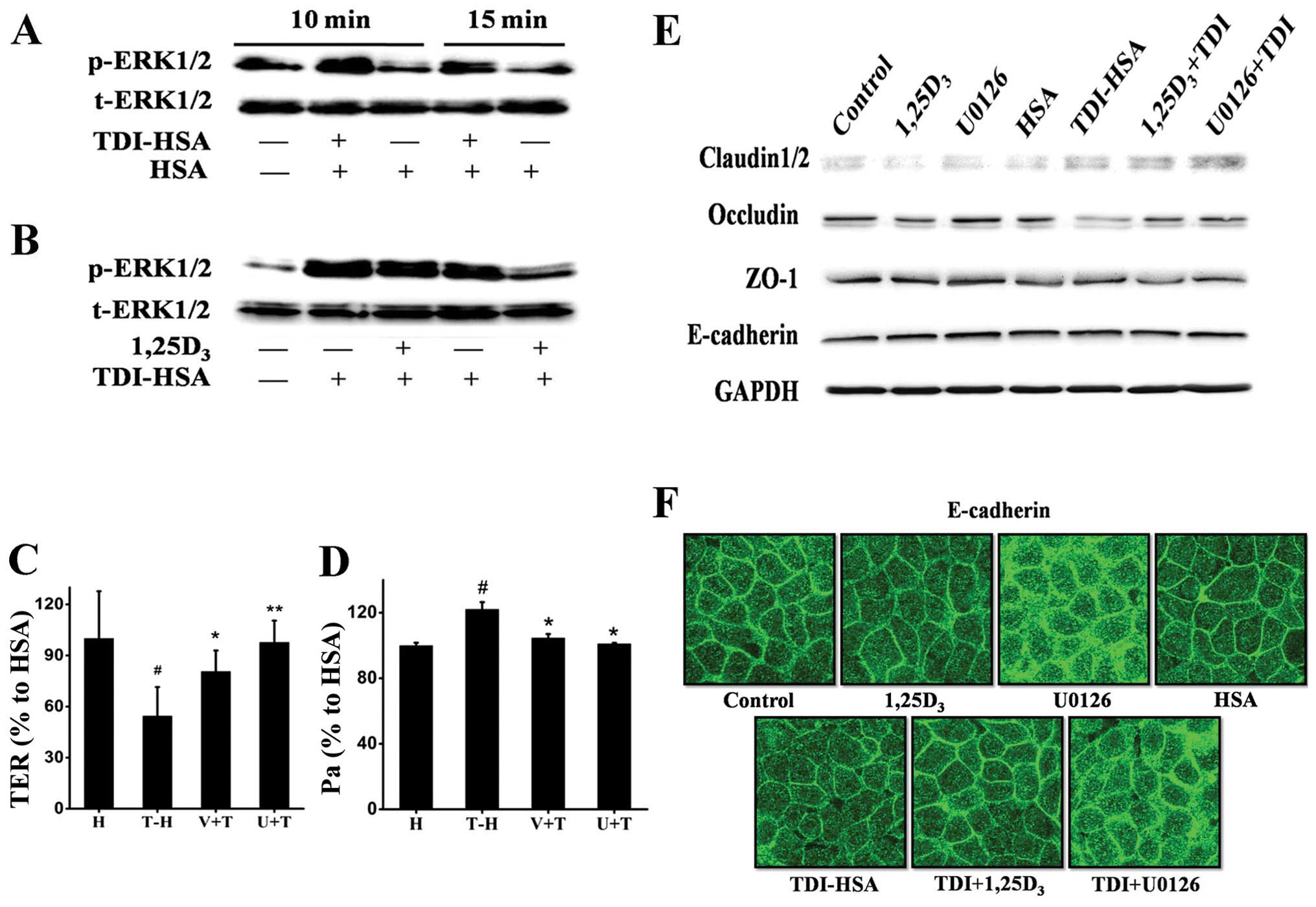
1,25D3 prevents TDI-induced
epithelial barrier disruption and inhibits ERK1/2 phosphorylation
in vitro
In line with the aforementioned results using the
mice with TDI-induced asthma, we observed an increase in the levels
of p-ERK1/2 when the 16HBE cells were stimulated with TDI-HSA
(Fig. 6A); this effect was
reversed by treatment with 1,25D3 (Fig. 6B), suggesting that the ERK1/2
pathway may be involved in this process. Thus, we compared the
effects of 1,25D3 with those of the selective ERK1/2
inhibitor, U0126, on the maintenance of airway epithelial
integrity. Treatment with both 1,25D3 and U0126
significantly increased TER (Fig.
6C), decreased FITC-Dx permeability (Fig. 6D), elevated the protein expression
of occludin and claudin-2 (Fig.
6E), and inhibited the delocalization of E-cadherin (Fig. 6F).
Discussion
To the best of our knowledge, this is the first
study to demonstrate that 1,25D3 alleviates airway
inflammation and prevents epithelial barrier disruption induced by
TDI.
TDI is a small molecular compound widely used in the
production of rigid or flexible polyurethane foam, as well as
hardeners in urethane spray paints and adhesives. It is one of the
most common causes of occupational asthma in many industrialized
areas (14). TDI-induced asthma
is characterized by AHR, Th2-dominated airway inflammation and
granulocytic infiltration (15,16) and is often associated with a poor
prognosis. Airway inflammation tends to persist despite inhaled
steroid medication even after the cessation of the exposure
(17,18). Although there is debate concerning
its etiopathogenesis, in our study, mice with TDI-induced asthma
displayed similar characteristics as those of affected patients:
AHR and inflammatory cell infiltration, higher levels of serum IgE,
as well as an imbalanced Th1/Th2 response. Supplementation with
1,25D3 significantly ameliorated airway reactivity and
the allergic airway inflammation induced by TDI. These results are
in accordance with those of the studies by Gorman et al and
Agrawal et al using a model of ovalbumin (OVA)-induced
asthma (19,20).
Vitamin D deficiency or lower serum
1,25D3 levels have been linked to a higher morbidity due
to asthma, poor disease control, a greater risk of exacerbation and
less sensitive responses to steroids (6,21).
The mechanisms involved remain incompletely understood, although
several have been suggested. Active vitamin D generated in the
pulmonary milieu leads to an increased recruitment of macrophages
(22) and an enhanced production
of cathelicidin (23), therefore
potentiating host defenses against alien microorganisms, gases and
allergens. Vitamin D has also been found to modulate regulatory T
cells (Tregs), an important regulator of asthma pathogenesis
(24). Vitamin D (alone or with
glucocorticoids) has been reported to promote the differentiation
of naive T cells into IL-10-secreting Tregs (25,26). The addition of vitamin D to cell
cultures has been shown to increase the glucocorticoid-induced
secretion of IL-10 by Tregs (27). In human T cells, vitamin D
downregulates dendritic cell OX40 ligand (OX40L), which is required
for Th2 priming, thus resulting in compromised Th2 cytokine release
(28). However, other researchers
have found that vitamin D inhibits the proliferation of
CD4-positive T cells and reduces the production of Th1 cytokines
(29). On the basis of these
observations, it is postulated that the susceptibility to asthma
may be enhanced in individuals with suboptimal levels of vitamin
D.
We have previously reported that TDI damages TJs and
induces E-cadherin delocalization (7,8),
both of which are critical for the maintenance of airway epithelial
integrity, as well as proper immunological responses against
environmental insults (2,30). In agreement with other studies on
the corneal and colon epithelium (11,31), in this study, mice administered
1,25D3 showed a better arrangement of E-cadherin and
ZO-1 at the adjacent epithelial cell-cell contact sites compared
with the vehicle-treated mice challenged with TDI. Subsequent in
vitro experiments using 16HBE cells confirmed the role of
1,25D3 in strengthening airway epithelial barrier
function. Treatment with 1,25D3 reversed the decrease in
TER and the increase in FITC-Dx permeability, which was paralleled
with the relatively unaltered expression of E-cadherin and ZO-1,
but a stronger staining of E-cadherin at the epithelial junctions,
accompanied by upregulated protein levels of occludin and
claudin-1/2. As we measured these protein levels in whole cell
lysates rather than harvesting membrane fractions to determine the
surface labeling of proteins, the paradoxical results of western
blot analysis of claudin-1/2 and occludin did not breach our in
vivo findings. Further studies are required, including more
quantitative measurements of the delocalization of junction
proteins.
The ERK pathway was previously proven to be involved
in the TDI-induced redistribution of E-cadherin by using the ERK
inhibitor, PD98095 (8). This was
verified in the present study using another ERK inhibitor (U0126)
in vitro. Pre-treatment with U0126 not only attenuated
E-cadherin redistribution, but also significantly increased TER and
decreased FITC-Dx permeability, further supporting the notion that
ERK1/2 activation is of great importance in TDI-induced epithelial
barrier disruption. We continued to determine whether the
phosphorylation of ERK is diminished by vitamin D. Intriguingly,
1,25D3 exerted potent suppressive effects on ERK1/2
activation both in vivo and in vitro, indicating that
the inhibition of ERK1/2 activation may be responsible for the
protective effects of vitamin D. Further investigations are
warranted to confirm this hypothesis.
In conclusion, the results from the present study
demonstrate that 1,25(OH)2D3 prevents
TDI-induced airway epithelial barrier disruption, and that the
inhibition of the ERK pathway may be involved in this process.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (81270087, 81270089, 81300029
and 81470228), the National Program on Key Basic Research Project
(973 program, 2012CB518203), the Special Project on the Integration
of Industry, Education and Research of Guangdong (2012B091100153),
and the Science and Technology Program of Guangdong
(2011B031800245).
References
|
1
|
Holgate ST: The airway epithelium is
central to the pathogenesis of asthma. Allergol Int. 57:1–10. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nawijn MC, Hackett TL, Postma DS, van
Oosterhout AJ and Heijink IH: E-cadherin: Gatekeeper of airway
mucosa and allergic sensitization. Trends Immunol. 32:248–255.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Boer WI, Sharma HS, Baelemans SM,
Hoogsteden HC, Lambrecht BN and Braunstahl GJ: Altered expression
of epithelial junctional proteins in atopic asthma: Possible role
in inflammation. Can J Physiol Pharmacol. 86:105–112. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiao C, Puddicombe SM, Field S, et al:
Defective epithelial barrier function in asthma. J Allergy Clin
Immunol. 128:549–556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Post S, Nawijn MC, Hackett TL, Baranowska
M, Gras R, van Oosterhout AJ and Heijink IH: The composition of
house dust mite is critical for mucosal barrier dysfunction and
allergic sensitisation. Thorax. 67:488–495. 2012. View Article : Google Scholar
|
|
6
|
Paul G, Brehm JM, Alcorn JF, Holguín F,
Aujla SJ and Celedón JC: Vitamin D and asthma. Am J Respir Crit
Care Med. 185:124–132. 2012. View Article : Google Scholar :
|
|
7
|
Zhao H, Peng H, Cai SX, Li W, Zou F and
Tong W: Toluene diisocyanate enhances human bronchial epithelial
cells’ permeability partly through the vascular endothelial growth
factor pathway. Clin Exp Allergy. 39:1532–1539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song J, Zhao H, Dong H, Zhang D, Zou M,
Tang H, Liu L, Liang Z, Lv Y, Zou F and Cai S: Mechanism of
E-cadherin redistribution in bronchial airway epithelial cells in a
TDI-induced asthma model. Toxicol Lett. 220:8–14. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Son M, Lee M, Kim YT, Youn JK and Park H:
Heterogeneity of IgE response to TDI-HSA conjugates by ELISA in
toluene diisocyanate (TDI)-induced occupational asthma (OA)
patients. J Korean Med Sci. 13:147–152. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wan H, Winton HL, Soeller C, Stewart GA,
Thompson PJ, Gruenert DC, Cannell MB, Garrod DR and Robinson C:
Tight junction properties of the immortalized human bronchial
epithelial cell lines Calu-3 and 16HBE14o-. Eur Respir J.
15:1058–1068. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao H, Zhang H, Wu H, Li H, Liu L, Guo J,
Li C, Shih DQ and Zhang X: Protective role of 1,25(OH)2 vitamin
D3 in the mucosal injury and epithelial barrier
disruption in DSS-induced acute colitis in mice. BMC Gastroenterol.
12:572012. View Article : Google Scholar
|
|
12
|
Petecchia L, Sabatini F, Varesio L,
Camoirano A, Usai C, Pezzolo A and Rossi GA: Bronchial airway
epithelial cell damage following exposure to cigarette smoke
includes disassembly of tight junction components mediated by the
extracellular signal-regulated kinase 1/2 pathway. Chest.
135:1502–1512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sekiyama A, Gon Y, Terakado M, Takeshita
I, Kozu Y, Maruoka S, Matsumoto K and Hashimoto S: Glucocorticoids
enhance airway epithelial barrier integrity. Int Immunopharmacol.
12:350–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tarlo SM and Lemiere C: Occupational
asthma. N Engl J Med. 370:640–649. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ban M, Morel G, Langonné I, Huguet N,
Pépin E and Binet S: TDI can induce respiratory allergy with
Th2-dominated response in mice. Toxicology. 218:39–47. 2006.
View Article : Google Scholar
|
|
16
|
De Vooght V, Smulders S, Haenen S, Belmans
J, Opdenakker G, Verbeken E, Nemery B, Hoet PH and Vanoirbeek JA:
Neutrophil and eosinophil granulocytes as key players in a mouse
model of chemical-induced asthma. Toxicol Sci. 131:406–418. 2013.
View Article : Google Scholar
|
|
17
|
Piirilä PL, Meuronen A, Majuri ML,
Luukkonen R, Mäntylä T, Wolff HJ, Nordman H, Alenius H and Laitinen
A: Inflammation and functional outcome in diisocyanate-induced
asthma after cessation of exposure. Allergy. 63:583–591. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Talini D, Novelli F, Bacci E, Costa F,
Dente FL, Di Franco A, Malagrinò L, Vagaggini B and Paggiaro P:
Mild improvement in symptoms and pulmonary function in a long-term
follow-up of patients with toluene diisocyanate-induced asthma. Int
Arch Allergy Immunol. 161:189–194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gorman S, Weeden CE, Tan DH, Scott NM,
Hart J, Foong RE, Mok D, Stephens N, Zosky G and Hart PH:
Reversible control by vitamin D of granulocytes and bacteria in the
lungs of mice: An ovalbumin-induced model of allergic airway
disease. PLoS One. 8:e678232013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Agrawal T, Gupta GK and Agrawal DK:
Vitamin D supplementation reduces airway hyperresponsiveness and
allergic airway inflammation in a murine model. Clin Exp Allergy.
43:672–683. 2013.PubMed/NCBI
|
|
21
|
Samrah S, Khatib I, Omari M, Khassawneh B,
Momany S, Daoud A, Malkawi M and Khader Y: Vitamin D deficiency and
level of asthma control in women from North of Jordan: A
case-control study. J Asthma. 51:832–838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Griffin MD, Xing N and Kumar R: Vitamin D
and its analogs as regulators of immune activation and antigen
presentation. Annu Rev Nutr. 23:117–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Herr C, Shaykhiev R and Bals R: The role
of cathelicidin and defensins in pulmonary inflammatory diseases.
Expert Opin Biol Ther. 7:1449–1461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hawrylowicz CM and O’Garra A: Potential
role of interleukin-10-secreting regulatory T cells in allergy and
asthma. Nat Rev Immunol. 5:271–283. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Urry Z, Xystrakis E, Richards DF, McDonald
J, Sattar Z, Cousins DJ, Corrigan CJ, Hickman E, Brown Z and
Hawrylowicz CM: Ligation of TLR9 induced on human IL-10-secreting
Tregs by 1alpha,25-dihydroxyvitamin D3 abrogates
regulatory function. J Clin Invest. 119:387–398. 2009.PubMed/NCBI
|
|
26
|
Barrat FJ, Cua DJ, Boonstra A, Richards
DF, Crain C, Savelkoul HF, de Waal-Malefyt R, Coffman RL,
Hawrylowicz CM and O’Garra A: In vitro generation of interleukin
10-producing regulatory CD4(+) T cells is induced by
immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and
Th2-inducing cytokines. J Exp Med. 195:603–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xystrakis E, Kusumakar S, Boswell S, et
al: Reversing the defective induction of IL-10-secreting regulatory
T cells in glucocorticoid-resistant asthma patients. J Clin Invest.
116:146–155. 2006. View Article : Google Scholar
|
|
28
|
Kreindler JL, Steele C, Nguyen N, et al:
Vitamin D3 attenuates Th2 responses to Aspergillus
fumigatus mounted by CD4+ T cells from cystic fibrosis
patients with allergic bronchopulmonary aspergillosis. J Clin
Invest. 120:3242–3254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mahon BD, Wittke A, Weaver V and Cantorna
MT: The targets of vitamin D depend on the differentiation and
activation status of CD4 positive T cells. J Cell Biochem.
89:922–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Georas SN and Rezaee F: Epithelial barrier
function: At the front line of asthma immunology and allergic
airway inflammation. J Allergy Clin Immunol. 134:509–520. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yin Z, Pintea V, Lin Y, Hammock BD and
Watsky MA: Vitamin D enhances corneal epithelial barrier function.
Invest Ophthalmol Vis Sci. 52:7359–7364. 2011. View Article : Google Scholar : PubMed/NCBI
|