Introduction
Colorectal cancer (CRC) is among the most fatal
forms of solid tumor in men and women worldwide (1), with over 96,000 new cases of colon
cancer and 40,000 new cases of rectal cancer diagnosed annually in
the US (2). While the majority of
CRC cases are sporadic, 15–25% of patients have a family history
(3,4), and 5% are diagnosed with inherited
CRC syndrome (5). A number of
genes have been implicated in the pathogenesis of CRC, such as
tumor-suppressor genes (APC, TP53 and CDKN2A),
proto-oncogenes (KRAS and HRAS) and DNA repair genes
(MUTYH) (6); however,
specific mutations in these genes have not been identified in
numerous CRC patients.
Hereditary colorectal polyposis includes a range of
disorders passed on through autosomal dominant inheritance, and is
divided into Lynch syndrome and familial adenomatous polyposis
(FAP) (7). Lynch syndrome is
characterized by the absence of polyposis (8), a positive family history and high
risk for developing CRC, and a predisposition for extracolonic
malignancies (such as endometrial, ovarian and gastric carcinomas)
(9). Patients suspected of having
Lynch syndrome are first tested for germline mutations in the
mismatch repair gene. Patients without polyposis or polyposis
family history, and not presenting Lynch syndrome should undergo
genetic counseling to determine the cancer risk (1).
Polyposis syndrome is one of the most common
syndromes associated with familial CRC, and is involved with a
number of diseases, including FAP, mutY Homolog (E. coli)
(MUTYH)-associated polyposis (MAP), Peutz-Jeghers syndrome and
juvenile polyposis (10,11). However, the majority of the
familial CRC cases do not present polyposis, and potentially
associated gene mutations are largely unknown (12). Adenomatous polyposis coli (APC), a
key regulator of β-catenin in the Wnt/β-catenin signaling pathway,
has a critical role in several fundamental cell processes,
including cell division and signal transduction, particularly in
tumor suppression (13,14). Several mutations and deletions, as
well as promoter methylation, have been identified in the
APC gene (15). Of
significance, a number of those genetic or epigenetic changes have
also been described in FAP syndrome, and more than two-thirds of
CRC and adenomas have somatic mutations in the APC gene
(16,17). In addition to CRC, mutations of
APC have been reported in other tumors, including cancers in
the liver (4), stomach (5–7),
lung (8), breast (9) and the brain (cerebellar
medulloblas-toma) (10). However,
the role of these types of mutations in the development of tumors
has not been fully elucidated (11–13). The present study reports a novel
mutation on exon 15 of the APC gene in four generations of a
Chinese family with FAP and 200 sporadic cases of adenomatous
polyposis, and furthermore, suggests a potential mechanism by which
this mutation contributes to the pathogenesis of CRC.
Materials and methods
Study population and DNA collection
The members of a four-generational Chinese Han
family with familial adenomatous polyposis (FAP) (Fig. 1), 200 sporadic adenomatous
polyposis cases and 220 normal controls (Table I) were included in this study,
which was conducted at the Second Affiliated Hospital of Harbin
Medical University (Harbin, China). Written informed consent was
obtained from each participant (or guardian for all the
participants <18 years of age) and the study was reviewed and
approved by the Ethics Committee of Harbin Medical University,
consistent with the 1975 Declaration of Helsinki. The medical
history was recorded in detail for all the enrolled participants.
Each patient received physical and enteroscopic examinations.
Genomic DNA was extracted from peripheral blood leukocytes of each
participant using standard protocols.
 | Table IClinical characteristics of the
population used for polymorphism-association analyses. |
Table I
Clinical characteristics of the
population used for polymorphism-association analyses.
| Parameter | Sporadic
adenomatous polyposis | Control |
|---|
| Sample, n | 200 | 220 |
| Male/female, n | 120/80 | 105/115 |
| Age, years | 58.65±12.13 | 59.36±4.21 |
DNA analysis
The exons and splicing sites of the APC and
MUTYH genes were amplified by polymerase chain reaction
(PCR) with the primers (data not shown), and the PCR products were
sequenced using standard protocols (18) for mutational analysis.
Statistical analysis of
disease-associated polymorphisms
Relevant polymorphisms were determined by DNA
sequencing for all the family members. The prevalence of these
polymorphisms among 200 sporadic adenomatous polyposis cases and
220 normal controls was subsequently analyzed. Statistical analyses
were performed using χ2 tests to calculate the odds
ratios and P-values using SPSS software (version 19.0; IBM Corp.,
Armonk, NY, USA).
Multiple sequence alignment and analysis
of protein models
From the NCBI website (http://www.ncbi.nlm.nih.gov/), the APC and MUTYH
protein sequences of various species were obtained and
multiple-sequence alignments of the proteins were conducted using
Vector NTI software (Life Technologies, Grand Island, NY, USA). The
protein structures of the mutant and wild-type proteins were
predicted and analyzed by Swiss-model software (version 3.5)
(19–22), and DNAMAN software (Lynnon Corp.,
Quebec, Canada).
Results
Clinical characteristics
The proband was a 32-year-old male (II:5; Fig. 1), admitted for diarrhea and weight
reduction over the course of one year. The medical history of the
patient revealed similar clinical features for ~5 years, however,
the symptoms were treated without systematic examination for a
diagnosis. Physical examination showed that the abdomen of the
patient was slightly distended and nontender. Blood analysis showed
the presence of a significant anemia; however, there was no
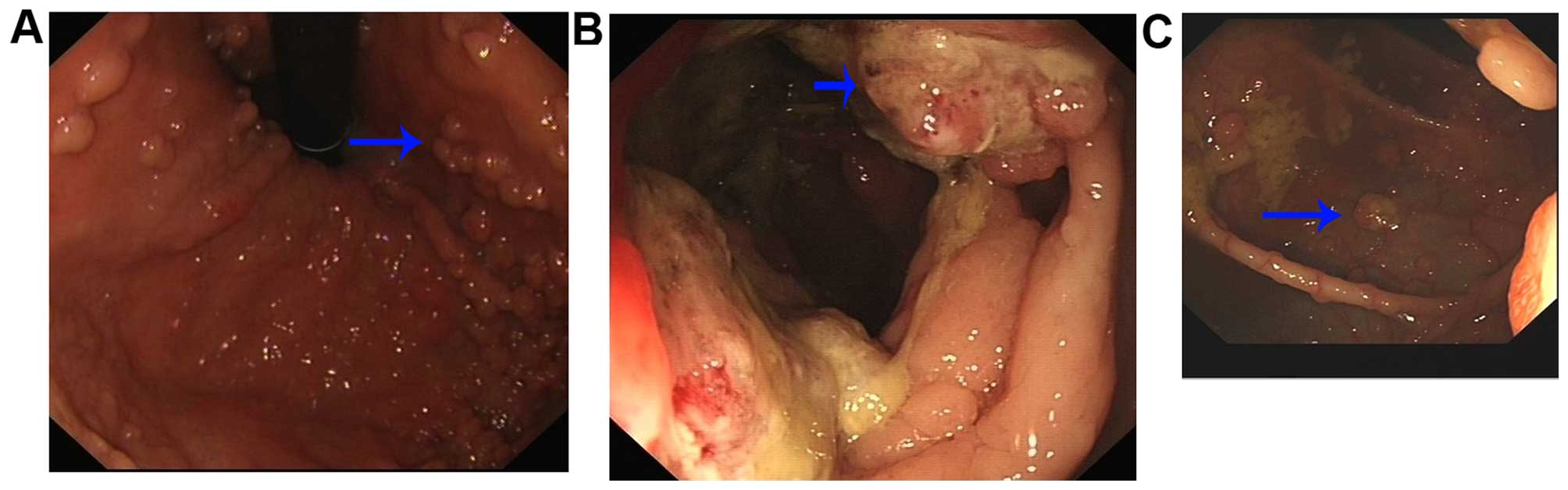
bleeding or clots on rectal examination. Colonoscopy revealed
numerous polyps (>100) measuring between 0.2–2 cm in diameter
along the colon and rectum, confirmed as tubular adenoma with
low-grade dysplasia. A mass was observed in the transverse colon
measuring 3.0×3.0 cm. Biopsy of the polyp revealed typical
adenomatous polyp features, and biopsy of the tumor demonstrated
moderately differentiated adenocarcinoma. Endoscopic examination of
the upper gastrointestinal system showed multiple gastric polyps in
the fundus and upper body. Pathological examination confirmed
fundic gland polyps. A computed tomographic scan also revealed
tumors in the transverse colon; however, there was no evidence of
liver or lymph node metastases.
In this four-generation family, there were 7
affected individuals with FAP syndrome that showed numerous polyps
along the colon and rectum (Fig.
2); 1 affected member with endometrial cancer, 3 with the
APC mutation but no corresponding clinical features, and 42
unaffected individuals (Table
II). All the diagnoses were confirmed by three colorectal
cancer specialists. There was no history of other systemic
abnormalities in the family.
| Table IIClinical features and gene
polymorphism of the affected individuals in the family. |
Table II
Clinical features and gene
polymorphism of the affected individuals in the family.
| Patient
identity | Gender | Age, years | Onset age,
years | Risk factor | Onset symptoms | Location | Age at death,
years | CEA (ng/ml) | CA199 (U/ml) | TNM stage | Pathological
type | APC
131564T>C | MUTYH
1126G>C |
|---|
| I:1 | M | 84 | 82 | Smoking | Bowel
obstruction | Caecum | 84 | 2.13 | 4.65 | IIIa | Adenocarcinoma | / | / |
| I:2 | F | 79 | 75 | Smoking | Hematochezia | Rectum | / | 5.34 | 3.21 | Νo | Adenocarcinoma | T/C | G/C |
| II:1 | M | 58 | 54 | No | Vaginal
bleeding | Uterus | / | / | / | IIa | Endometrioid
carcinoma | T/C | G/G |
| II:3 | M | 55 | 50 | Smoking | Abdominal pain | Rectum | 55 | 4.31 | 10.2 | IIIb | Mucinous
adenocarcinoma | / | / |
| II:5 | M | 52 | 48 | Smoking | Abdominal pain | Ascending
colon | 52 | 6.45 | 5.75 | / | Adenocarcinoma | T/C | G/C |
| II:7 | M | 39 | 22 | Smoking | Abdominal pain | Sigmoid colon | / | 3.46 | 2.47 | / | Intraepithelial
neoplasia | T/C | G/G |
| II:8 | M | 30 | 29 | Smoking | Anemia | Caecum | / | 3.42 | 3.21 | IIIa | Adenocarcinoma | T/C | G/C |
| III:4 | F | 25 | No | No | / | / | / | / | / | / | / | T/C | G/C |
| III:5 | F | 30 | 29 | Smoking | Hematochezia | Rectum | / | 3.45 | 5.87 | IIb | Adenocarcinoma | T/C | G/C |
| III:21 | M | 24 | No | No | / | / | / | / | / | / | / | T/C | G/G |
| III:25 | F | 8 | No | No | / | / | / | / | / | / | / | T/C | G/C |
DNA analysis
Sanger sequencing of the amplified fragments in two
affected family members identified a single base alteration,
131564T>C (Fig. 3), in exon 15
of the APC gene (GI:324) located at 5q21-q22, resulting in
the substitution of Val to Ala at codon 1125 (p.1125Val>Ala),
and a single base alteration, 1126G>C (Fig. 3) in exon 12 of the MUTYH
gene (GI:4595) located at 1p34.1, resulting in the substitution of
Gln to His at codon 324 (p.324Gln>His). The remaining coding
sequence of the two genes showed no other changes.
Further sequence analysis revealed that the
131564T>C alteration in the APC gene was co-segregated
with all the affected individuals in the family (except III:4, 21
and 25; analyzed in the discussion). However, the 1126G>C
alteration in the MUTYH gene was not co-segregated with any
affected individuals in the family (data not shown).
Statistical analysis of the polymorphisms
associated with disease
To further test any possible associations between
the genetic mutations and CRC, polymorphism-association analyses
were conducted and the 131564T>C variation in the APC
gene was clearly associated with the risk of CRC (P=0.018<0.05);
however, there was no statistical significance between the
1126G<C variation in the MUTYH gene and CRC (Tables III and IV). The Hardy-Weinberg equilibrium test
was also conducted for the CRC patients and the control population,
and identified that they were in line with the Hardy-Weinberg
equilibrium.
| Table IIIGenotype and allele frequency of the
131564T>C mutation and 1126G>C variation in 200 Chinese Han
sporadic adenomatous polyposis patients and 220 non-CRC
controls. |
Table III
Genotype and allele frequency of the
131564T>C mutation and 1126G>C variation in 200 Chinese Han
sporadic adenomatous polyposis patients and 220 non-CRC
controls.
| Gene/variation | SAP, frequency
(%) | Control, frequency
(%) |
|---|
|
MUTYH/1126G<C | | |
| Genotype | | |
| G/G | 75 (37.5) | 81 (36.8) |
| G/C | 105 (52.5) | 104 (47.3) |
| C/C | 20 (10.0) | 35 (15.9) |
| Allele | | |
| G | 255 (63.8) | 266 (60.5) |
| C | 145 (36.3) | 174 (39.5) |
|
APC/131564T>C | | |
| Genotype | | |
| T/T | 195 (97.5) | 220 (100.0) |
| T/C | 5 (2.5) | 0 (0.0) |
| C/C | 0 (0.0) | 0 (0.0) |
| Allele | | |
| T | 395 (98.8) | 440 (100.0) |
| C | 5 (1.3) | 0 (0.0) |
| Table IVAssociations with the risk of
sporadic adenomatous polyposis in the Chinese populations with the
131564T>C mutation within APC, but not the 1126G>C
variation in MUTYH. |
Table IV
Associations with the risk of
sporadic adenomatous polyposis in the Chinese populations with the
131564T>C mutation within APC, but not the 1126G>C
variation in MUTYH.
| Gene/variation | Type | Pearson
χ2
| Pearson's R
|
|---|
| Value | Min. counta | df | Asymp. sig.
(2-sided) | Value | Asymp. std.
errorb | Approx. Tc | Approx. sig |
|---|
|
MUTYH/1126G<C | Genotype | 3.382a | 26.19 | 2 | 0.184 | 0.049 | 0.048 | 1.011 | 0.313d |
| Allele | 0.966a | 151.90 | 1 | 0.326 | 0.034 | 0.034 | 0.982 | 0.326d |
|
APC/131564T>C | Genotype | 5.566a | 2.38 | 1 | 0.018 | −0.115 | 0.026 | −2.369 | 0.018d |
| Allele | 5.533a | 2.38 | 1 | 0.019 | −0.081 | 0.018 | −2.357 | 0.019d |
Evolutionary conservation of protein
across the species
APC and MUTYH protein sequences were compared across
multiple species including birds, fish, rodents and primates.
Multiple-sequence alignment analysis showed that with the exception
of Saccoglossus kowalevskii, Danio rerio and
Xenopus laevis, the 1125Val residue in the APC protein was
highly conserved, but the 324Gln residue in the MUTYH protein was
only conserved in rodents and primates (Fig. 4).
3D modeling of protein structure
The secondary structures, hydrophobicity and
hydrophilicity of the wild and mutant proteins were also compared.
The online bioinformatics Swiss-model software (version 3.5) was
used to predict the wild-type and mutant APC protein structure. The
p.1125Val>Ala mutation made minor changes in ~10 amino acids
within the secondary structure. For hydrophilicity, ~700 local
amino acids were changed in the mutant APC protein; however, the
hydrophobicity exhibited fewer changes (data not shown). The
p.1125Val>Ala mutation also exerted a minor effect on the
tertiary structure of the protein (Fig. 5). The degree of change caused by
the p.324Gln>His variation in the MUTYH protein was less than
the changed caused by the p.1125Val>Ala variation to the APC
protein (data not shown).
Discussion
Familial adenomatous polyposis (FAP) is a disease of
autosomal dominant inheritance, with the main clinical
manifestations consisting of multiple adenomatous polyps formed in
the colon and rectum of affected patients (23). Approximately 80% of sporadic
colorectal tumors are caused by mutations in the APC gene
(24), and recently, two
non-conservative mutations, Y165C and G382D, were identified in the
MUTYH gene (25). The
MUTYH gene encodes a DNA glycosylase that is involved in the
repair of oxidative DNA damage. These MUTYH gene mutations
were shown to cause an increased tendency of somatic CG→AT
transversion in the APC gene in several colorectal adenoma
or carcinoma patients (26).
Further research revealed that 7–10% of FAP patients exhibit
MUTYH mutations (27,28), several of which can enhance the
spontaneous mutator phenotype resulting in the accumulation of
8-oxoguanine (oxoG) DNA in response to oxidative stress (29). These two MUTYH mutations, Y165C
and G382D, have been shown to reduce the activity of mutY in
removing A from G:A mismatches in E. coli, and have also
demonstrated a decrease in activity of the human MUTYH
enzyme in the excision of A opposite 8-oxoG, which ultimately led
to the formation of tumors (30,31). This suggests that the MUTYH
gene has an important role in the development of FAP,
APC-associated colorectal tumors and the generation of
APC mutations.
In the present study, a c.131564T>C
(p.Val1125Ala) mutation in the APC gene was identified,
which was co-segregated with disease in a multigenerational family
afflicted with FAP. This mutation was not found in unaffected
family members or in 220 random subjects selected from a normal
population that served as a control group. Notably, 5 patients out
of 200 with sporadic adenomatous polyposis also had the
p.Val1125Ala mutation. Taken together, the frequency of this
APC mutation was high in the sporadic adenomatous polyposis
population, and the mutation was possibly the main cause of disease
in the family studied.
The way in which one amino acid substitution in the
APC protein can lead to the phenotypic changes observed in FAP is
not fully understood. The present results demonstrate that the
p.1125Val>Ala mutation results in minor changes in the secondary
and tertiary structure, as well as hydrophilicity. The region of
the APC protein from codon 1265 to 2035 is the binding site for
β-catenin, and is essential for β-catenin degradation (32,33). Mutations in this functional domain
can cause changes in β-catenin binding, which is believed to have
an important role in the pathogenesis of FAP (34). FAP patients with mutations in this
subunit (particularly beyond codon 1309 or 1444) are more likely to
develop desmoid disease, which is more severe than patients with
mutations in other regions of the APC gene (33,35). Around 21% of patients with desmoid
tumors had APC mutations downstream of the 1444 codon, and
similar mutations were identified in only 4.1% of patients with FAP
(33). In the present study, the
mutation identified in the Chinese Han family was in the 1125 codon
(downstream of the 1444 codon), and the main clinical features of
the patients were adenocarcinoma and numerous polyps along the
colon and rectum. Therefore, this suggests that the 1125 codon is
not as important for the function of the APC protein as codon
1265–2035, which is the binding site for β-catenin (32,33).
A c.1126G>C (p.324Gln>His) variation in the
MUTYH gene was also identified. The majority of CRC patients
with MUTYH mutations exhibit fewer polyps, and in certain
cases have no polyps; and only extremely few cases have >500
polyps (23). However, certain
MUTYH mutation CRC patients may develop secondary cancers in
the skin, ovary, bladder and breast. In the present study, one
patient in the family had endometrial cancer; however, the majority
presented with adenocarcinoma and numerous polyps (>100) along
the colon and rectum. Furthermore, the c.1126G>C
(p.324Gln>His) variation in the MUTYH gene was not
co-segregated with the studied family, and was also not associated
with sporadic adenomatous polyposis disease. Taken together, the
effects of the c.1126G>C (p.324Gln>His) mutation in the
MUTYH gene are negligible with regards to cancer development
in this family.
The present clinical investigation showed that the
affected family members with the T/C-APC mutation were
either of relatively old age or were reported smokers. The average
age of onset of the disease is 48 years, ranging from 21 to 70
years (36). Notably, certain
younger APC-mutation carriers have no clinical features of
the disease, suggesting that age may be a potential risk factor for
FAP. While certain risk factors can increase the chance of
developing colorectal polyps or colorectal cancer, such as
increased age (37) and smoking
(38), the development of CRC
from adenomatous polyps can be prevented through intensive
colorectal screening (39).
In conclusion, a Chinese FAP family with
adenocarcinoma and numerous polyps along the colon and rectum, in
which several gene variants that may have a role in the
pathogenesis of this disease were identified, was reported. The
c.131564T>C (p.Val1125Ala) mutation in the APC gene was
co-segregated with the patients with the disease in this family,
and was also directly associated with sporadic adenomatous
polyposis disease.
Acknowledgments
The authors would like to thank the patients and the
family members for their cooperation and participation in the
present study. The study was supported by a grant from the
Heilongjiang Innovation Research Foundation for Graduate Studies
(no. YJSCX2014-10HYD); grants of the National Natural Science
Foundation of China (nos. NSFC81271786, 81110378, 30970119,
81030029 and 81272706); a grant from the Natural Science Foundation
of Heilongjiang Province (no. QC2013C086); and a grant from the
Science Foundation of Health Department of Heilongjiang Province
(no. 2012-452).
References
|
1
|
Knopperts AP, Nielsen M, Niessen RC, Tops
CM, Jorritsma B, Varkevisser J, Wijnen J, Siezen CL, Heine-Bröring
RC, van Kranen HJ, et al: Contribution of biallelic germline MUTYH
mutations to early-onset and familial colorectal cancer and to low
number of adenomatous polyps: Case-series and literature review.
Fam Cancer. 12:43–50. 2013. View Article : Google Scholar
|
|
2
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lovett E: Family studies in cancer of the
colon and rectum. Br J Surg. 63:13–18. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stephenson BM, Finan PJ, Gascoyne J,
Garbett F, Murday VA and Bishop DT: Frequency of familial
colorectal cancer. Br J Surg. 78:1162–1166. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tops CM, Wijnen JT and Hes FJ:
Introduction to molecular and clinical genetics of colorectal
cancer syndromes. Best Pract Res Clin Gastroenterol. 23:127–146.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Järvinen HJ: Hereditary cancer: Guidelines
in clinical practice. Colorectal cancer genetics. Ann Oncol.
15(Suppl 4): iv127–iv131. 2004.PubMed/NCBI
|
|
7
|
Hampel H, Frankel WL, Martin E, Arnold M,
Khanduja K, Kuebler P, Clendenning M, Sotamaa K, Prior T, Westman
JA, et al: Feasibility of screening for Lynch syndrome among
patients with colorectal cancer. J Clin Oncol. 26:5783–5788. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Umar A, Boland CR, Terdiman JP, Syngal S,
de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ,
Hamelin R, et al: Revised Bethesda Guidelines for hereditary
nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite
instability. J Natl Cancer Inst. 96:261–268. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lynch HT and Lynch JF: What the physician
needs to know about Lynch syndrome: an update. Oncology (Williston
Park). 19:455–464. 466–469. 2005.
|
|
10
|
Farrington SM, Tenesa A, Barnetson R,
Wiltshire A, Prendergast J, Porteous M, Campbell H and Dunlop MG:
Germline susceptibility to colorectal cancer due to base-excision
repair gene defects. Am J Hum Genet. 77:112–119. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cleary SP, Cotterchio M, Jenkins MA, Kim
H, Bristow R, Green R, Haile R, Hopper JL, LeMarchand L, Lindor N,
et al: Germline MutY human homologue mutations and colorectal
cancer: A multisite case-control study. Gastroenterology.
136:1251–1260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aaltonen LA, Salovaara R, Kristo P,
Canzian F, Hemminki A, Peltomäki P, Chadwick RB, Kääriäinen H,
Eskelinen M, Järvinen H, et al: Incidence of hereditary
nonpolyposis colorectal cancer and the feasibility of molecular
screening for the disease. N Engl J Med. 338:1481–1487. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ,
Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P,
et al: Localization of the gene for familial adenomatous polyposis
on chromosome 5. Nature. 328:614–616. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schneikert J and Behrens J: The canonical
Wnt signalling pathway and its APC partner in colon cancer
development. Gut. 56:417–425. 2007. View Article : Google Scholar
|
|
15
|
Hanson CA and Miller JR: Non-traditional
roles for the Adenomatous Polyposis Coli (APC) tumor suppressor
protein. Gene. 361:1–12. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nagase H and Nakamura Y: Mutations of the
APC (adenomatous polyposis coli) gene. Hum Mutat. 2:425–434. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miyoshi Y, Nagase H, Ando H, Horii A,
Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T and Nakamura Y:
Somatic mutations of the APC gene in colorectal tumors: Mutation
cluster region in the APC gene. Hum Mol Genet. 1:229–233. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan ZX, Li FF, Qu YY, Liu J, Liu GR, Zhou
J, Zhu YL and Liu SL: Identification of a known mutation in Notch 3
in familiar CADASIL in China. PLoS One. 7:e365902012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biasini M, Bienert S, Waterhouse A, Arnold
K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli
L, et al: SWISS-MODEL: Modelling protein tertiary and quaternary
structure using evolutionary information. Nucleic Acids Res.
42:W252–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Arnold K, Bordoli L, Kopp J and Schwede T:
The SWISS-MODEL workspace: A web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar
|
|
21
|
Kiefer F, Arnold K, Künzli M, Bordoli L
and Schwede T: The SWISS-MODEL Repository and associated resources.
Nucleic Acids Res. 37:D387–D392. 2009. View Article : Google Scholar :
|
|
22
|
Guex N, Peitsch MC and Schwede T:
Automated comparative protein structure modeling with SWISS-MODEL
and Swiss-PdbViewer: A historical perspective. Electrophoresis.
30(Suppl 1): S162–S173. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barrow P, Khan M, Lalloo F, Evans DG and
Hill J: Systematic review of the impact of registration and
screening on colorectal cancer incidence and mortality in familial
adenomatous polyposis and Lynch syndrome. Br J Surg. 100:1719–1731.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Markkanen E, Dorn J and Hübscher U: MUTYH
DNA glycosylase: The rationale for removing undamaged bases from
the DNA. Front Genet. 4:182013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Al-Tassan N, Chmiel NH, Maynard J, Fleming
N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS,
Sampson JR, et al: Inherited variants of MYH associated with
somatic G:C→T:A mutations in colorectal tumors. Nat Genet.
30:227–232. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jones S, Emmerson P, Maynard J, Best JM,
Jordan S, Williams GT, Sampson JR and Cheadle JP: Biallelic
germline mutations in MYH predispose to multiple colorectal adenoma
and somatic G:C→T:A mutations. Hum Mol Genet. 11:2961–2967. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Filipe B, Baltazar C, Albuquerque C,
Fragoso S, Lage P, Vitoriano I, Mão de Ferro S, Claro I, Rodrigues
P, Fidalgo P, et al: APC or MUTYH mutations account for the
majority of clinically well-characterized families with FAP and
AFAP phenotype and patients with more than 30 adenomas. Clin Genet.
76:242–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pezzi A, Roncucci L, Benatti P, Sassatelli
R, Varesco L, Di Gregorio C, Venesio T, Pedroni M, Maffei S,
Reggiani Bonetti L, et al: Relative role of APC and MUTYH mutations
in the pathogenesis of familial adenomatous polyposis. Scand J
Gastroenterol. 44:1092–1100. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruggieri V, Pin E, Russo MT, Barone F,
Degan P, Sanchez M, Quaia M, Minoprio A, Turco E, Mazzei F, et al:
Loss of MUTYH function in human cells leads to accumulation of
oxidative damage and genetic instability. Oncogene. 32:4500–4508.
2013. View Article : Google Scholar
|
|
30
|
Chmiel NH, Golinelli MP, Francis AW and
David SS: Efficient recognition of substrates and substrate analogs
by the adenine glycosylase MutY requires the C-terminal domain.
Nucleic Acids Res. 29:553–564. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pope MA and David SS: DNA damage
recognition and repair by the murine MutY homologue. DNA Repair
(Amst). 4:91–102. 2005. View Article : Google Scholar
|
|
32
|
Sturt NJ, Gallagher MC, Bassett P, Philp
CR, Neale KF, Tomlinson IP, Silver AR and Phillips RK: Evidence for
genetic predisposition to desmoid tumours in familial adenomatous
polyposis independent of the germline APC mutation. Gut.
53:1832–1836. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schiessling S, Kihm M, Ganschow P, Kadmon
G, Büchler MW and Kadmon M: Desmoid tumour biology in patients with
familial adenomatous polyposis coli. Br J Surg. 100:694–703. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Polakis P: The adenomatous polyposis coli
(APC) tumor suppressor. Biochim Biophys Acta. 1332:F127–F147.
1997.PubMed/NCBI
|
|
35
|
Bertario L, Russo A, Sala P, Eboli M,
Giarola M, D'amico F, Gismondi V, Varesco L, Pierotti MA and Radice
P; Hereditary Colorectal Tumours Registry: Genotype and phenotype
factors as determinants of desmoid tumors in patients with familial
adenomatous polyposis. Int J Cancer. 95:102–107. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ali M, Kim H, Cleary S, Cupples C,
Gallinger S and Bristow R: Characterization of mutant MUTYH
proteins associated with familial colorectal cancer.
Gastroenterology. 135:499–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
U.S. Preventive Services Task Force:
Screening for colorectal cancer: U.S. Preventive Services Task
Force recommendation statement. Ann Intern Med. 149:627–637. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Morrison DS, Batty GD, Kivimaki M, Davey
Smith G, Marmot M and Shipley M: Risk factors for colonic and
rectal cancer mortality: Evidence from 40 years' follow-up in the
Whitehall I study. J Epidemiol Community Health. 65:1053–1058.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Levin B, Smith RA, Feldman GE, Colditz GA,
Fletcher RH, Nadel M, Rothenberger DA, Schroy PS III, Vernon SW and
Wender R; National Colorectal Cancer Roundtable: Promoting early
detection tests for colorectal carcinoma and adenomatous polyps: a
framework for action: the strategic plan of the National Colorectal
Cancer Roundtable. Cancer. 95:1618–1628. 2002. View Article : Google Scholar : PubMed/NCBI
|