Introduction
Autophagy is a conserved eukaryotic cellular
degradative process that is indispensible in cell homeostasis
(1). Autophagy ameliorates
cellular damage during stress conditions by eliminating damaged
cellular machinery, aged organelles and unwanted macromolecules,
and recycling them to produce essential nutrients and energy for
reuse (2,3). On the contrary, failure in any part
of the autophagic machinery leads to the development of a variety
of diseases, such as cancer (4–6),
neurodegenerative diseases (7),
heart diseases (8) and
autoimmunity diseases (9).
Consequently, the identification and/or development of
autophagy-modulating compounds has become a new research goal for
the treatment of these diseases.
The orexin peptides (orexin A and B) and their
receptors (orexin receptor type 1 and 2) are involved in the
regulation of numerous physiological processes, such as
sleep/wakefulness (10),
breathing (11), the reward
system (12) and drug addiction
(12,13). Previous research has reported that
orexin A markedly induces apoptosis, resulting in a reduction in
cell growth in various cancer cell lines. In a previous study,
orexin A promoted the apoptosis of HT29-D4 colon cancer cells, but
not that of normal colonic epithelial cells (14). Upon treatment with orexins,
Chinese hamster ovary (CHO) cells transfected with orexin receptor
type 1 (OX1R) cDNA underwent growth suppression and apoptosis, as
did SK-N-MC neuroblastoma cells transfected with endogenous OX1R
(14). In addition, it has been
observed that OX1R receptors induced cell death via the p38
mitogen-activated protein kinase (MAPK), but independently of p53
and caspase activation in CHO cells (15). In another study, rat C6 glioma
cells expressing both OX1R and orexin receptor type 2 (OX2R) were
treated with orexin A, which suppressed the growth of the cells
through a caspase-dependent mechanism (16). However, programmed cell death is
generally divided into apoptosis, autophagy and necrosis (17). Studies have mainly focused on the
promotion of cell apoptosis by orexin A (14,16); however, to date and to the best of
our knowledge, no study has addressed the theory that orexin A can
induce autophagy in cancer cells. Thus, in the present sudy, we
investigated the effects of orexin A on autophagy in HCT-116 human
colon cancer cells.
The extracellular signal-regulated kinase (ERK)
controls various cellular processes, including autophagy. It has
been shown that growth factor increases the interaction of the ERK
cascade components with autophagy-related (ATG) proteins in both
the cytosol and nucleus. ERK and its upstream kinase, MAPK/ERK
kinase (MEK), localize to the extra-luminal face of autophagosomes,
and lipidation of the autophagic protein, microtubule-associated
protein-1 light chain 3 (LC3) upregulates ERK phosphorylation
(18). The enhanced activity of
ERK1/2 has been implicated in controlling the induction of
autophagy in mammalian cancer cells (19). Based on these previous findings,
in this study, we aimed to determine whether the MEK/ERK pathway
plays a role in orexin A-induced autophagy.
In the present study, we hypothesized that a
membrane-associated pathway is involved in orexin A-induced
autophagy, and to prove this hypothesis, we used HCT-116 human
colon cancer cells as a model. We observed that orexin A induced
autophagy in the HCT-116 cells. Moreover, it was demonstrated that
in the presence of an ERK inhibitor and an autophagy inhibitor, the
orexin A-induced autophagy was inhibited, which indicated that
orexin A induced autophagy through the ERK pathway. The findings of
our study demonsrate a different mechanism responsible for the
activation of the autophagic pathway induced by orexin A.
Materials and methods
Reagents
Orexin A was purchased from Sigma-Aldrich (St.
Louis, MO, USA). RPMI-1640 medium and fetal bovine serum (FBS) were
purchased from Gibco-BRL (Grand Island, NY, USA). Acridine orange
(AO) and the autophagy inhibitor, chloroquine, were obtained from
Sigma-Aldrich. The ERK inhibitor, U0126, was purchased from
Beyotime Biotechnology (Jiangsu, China). Anti-actin (#sc-1616) and
anti-ERK (#sc-292838) antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-Beclin-1 antibody
(#B6061) was purchased from Sigma-Aldrich. Anti-LC3 (#L7543) and
anti-p-ERK (#4370s) antibodies were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
Cell culture
HCT-116 human colon cancer cells were obtained from
the American Type Culture Collection (ATCC, Manassas, VA, USA) and
were maintained in RPMI-1640 medium supplemented with FBS (10%
w/v), L-glutamine (2 mM), penicillin (50 µg/ml) and
streptomycin (100 µg/ml) (Beyotime Biotechnology). The cells
were incubated in a humidified atmosphere containing 5% carbon
dioxide (CO2) at 37°C. The cells were subcultured every
2–3 days to maintain the logarithmic phase of their growth.
Cell viability
The HCT-116 cells were seeded (2×103
cells/well) on a 96-well plate and cultured for 24 h. Following
incubation in serum-free RPMI-1640 medium supplemented with with
orexin A (0, 10−9, 10−8 and 10−7
M) for 24 h, thiazolyl blue tetrazolium blue (MTT) solution (0.5
mg/ml) was added (Sigma-Aldrich). After 3 h, the culture medium was
removed and formazan crystals that had formed were dissolved using
100 µl dimethyl sulfoxide (Merck KGaA, Darmstadt, Germany).
Optical density was measured using a plate reader (SpectraMax Plus
384 Microplate Reader; Molecular Devices, Ismaning, Germany) at 570
and 650 nm (reference wave/length).
Annexin V/propidium iodide (PI) assays
for the measurement of apoptosis
The number of apoptotic cells was quantified using
the Annexin V/PI Apoptosis Detection kit and the cells were
evaluated to determine apoptosis using a BD Accuri™ C6 Flow
Cytometer according to the manufacturer's instructions (BD
Pharmingen, San Diego, CA, USA). The cells were treated with
various concentrations of orexin A in serum-free medium for 48 h.
The cells (1×105) were then washed twice with
phosphate-buffered saline (PBS) and stained with Annexin
V-fluorescein isothiocyanate (FITC; 5 µl) and PI (10
µl) in 500 µl binding buffer for 15 min at room
temperature in the dark. The rate of apoptosis was determined by
counting the number of cells stained by FITC-labeled Annexin V by
fluorescence-activated cell sorting (FACS) analysis. The early
apoptotic cells were identified as PI-negative and FITC-Annexin
V-positive; cells that were in late apoptosis or already dead were
PI- as well as FITC-Annexin V-positive.
Transmission electron microscopy
The cells were treated and collected by
trypsinization, fixed with 2.5% phosphate-buffered glutaraldehyde,
and post-fixed in 1% phosphate-buffered osmium tetroxide. The cells
were embedded, sectioned, double stained with uranyl acetate and
lead citrate, and analyzed using a JEM-1200EX transmission electron
microscope (TEM; JEOL, Tokyo, Japan).
AO staining
AO (0.1 mg/ml) was added to the cells following
treatment with various concentrations of orexin A (0,
10−9, 10−8 and 10−7 M) for 24 h,
or to the untreated controls cells for a period of 20 min in the
dark at 37°C. The cells were washed twice with PBS. The cells were
then examined under a fluorescence microscope (Olympus Optical Co.,
Hamburg, Germany).
Flow cytometric quantification of acidic
vesicular organelles (AVOs)
For the quantification of AVOs, flow cytometry was
used following the staining of the cells with AO. AO is a weak base
that accumulates in the acidic spaces and imparts bright red
fluorescence [punctate staining (dots)] in the cytoplasm, which is
detected under a fluorescence microscope. The intensity of the red
fluorescence is proportional to the degree of acidity. Thus, the
formation of AVOs can be quantified. Briefly, the HCT-116 cells
were harvested following treatment with various concentrations of
orexin A (0, 10−9, 10−8 and 10−7
M) for 24 h. The cell pellet was collected in an Eppendorf tube,
and the cells were resuspended in PBS (1 ml). The staining of the
cells was performed with AO (0.1 mg/ml) for 20 min in the dark at
37°C. The cells were centrifuged at 1,000 rpm for 5 min; the cell
pellet was then rinsed twice with PBS, resuspended in PBS (500
µl), and analyzed by flow cytometry using the PI staining
assay. Cell death was determined by PI staining.
Protein preparations and western blot
analysis
The HCT-116 cells were washed with cold PBS and
harvested in radioimmunoprecipitation assay buffer (Beyotime
Biotechnology) containing protease inhibitors, namely
phenylmethylsulfonyl fluoride (Beijing, Jiangsu, China) and
phosphatase inhibitors (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China). The cell lysates were incubated on ice for 30 min, and were
then collected and centrifuged at 12,000 × g for 10 min at 4°C. The
supernatants were collected and mixed with 5X loading buffer, and
were denatured by boiling for 10 min. The samples were separated by
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto polyvinylidene fluoride membranes
at 60 V for 2.5 h in a transfer buffer containing Tris (20 mM)
(bioWORLD, Dublin, OH, USA), 150 mM glycine (Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) and methanol
(20%) (Liaoning Xinxing Chemical Group Co., Ltd., Liaoning, China).
The membranes were incubated in non-fat dry milk for 120 min at
room temperature, and were washed thrice with Tris-buffered saline
with Tween 20 (TBST) for 30 min. The membranes were incubated with
primary antibodies in TBST overnight at 4°C. The membranes were
then washed and incubated with horseradish peroxidase-conjugated
anti-species secondary antibody (A0208; Beyotime Biotechnology) for
1.5 h at room temperature, and were washed thrice with TBST for 30
min. Proteins were visualized using the BeyoECL plus kit (Beyotime
Biotechnology).
Statistical analysis
Data are expressed as the means ± standard error of
the mean (SEM), and the differences between the means were analyzed
by one-way analysis of variance (ANOVA). A P-value <0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was performed using the Statistical Package
for the Social Sciences (SPSS) 15.0 software package (SPSS, Inc.,
Chicago, IL, USA).
Results
Orexin A inhibits cell viability and
induces apoptosis
The HCT-116 cells were treated with various
concentrations of orexin A (0, 10−7, 10−8 and
10−9 M). The cells were serum-starved for 24 h prior to
exposure to the test compounds in order to avoid interaction with
growth factors and other mediators present in the serum. The
results from the MTT assay revealed that orexin A inhibited cell
growth in a dose-dependent manner. Treatment with orexin A at the
concentrations of 10−8 and 10−7 M resulted in
a significant decrease in the viability of the HCT-116 cells as
compared to the untreated control (P<0.05; Fig. 1A). The results of flow cytometry
indicated a significant increase in apoptosis in the cells treated
with orexin A for 24 h. Thus, the present study demonstrated that
treatment with orexin A (10−7 M) increased the cell
apoptotic rate by 1.5-fold, as compared to the untreated control
(P<0.05; Fig. 1B). These
findings indicate that orexin A inhibits HCT-116 cell viability by
inducing apoptosis.
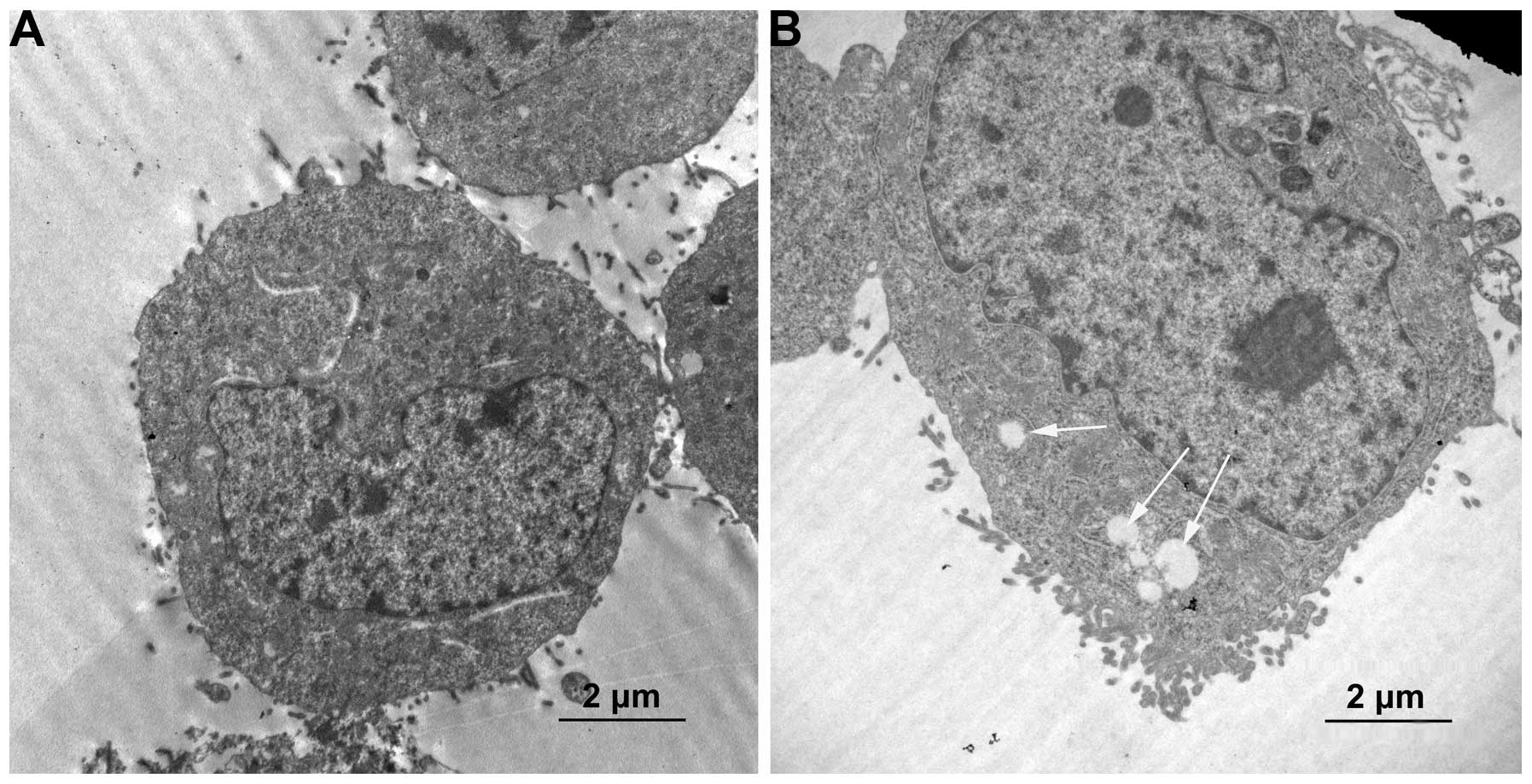
Formation of autophagic vacuoles and AVOs
in HCT-116 human colon cancer cells
To further elucidate the anticancer effects of
orexin A, we examined other cellular responses associated with cell
death following treatment of the cells with orexin A. In contrast
to the untreated control group, numerous microscopic vacuoles were
observed in the HCT-116 cells treated with orexin A
(10−7 M) (Fig. 2).
Since the formation of double-membrane autophagic vacuoles is a
characteristic of autophagy, we focused on examining the
ultrastructural details of the vacuoles using a TEM. As shown in
Fig. 2A, various
membrane-associated vacuoles were observed in the cytoplasm of the
HCT-116 cells treated with orexin A. To further characterize the
membrane-associated vacuoles, AO staining was used to analyze the
formation of AVOs, which is a characteristic of autophagy. Orexin A
induced the formation of orange AVOs in a dose-dependent manner in
the HCT-116 cells, whereas the cells in the untreated control group
primarily exhibited green fluorescence, indicating a lack of AVOs
(Fig. 3A–D). The orexin A-induced
formation of AVOs was quantified by flow cytometry after staining
the cells with AO. The results from the flow cytometry demonstrated
a dose-dependent increase in the percentage of AVOs from 0.8 to 21%
(Fig. 3E–H).
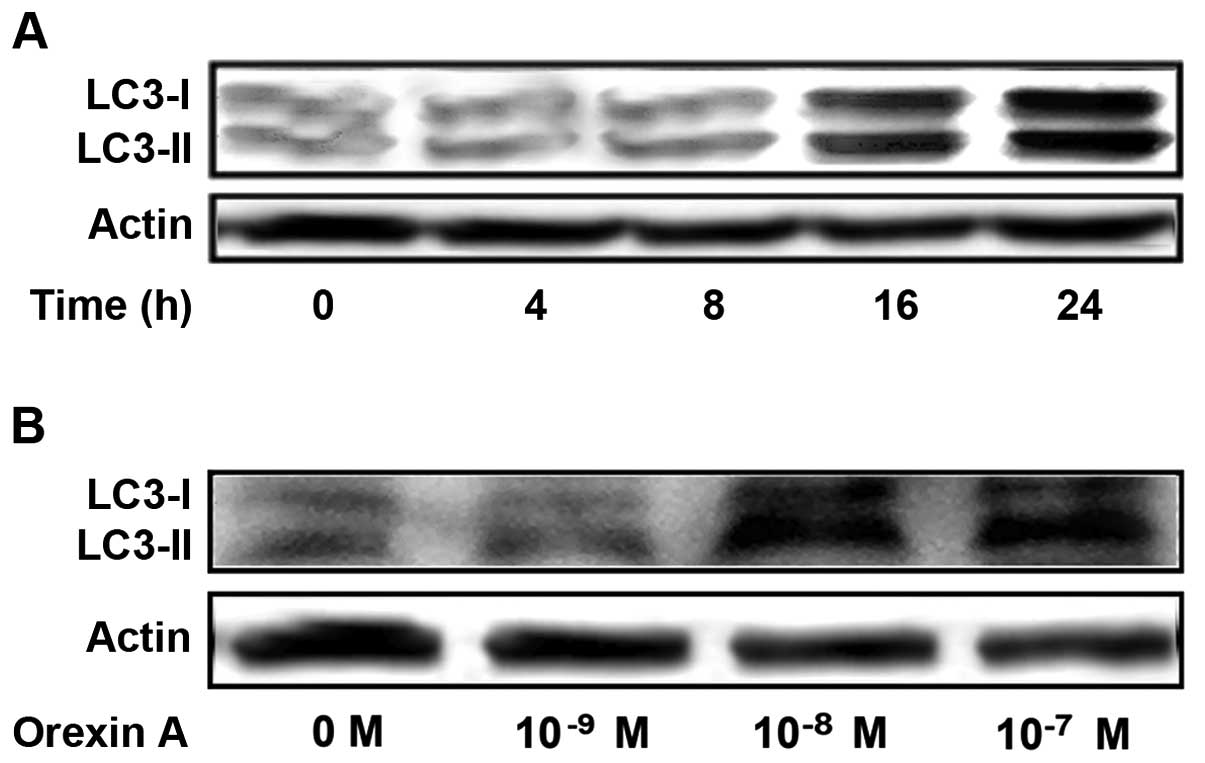
Orexin A induces LC3-II accumulation
We demonstrated that orexin A induced autophagy in
HCT-116 cells, an observation which was based on the
ultrastructural changes in the cells as observed by a TEM and the
visualization of acridine orange-labeled autophagic vacuoles under
a fluorescence microscope. Thus, we further examined the expression
levels of LC3, which exists in cells in two forms: LC3-I and
LC3-II. LC3-I resides in the cytoplasm and conjugates with
phosphatidylethanolamine to form LC3-II, which is closely
associated with autophagosome membranes and serves as a reliable
marker for autophagy. Western blot analysis of the proteins
obtained from the orexin A-treated cells revealed the presence of
two bands. Weak bands corresponding to LC3-I and LC3-II were
observed in the untreated cells. When the cells were treated with
orexin A (10−7 M), the expression levels of LC3-II were
significantly increased in a time-dependent manner (Fig. 4A). The exposure of the HCT-116
cells to orexin A for 24 h also resulted in a
concentration-dependent increase in the LC3-II protein levels
(Fig. 4B).
Orexin A upregulates the expression of
Beclin-1 in the HCT-116 cells
Based on the above-mentioned results, we speculated
that orexin A would induce HCT-116 cell death through the apoptosis
and autophagic pathways simultaneously. To gain better insight into
the molecular mechanisms underlying the autophagy and apoptosis
induced by orexin A, the expression levels of Beclin-1, which plays
a key role in autophagy in HCT-116 cells treated with orexin A,
were measured by western blot analysis. As shown in Fig. 5, orexin A upregulated the
expression of Beclin-1 in a dose- and time-dependent manner. These
findings indicate that the upregulation of Beclin-1 may contribute
to the induction of autophagy caused by orexin A.
Orexin A induces autophagy in HCT-116
cells through the ERK signaling pathway
In order to confirm the involvement of the ERK
signaling pathway in orexin A-indcued autophagy in HCT-116 cells,
the protein expression levels of LC3 and Beclin-1 were measured by
western blot analysis. The results revealed that treatment with
10−7 M orexin A significantly increased the expression
levels of LC3-II and Beclin-1 (Fig.
6). However, this effect was blocked by treatment wiht
10−5 M of the ERK antagonist, U0126, or with 20
µmol/l chloroquine (autophagy inhibitor) (Fig. 6). Moreover, the expression levels
of LC3-II and Beclin-1 were not significantly altered when the
cells were treated with the ERK antagonist, U0126, or chloroquine
without orexin A treatment (Fig.
6). These data suggest that ERK participates in the autophagy
of HCT-116 cells induced by orexin A.
It is well known that the ERK signaling pathway is
involved in cell autophagy and apoptotic signaling; therefore, the
we also wished to investigate whether the treatment of HCT-116
cells with orexin A induces the activation of ERK. Our results
reavealed a 1.5-fold increase in p-ERK protein expression in the
HCT-116 cells treated with 10−7 M orexin A compared to
the untreated control cells (Fig.
6). However, the total ERK (t-ERK) levels remained unaffected
by the aforementioned treatment. Moreover, treatment with
10−5 M of the ERK antagonist, U0126, or with 20
µmol/l chloroquine (autophagy inhibitor) abolished the
relative increase in ERK activation in response to orexin A
independently (Fig. 6).
Discussion
It has previously been demonstrated that orexin A is
involved in a wide range of biological activities (10–13). In particular, orexin A has been
reported to exert anti-proliferative and apoptotic effects on
cancer cells (20,21). Programmed cell death is generally
divided into apoptosis, autophagy and necrosis. Previous studies
have focused on the promotion of cell apoptosis by orexin A
(14,16), and a few have reported on the
promotion of autophagy by orexin A. To the best of our knowlege,
there is no study available to date addressing the theory that
orexin A induces autophagy in cancer cells. In this study, we
investigated the effects of orexin A on autophagy in HCT-116 human
colon cancer cells.
Autophagy can be induced under various
circumstances, including aging, nutrient deprivation, as well as
chemical therapy and radiation cancer therapy (3,22).
Autophagy is a process of catabolism through which macromolecules
and organelles are recycled using lysosomal degradation machinery
(3,23). Autophagy begins with the formation
of double-membraned vesicles known as autophagosomes, which undergo
acidification after maturation and subsequently fuse with lysosomes
to form autophagolysosomes (24).
Autophagy likely plays different roles in the occurrence and
progression of cancer, while it also potentially promotes or
inhibits cell proliferation at different stages of tumor growth
(25). For example, autophagy
plays a protective role in tumor cells via the degradation of
organelles under conditions of nutritional deficiency. Conversely,
autophagy can also inhibit tumor growth via Beclin-1, UV radiation
resistance associated gene (UVRAG), Bif and ATG proteins (26). In the present study, we observed
that orexin A inhibited the proliferation and induced the apoptosis
of HCT-116 cells. Moreover, treatment of the cells with orexin A
induced changes to the cells which are characteristic of autophagy,
such as the formation of cytoplasmic autophagic vacuoles and
autophagosomes. It was evident that AVOs formed following the
exposure of the cells to orexin A, which was shown by AO staining.
The formation of autophagosomes was further confirmed by
transmission electron microscopy. Additionally, the conversion of
LC3-I to LC3-II induced by orexin A was also shown by western blot
analysis, and we also noted that LC3 expression increased in a
dose- and time-dependent manner following treatment with orexin A.
These specific changes of LC3 have been characterized as an
autophagosomal marker in mammalian autophagy. Our results
demonstrated that orexin A induced autophagy in HCT-116 human colon
cancer cells.
We further examined the protein expression levels of
Beclin-1 in the HCT-116 cells. Beclin-1 is a part of the class III
phosphatidylinositol 3-kinase (PI3K) complex, which is essential
for the initiation of the early stages of autophagy. Beclin-1
expression is commonly decreased or absent in various cancer cells,
such as human breast carcinoma, ovarian cancer, brain tumors,
cervical cell carcinoma and gastric cancer (27–31). These findings demonstrate that
Beclin-1 is a critical component of mammalian autophagy. In the
present study, treatment of the HCT-116 cells with orexin A
increased the expression of Beclin-1 in a dose-dependent manner,
indicating that Beclin-1 is involved in orexin A-induced autophagy
and plays an important role in orexin A-mediated anticancer
activities.
ERK is a versatile protein kinase that regulates a
number of cellular functions. It is considered that the magnitude
and duration of ERK1/2 activity determine its cellular function
(32). It has also been reported
that the activation of the ERK1/2 pathway increases the expression
of autophagic genes, which can lead to an increase in the
autophagic flux (33,34). An increase in p-ERK 1/2 and ATG
protein LC3-II expression has also been observed in human glioma
cells treated with interferon β (IFN-β). The autophagy induced by
IFN-β was suppressed when p-ERK1/2 was inhibited by treatment with
U0126 (35). U0126 is a
chemically synthesized organic compound, which inhibits the
catalytic activity of the activating enzyme through non-competitive
binding to MEK, thereby decreasomg the phosphorylation of ERK1/2
(36,37). Certain antitumor drugs induce
autophagy and activate the ERK1/2 signaling pathways in human lung
cancer cells (38). In the
present study, we found that orexin A significantly enhanced the
phosphorylation of ERK, and we also noted that the p-ERK inhibitor,
U0126, reduced the orexin A-induced increase in LC3-II expression.
To further investigate the effects of orexin A on cell autophagy
through the ERK pathway, we used the autophagy inhibitor,
chloroquine as a reference. Chloroquine blocks the process of
autophagy by blocking autophagic bodies fused with lysosomes, and
thus cells cannot be provided with raw materials or energy
(39). Our results revealed that
the ERK inhibitor and autophagy inhibitor significantly inhibited
the promoting effects of orexin A on autophagy, indicating that
orexin A induced autophagy through the ERK pathway.
In conclusion, in the present study, we explored the
antitumor effects and the underlying mechanisms of action of orexin
A on colon cancer cells. Our data lay the experimental foundation
for the better development and clinical application of orexin
A.
Acknowledgments
The authors would like to thank The China Medical
University Affiliated Hospital Laboratory Center for kindly
providing the equipment. This study was supported by the National
Natural Science Foundation of China (grant nos. 81470998, 81071460
and 81271996) and the Natural Science Foundation of Liaoning
Province (grant no. 201202292).
References
|
1
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koneri K, Goi T, Hirono Y, Katayama K and
Yamaguchi A: Beclin 1 gene inhibits tumor growth in colon cancer
cell lines. Anticancer Res. 27:1453–1457. 2007.PubMed/NCBI
|
|
5
|
Mathew R, Karp CM, Beaudoin B, Vuong N,
Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mathew R, Kongara S, Beaudoin B, Karp CM,
Bray K, Degenhardt K, Chen G, Jin S and White E: Autophagy
suppresses tumor progression by limiting chromosomal instability.
Genes Dev. 21:1367–1381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wong E and Cuervo AM: Autophagy gone awry
in neurodegenerative diseases. Nat Neurosci. 13:805–811. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie M, Morales CR, Lavandero S and Hill
JA: Tuning flux: autophagy as a target of heart disease therapy.
Curr Opin Cardiol. 26:216–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Taneike M, Yamaguchi O, Nakai A, Hikoso S,
Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, et al:
Inhibition of autophagy in the heart induces age-related
cardiomyopathy. Autophagy. 6:600–606. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakurai T, Mieda M and Tsujino N: The
orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci.
1200:149–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gestreau C, Bévengut M and Dutschmann M:
The dual role of the orexin/hypocretin system in modulating
wakefulness and respiratory drive. Curr Opin Pulm Med. 14:512–518.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aston-Jones G, Smith RJ, Sartor GC,
Moorman DE, Massi L, Tahsili-Fahadan P and Richardson KA: Lateral
hypothalamic orexin/hypocretin neurons: a role in reward-seeking
and addiction. Brain Res. 1314:74–90. 2010. View Article : Google Scholar
|
|
13
|
Kodadek T and Cai D: Chemistry and biology
of orexin signaling. Mol Biosyst. 6:1366–1375. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rouet-Benzineb P, Rouyer-Fessard C, Jarry
A, Avondo V, Pouzet C, Yanagisawa M, Laboisse C, Laburthe M and
Voisin T: Orexins acting at native OX(1) receptor in colon cancer
and neuroblastoma cells or at recombinant OX(1) receptor suppress
cell growth by inducing apoptosis. J Biol Chem. 279:45875–45886.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ammoun S, Lindholm D, Wootz H, Akerman KE
and Kukkonen JP: G-protein-coupled OX1 orexin/hcrtr-1 hypocretin
receptors induce caspase-dependent and -independent cell death
through p38 mitogen-/stress-activated protein kinase. J Biol Chem.
281:834–842. 2006. View Article : Google Scholar
|
|
16
|
Biegańska K, Sokołowska P, Jöhren O and
Zawilska JB: Orexin A suppresses the growth of rat C6 glioma cells
via a caspase-dependent mechanism. J Mol Neurosci. 48:706–712.
2012. View Article : Google Scholar
|
|
17
|
Quan W, Lim YM and Lee MS: Role of
autophagy in diabetes and endoplasmic reticulum stress of
pancreatic β-cells. Exp Mol Med. 44:81–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Martinez-Lopez N, Athonvarangkul D,
Mishall P, Sahu S and Singh R: Autophagy proteins regulate ERK
phosphorylation. Nat Commun. 4:27992013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ellington AA, Berhow MA and Singletary KW:
Inhibition of Akt signaling and enhanced ERK1/2 activity are
involved in induction of macroautophagy by triterpenoid B-group
soyasaponins in colon cancer cells. Carcinogenesis. 27:298–306.
2006. View Article : Google Scholar
|
|
20
|
Alexandre D, Hautot C, Mehio M, Jeandel L,
Courel M, Voisin T, Couvineau A, Gobet F, Leprince J, Pfister C, et
al: The orexin type 1 receptor is overexpressed in advanced
prostate cancer with a neuroendocrine differentiation, and mediates
apoptosis. Eur J Cancer. 50:2126–2133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Esmaeili-Mahani S, Vazifekhah S,
Pasban-Aliabadi H, Abbasnejad M and Sheibani V: Protective effect
of orexin-A on 6-hydroxydopamine-induced neurotoxicity in SH-SY5Y
human dopaminergic neuroblastoma cells. Neurochem Int. 63:719–725.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Awan MU and Deng Y: Role of autophagy and
its significance in cellular homeostasis. Appl Microbiol
Biotechnol. 98:5319–5328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mizushima N and Komatsu M: Autophagy:
renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maiuri MC, Tasdemir E, Criollo A, Morselli
E, Vicencio JM, Carnuccio R and Kroemer G: Control of autophagy by
oncogenes and tumor suppressor genes. Cell Death Differ. 16:87–93.
2009. View Article : Google Scholar
|
|
25
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Helgason GV, Holyoake TL and Ryan KM: Role
of autophagy in cancer prevention, development and therapy. Essays
Biochem. 55:133–151. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aita VM, Liang XH, Murty VV, Pincus DL, Yu
W, Cayanis E, Kalachikov S, Gilliam TC and Levine B: Cloning and
genomic organization of beclin 1, a candidate tumor suppressor gene
on chromosome 17q21. Genomics. 59:59–65. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miracco C, Cosci E, Oliveri G, Luzi P,
Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M,
Malagnino V, et al: Protein and mRNA expression of autophagy gene
Beclin 1 in human brain tumours. Int J Oncol. 30:429–436.
2007.PubMed/NCBI
|
|
30
|
Wang ZH, Xu L, Duan ZL, Zeng LQ, Yan NH
and Peng ZL: Beclin 1-mediated macroautophagy involves regulation
of caspase-9 expression in cervical cancer HeLa cells. Gynecol
Oncol. 107:107–113. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Furuya D, Tsuji N, Yagihashi A and
Watanabe N: Beclin 1 augmented cisdiamminedichloroplatinum induced
apoptosis via enhancing caspase-9 activity. Exp Cell Res.
307:26–40. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Subramaniam S and Unsicker K: ERK and cell
death: ERK1/2 in neuronal death. FEBS J. 277:22–29. 2010.
View Article : Google Scholar
|
|
33
|
Dai JP, Zhao XF, Zeng J, Wan QY, Yang JC,
Li WZ, Chen XX, Wang GF and Li KS: Drug screening for autophagy
inhibitors based on the dissociation of Beclin1-Bcl2 complex using
BiFC technique and mechanism of eugenol on anti-influenza A virus
activity. PLoS One. 8:e610262013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Niso-Santano M, Criollo A, Malik SA,
Michaud M, Morselli E, Mariño G, Lachkar S, Galluzzi L, Maiuri MC
and Kroemer G: Direct molecular interactions between Beclin 1 and
the canonical NFκB activation pathway. Autophagy. 8:268–270. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li Y, Zhu H, Zeng X, Fan J, Qian X, Wang
S, Wang Z, Sun Y, Wang X, Wang W and Ju D: Suppression of autophagy
enhanced growth inhibition and apoptosis of interferon-β in human
glioma cells. Mol Neurobiol. 47:1000–1010. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Favata MF1, Horiuchi KY, Manos EJ,
Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA,
Hobbs F, et al: Identification of a novel inhibitor of
mitogen-activated protein kinase kinase. J Biol Chem.
273:18623–18632. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ji RR, Befort K, Brenner GJ and Woolf CJ:
ERK MAP kinase activation in superficial spinal cord neurons
induces prodynorphin and NK-1 upregulation and contributes to
persistent inflammatory pain hypersensitivity. J Neurosci.
22:478–485. 2002.PubMed/NCBI
|
|
38
|
Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ and
Chiou HL: Dioscin-induced autophagy mitigates cell apoptosis
through modulation of PI3K/Akt and ERK and JNK signaling pathways
in human lung cancer cell lines. Arch Toxicol. 87:1927–1937. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|