Introduction
Acute myocardial infarction (AMI) is one of the most
severe fatal conditions worldwide (1). AMI is caused by the complete
blockage of the coronary artery which can lead to serious
complications following myocardial necrosis, including malignant
ventricular arrhythmia, heart failure, rupture of the heart and
even sudden cardiac death. Thus, AMI patients remain at a higher
risk of long-term mortality following discharge from hospitals
(2,3). Restoration of the blood supply to
the heart, termed reperfusion, is considered the most effective
treatment for patients with AMI (4,5).
However, reperfusion itself may aggravate the extent of myocardial
injury, which is termed myocardial ischemia-reperfusion injury
(MIRI) (6). The manifestations of
MIRI include myocardial stunning, severe fatal ventricular
arrhythmia, no reflow in coronary artery or even enlargement of
myocardial infarct size (4,7).
Although several methods, including ischemic preconditioning,
ischemic post-conditioning and remote ischemic preconditioning,
have been demonstrated to alleviate MIRI (8-10),
these procedures are limited in clinical practice due to medical
ethics. Therefore, identification of a novel drug to prevent and
mitigate MIRI that is more viable in clinical practice has become
the focus of clinical studies.
The process of MIRI is complex and involves multiple
mechanisms, including oxidative stress, inflammatory response and
apoptosis (11-13). The mitochondrial membrane
permeability transition pore (MPTP) is a transmembrane structure of
mitochondria. MPTP has been identified as a key modulator of MIRI
(14). Several studies have
identified that MPTP remains closed during myocardial ischemia, but
becomes open in the early stage of reperfusion (15). MPTP opening is usually induced by
increased cytosolic and mitochondrial matrix Ca2+
levels, accumulation of reactive oxygen species (ROS) and oxidative
stress (16-18). Blocking the opening of MPTP with
its specific inhibitor (cyclosporin A) prior or subsequent to
ischemia may attenuate MIRI in animals and humans (19,20). Therefore, MPTP is an important
therapeutic target for preventing MIRI.
Morin, or 3,5,7,2′,4′-pentahydroxyflavone, is a
natural flavonol compound (21).
Morin has multiple pharmacological effects, including
antioxidative, anti-inflammatory and anti-apoptotic functions,
scavenging of oxygen free radicals, inhibition of lipid
peroxidation and improving the activities of various oxidases
(22,23). Furthermore, morin is able to
scavenge reactive oxygen species (ROS) and decrease the level of
ROS (23). Therefore, we
hypothesized that morin may protect against MIRI by inhibiting the
MPTP opening. Taken together, the aim of the present study was to
determine the protective effect of morin on MIRI in cultured H9c2
cardiomyocytes and isolated rat hearts, and to additionally explore
the critical role of MPTP in the cardioprotective effects of
morin.
Materials and methods
Animals
A total of 135 healthy male SPF Wistar rats,
weighing 300±20 g, were purchased from Liaoning Changsheng
Biotechnology Co., (Shenyang, China). The rats were housed in a
quiet animal room maintained at 20–25°C with a relative humidity of
50–65%. The rats were provided fresh food and water ad
libitum and a 12-h light/dark cycle. The food was also
purchased from Liaoning Changsheng Biotechnology Co., Ltd. All rats
were treated and used according to the Guide for the Care and Use
of Laboratory Animals (Federal Register Doc. 2011-11490; National
Institutes of Health, Bethesda, MD, USA) (24). The experimental protocol was
approved by the Institutional Ethics Committee of China Medical
University.
Drugs
Morin (CAS, 480-16-0; purity, ≥98%) and
atractyloside (ATR; CAS, 102130-43-8, purity: ≥98%) were purchased
from Nantong Feiyu Biological Technology Co. Ltd. (Nantong, China).
The 2,3,5-triphenyltetrazolium chloride (TTC) was purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Morin and ATR
powder used in the animal experiments were dissolved in 10%
polyethylene glycol 400 (PEG400; Beijing SolarBio Technology Co.,
Ltd., Beijing, China) and normal saline, respectively, to prepare
solutions for intraperitoneal injections. Morin and ATR powders
used in the cell experiments were dissolved in dimethyl sulfoxide
(DMSO; Sigma-Aldrich; Merck KGaA).
Establishment of MIRI model in isolated
rat hearts
Preparation of the heart tissue was performed as
described previously (25). In
brief, rats were anesthetized with intraperitoneal injection of
pentobarbital sodium at a dose of 30 mg/kg. Each rat heart was
rapidly isolated following opening of the chest, and then placed
into an iced heparinized K-H solution (NaCl 0.15 mol/l, KCl 0.006
mol/l, CaCl2 0.002 mol/l, NaHCO3 0.002
mol/l), which was saturated with 100% oxygen. Subsequent to removal
of the redundant connective tissue of the heart, each isolated rat
heart was swiftly connected to the Langendorff apparatus through
the aorta with a 7-0 surgical suture and continuously perfused
using a K-H solution saturated with a mixed gas solution containing
95% O2 and 5% CO2 at the pressure of 75 mmHg
at 37°C. All isolated rat hearts were perfused with the K-H
solution using the perfusion apparatus and stabilized for 10 min
prior to the induction of ischemia, and then subjected to global
ischemia by stopping perfusion for 30 min, followed by the
induction of a 60 min reperfusion to induce MIRI.
Animal experimental protocol
The animal experiment consisted of two phases. In
the first phase, 75 rats were randomly divided into 5 groups with
15 rats/group as follows: i) the ischemia-reperfusion group (IR);
ii) the solvent group (vehicle): Rats were treated with 1.5 ml 10%
PEG400 by intraperitoneal injection once/day for a total of 5 days
prior to surgery; iii) the 10 mg/kg morin pretreatment group
(morin10): Rats were treated with 10 mg/kg morin by intraperitoneal
injection once/day for a total of 5 days prior to surgery; iv) the
20 mg/kg morin pretreatment group (morin20): Rats were treated with
20 mg/kg morin by intraperitoneal injection as aforementioned for
the morin10 group; and v) the 40 mg/kg morin pretreatment group
(morin40): Rats were treated with 40 mg/kg morin by intraperitoneal
injection as aforementioned for the morin10 group.
Based on the results at the first phase, morin at a
dose of 20 mg/kg was used to additionally explore the mechanism of
action. Therefore, in the second phase, 60 rats were randomly
divided into 4 groups with 15 rats per group, as follows: i) The IR
group; ii) the vehicle group; iii) the morin20 group; and iv) the
morin20 combined with ATR group (morin20+ATR): Following treatment
with 20 mg/kg morin the rats were additionally treated with ATR by
intraperitoneal injection 30 min prior to surgery at a dose of 5
mg/kg.
Cell culture
H9c2 cardiomyocytes (Shanghai Institutes for
Biological Sciences, China, Chinese Academy for Sciences, Shanghai,
China) were cultured in Low Glucose Dulbecco’s Modified Eagle’s
Medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal bovine serum (FBS; TBD, Tianjin, China) and 1%
penicillin/streptomycin (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) under humid conditions in incubators with 5%
CO2 at 37°C.
Cell experimental protocol
The H9c2 cells were subjected to
hypoxia/reoxygenation to mimic the MIRI model in vitro.
Specifically, cells were treated when the confluence of cells was
80-90%. The culture medium was removed and changed to Earle’s
medium (CaCl2 0.18 mmol/l, MgSO4,
7H2O 0.08 mmol/l, KCl 0.05 mmol/l, NaCl 11.43 mmol/l,
NaHCO3 2.62 mmol/l and NaH2PO4
0.10 mmol/l) without glucose and FBS, and cells were cultured in a
tri-gas incubator with 90% N2, 5% CO2 and 5%
O2 at 37°C for 12 h to induce hypoxia. Subsequently,
Earle’s medium was removed, and the cells were cultured for 1 h
with normal medium in incubators with 5% CO2 at 37°C to
re-oxygenate the samples.
Similar to the aforementioned animal experimental
protocol, cells were divided into six groups during the first phase
as follows: i) The control group (control): Cardiomyocytes were
cultured under normal conditions for 13 h; ii) the
hypoxia/reoxygenation group (HR): Cardiomyocytes were subjected to
hypoxia for 12 h followed by reoxygenation for 1 h; iii) the
solvent group (vehicle): Cardiomyocytes were pretreated with 0.1%
DMSO for 12 h followed by hypoxia/reoxygenation as described above
for the HR group; iv) the 12.5 µM morin pretreatment group
(morin12.5): Cardiomyocytes were pretreated with morin at a dose of
12.5 µM for 12 h, followed by hypoxia/reoxygenation as
described above for the HR group; v) the 25 µM morin
pretreatment group (morin25): Cardiomyocytes were pretreated with
morin at a dose of 25 µM for 12 h, followed by
hypoxia/reoxygenation as aforementioned for the HR group; and vi)
the 50 µM morin pretreatment group (morin50): Cardiomyocytes
were pretreated with morin at a dose of 50 µM for 12 h
followed by hypoxia/reoxygenation as aforementioned for the HR
group.
According to the result of the first phase, morin at
a dose of 25 µM was used to additionally explore the
mechanism of action. Cells were subsequently divided into four
groups in the second phase, as follows: i) The control group; ii)
the HR group; iii) the morin25 group; and iv) the 25 µM
morin combined with 20 µM ATR pretreatment group
(morin25+ATR): Cardiomyocytes were pretreated with morin at a dose
of 25 µM and ATR at a dose of 20 µM for 12 h followed
by hypoxia/reoxygenation as aforementioned for the HR group.
Cardiac function monitoring
Cardiac function parameters, including heart rate
and coronary flow, were measured at 10 min of stabilization, and at
30 and 60 min following reperfusion.
Hematoxylin & eosin (HE)
staining
The hearts were removed from the perfusion apparatus
at the end of reperfusion. The heart tissues were fixed in 4%
paraformaldehyde at room temperature for 24~72 h. Then, the tissues
were washed with flowing water for 4 h, and immersed in 70% ethanol
for 2 h, 80% ethanol overnight, 90% ethanol for 2 h, anhydrous
ethanol I for 1 h, and anhydrous ethanol II for 1 h at room
temperature to be dehydrated. Following dehydration the tissues
were paraffin embedded and the paraffin blocks cut into
5-µm-thick paraffin sections. The paraffin sections were
immersed in xylene I for 15 min, xylene II for 15 min, absolute
ethanol I for 5 min, absolute ethanol II for 5 min, 95% ethanol for
2 min, 85% ethanol for 2 min, 75% ethanol for 2 min, and distilled
water for 2 min at room temperature to be de-waxed and rehydrated.
They were then immersed in hematoxylin solution for 5 min, 1%
hydrochloric acid alcohol for 3 sec, and eosin solution for 3 min
at room temperature for staining. The pathological changes in heart
tissues were observed under a light microscope and images were
captured at magnification, ×400.
Measurement of the infarct size
The myocardial infarct size was measured by TTC
staining. The hearts were harvested as aforementioned, and the
heart samples were incubated at −80°C for 30 min. The frozen hearts
were cut into 1-2 mm thick sections from the apex to the bottom of
the hearts and incubated in a 1% TTC solution at 37°C for 30 min.
Subsequently, heart slices were washed with 1X PBS and fixed in 4%
paraformaldehyde at room temperature overnight. Images of the
stained slices were captured using a digital camera and analyzed
using Image J2x analysis software (National Institutes of Health).
The severity of the myocardial infarction was indicated by the
ratio of the infarct size to the total size.
Measurement of cell viability
Cell viability was determined by CCK-8 assay using
the Cell Counting Kit-8 (Dojindo Molecular Technologies, Kumamoto,
Japan) according to the manufacturer’s protocol. The absorbance
value was detected at a wavelength of 450 nm by ultramicro
microporous plate spectrophotometer (Biotek Instruments, Inc.,
Winooski, UT, USA).
Measurement of lactate dehydrogenase
(LDH) activity
LDH released from cardiomyocytes into the culture
medium was determined using the LDH assay kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
manufacturer’s protocol. The absorbance value was detected at a
wavelength of 450 nm by ultramicro microporous plate
spectrophotometer (Biotek Instuments, Inc.).
Measurement of apoptosis
Myocardial apoptosis was detected by terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
using and In Situ Cell Death Detection kit (Roche
Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s
protocol. The apoptotic cells were observed under a light
microscope and ≥3 fields of view at lease were captured at ×400
magnification. Image-Pro Plus version 6.0 (Media Cybernetics, Inc.,
Rockville, MD, USA) was used for cell counting, and SPSS version
17.0 (SPSS, Inc., Chicago, IL, USA) used for data analysis.
Cell apoptosis was determined by flow cytometry
analysis (LSRFortessa, BD Biosciences, Franklin Lakes, NJ, USA)
using the Annexin V-Fluorescein isothiocyanate (FITC)/Propidium
Iodide (PI) kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China)
to label the cells according to the manufacturer’s protocol. SPSS
version 17.0 (SPSS, Inc.) was used for data analysis.
Sensitivity of MPTP to calcium
The mitochondria were isolated from the heart
tissues using the Mitochondrial Extract kit (BestBio Co., Shanghai,
China) according to the manufacturer’s protocol. The sensitivity of
MPTP to calcium was determined using the Purified Mitochondrial
Membrane Pore Channel Colorimetric Assay kit (Shanghai Genmed
Pharmaceutical Technology Co., Ltd., Shanghai, China) according to
the manufacturer’s protocol.
Measurement of mitochondrial membrane
potential (ΔΨm)
The change in ΔΨm was determined using the
Mitochondrial Membrane Potential Assay kit with
5,5′,6,6′-Tetrachloro-1,1′, 3,3′-tetraethyl-imidacarbocyanine
iodide (JC-1; Beyotime Institute of Biotechnology, Haimen, China)
according to the manufacturer’s protocol. JC-1 is an ideal
fluorescent probe widely used for the detection of ΔΨm. When ΔΨm is
high, JC-1 accumulates in the matrix of mitochondria and forms
J-aggregates that produce red fluorescence. However, when ΔΨm
decreases, JC-1 cannot aggregate in the matrix of the mitochondria
and maintains a monomer that produces green fluorescence. The
fluorescence was detected under a fluorescence microscope and
images were captured at ×400 magnification. Image-Pro Plus version
6.0 (Media Cybernetics, Inc.) was used for cell counting.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from myocardium and
cardiomyocytes using TRIzol® reagent (Life Technologies;
Thermo Fisher Scientific, Inc.). RNA was transcribed into cDNA
using PrimeScript RT reagent kit with gDNA Eraser (Takara Bio,
Inc., Otsu Japan) according to the manufacturer’s protocol. The RNA
was heated at 37°C for 15 min and at 85°C for 5 sec, and then
cooled at 4°C to obtain cDNA. SYBR Premix Ex Taq II (Takara Bio,
Inc.) was used for qPCR amplification, according to the
manufacturer’s protocol. The cDNA was amplified under the following
four segments: Segment 1: 95°C for 5 min; segment 2: 8 cycles of
95°C for 30 sec, 60°C for 45 sec, 72°C for 20 sec; segment 3: 35
cycles of 95°C for 30 sec, 56°C for 45 sec, 72°C for 20 sec;
segment 4: 95°C for 1 min, 55°C for 30 sec, 95°C for 30 sec. All
oligonucleotide primer pairs and the reference primers (β-actin)
were designed by Sangon Biotech Co., Ltd. (Shanghai, China)
(Table I). Relative gene
expression was analyzed using the 2ΔΔCq method (26).
 | Table IPrimer sequences in the present
study. |
Table I
Primer sequences in the present
study.
| Gene | Primer sequences
(5′-3′) |
|---|
| Bax forward |
GGCGATGAACTGGACAACAA |
| Bax reverse |
CAGTTGAAGTTGCCGTCTGC |
| Bcl-2 forward |
CACGGTGGTGGAGGAACTCT |
| Bcl-2 reverse |
TCCACAGAGCGATGTTGTCC |
| Cytochrome c
forward |
AGGGTGTCGCCTCAAACCTA |
| Cytochrome c
reverse |
ACTGAAGCACGGGTGAGTCT |
| APAF-1 forward |
CAAGGACACAGACGGTGGAA |
| APAF-1 reverse |
TGAATCGCACTGACCAGCTT |
| Caspase-9
forward |
CAGGTGGAGGTCAGGTGTGA |
| Caspase-9
reverse |
TCCGTGAGAGAGGATGACCA |
| Caspase-3
forward |
CCATCCTTCAGTGGTGGACA |
| Caspase-3
reverse |
TTGAGGCTGCTGCATAATCG |
Western blot analysis
The myocardium and cardiomyocytes were homogenized
with radioimmunoprecipitation assay lysis buffer (Beyotime
Institute of Biotechnology). Following 15 min of centrifugation of
13,000 × g at 4°C, the supernatants containing proteins were
collected. Protein concentrations were measured using the BCA
protein concentration kit (Beyotime Institute of Biotechnology).
Subsequently, 30 µg protein was denatured at 100°C for 10
min, and then SDS-PAGE (12% separation gel and 5% stacking gel)
electrophoresis was performed for protein separation, and then the
proteins were transferred to a polyvinylidene fluoride (PVDF)
membrane. The PVDF membrane was blocked with 5% skim milk for 1 h
at room temperature, and then incubated with specific antibodies,
including anti-B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax;
cat. no. WL01637; 1:1,000; Shenyang Wan Biotechnology Co., Ltd.,
Shenyang, China), anti-Bcl-2 (cat. no. WL01556; 1:1,000; Shenyang
Wan Biotechnology Co., Ltd.), anti-cytochrome c (cat. no.
ab90529; 1:1,000; Abcam, Cambridge, UK), anti-apoptotic protease
activating factor-1 (APAF-1; cat. no. ab2001; 1:1,000; Abcam),
anti-cleaved caspase-9 (cat. no. 40503-1; 1:1,000; Signalway
Anitbody LLC, College Park, MD USA), anti-cleaved caspase-3 (cat.
no. ab2302; 1:1,000; Abcam) and anti-β-actin (cat. no. TA-09;
1:1,000; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.,
Beijing, China), at 4°C overnight. The following day, the PVDF
membrane was incubated with horseradish peroxidase labeled goat
anti-rabbit immunoglobulin G (cat. no. E030120-01; 1:4,000; EarthOx
Life Sciences, Millbrae, CA, USA) or goat anti-mouse immunoglobulin
G (cat. no. E030110-01; 1:4,000; EarthOx Life Sciences) at room
temperature for 30 min. Detection of protein bands was performed
using an enhanced chemiluminescence for western blotting kit
(Beyotime Institute of Biotechnology) according to the
manufacturer’s protocol. Relative densitometry was calculated using
Image J2x analysis software (National Institutes of Health).
Statistical analysis
All data were presented as the mean ± standard
deviation and analyzed using SPSS software version 17.0 (SPSS,
Inc., Chicago, IL, USA). Differences among ≥ three groups were
firstly evaluated using one-way analysis of variance, and if the
differences were significant, multiple comparison analysis was
performed using Fisher’s Least Significant Difference test.
Differences between two groups were evaluated using independent
sample t-test. P<0.05 were considered to indicate a
statistically significant difference.
Results
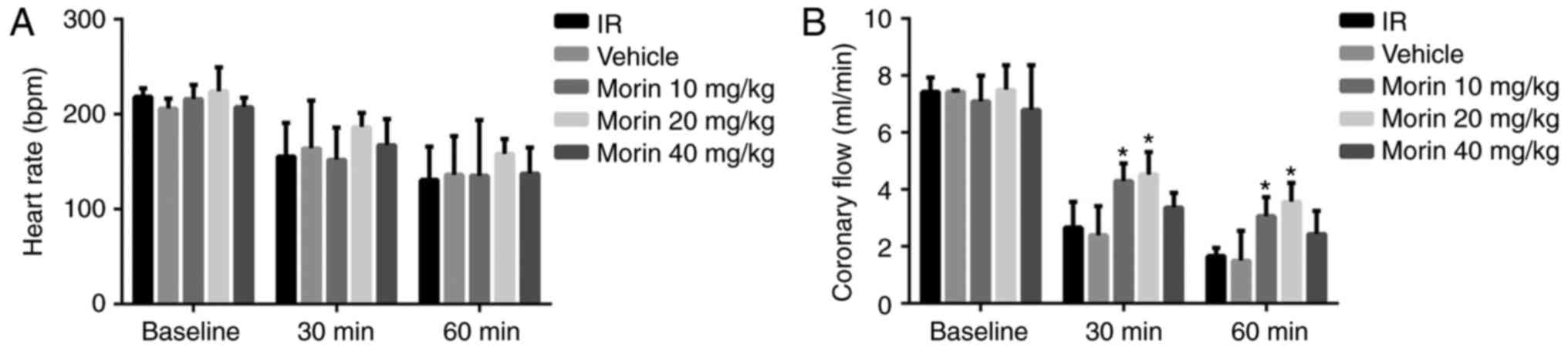
Effect of morin on cardiac function
No difference was observed in the heart rate across
all the groups (Fig. 1A).
Coronary circulation at 30 and 60 min of reperfusion in the morin10
and morin20 groups was significantly increased compared with the IR
group (Fig. 1B).
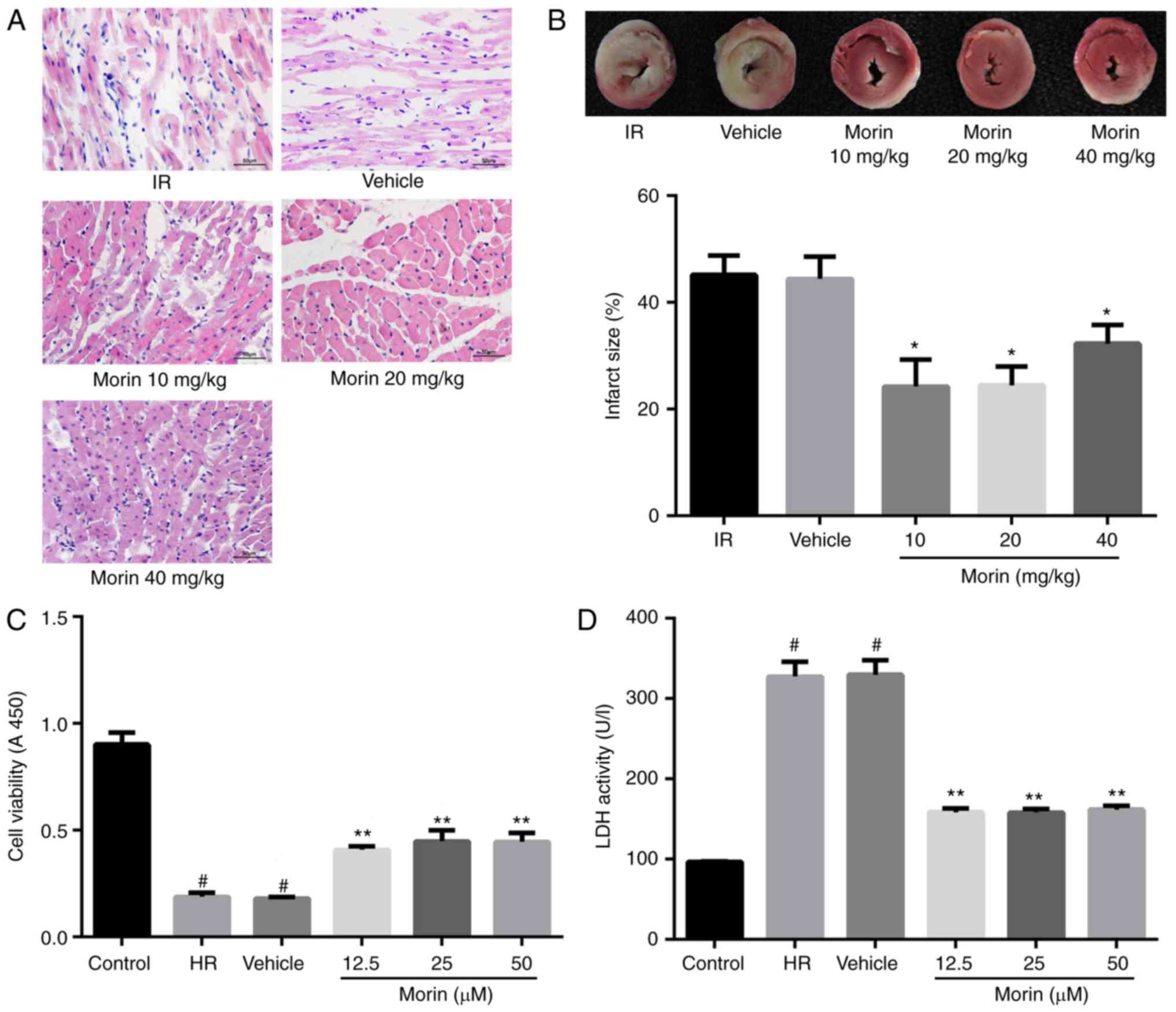
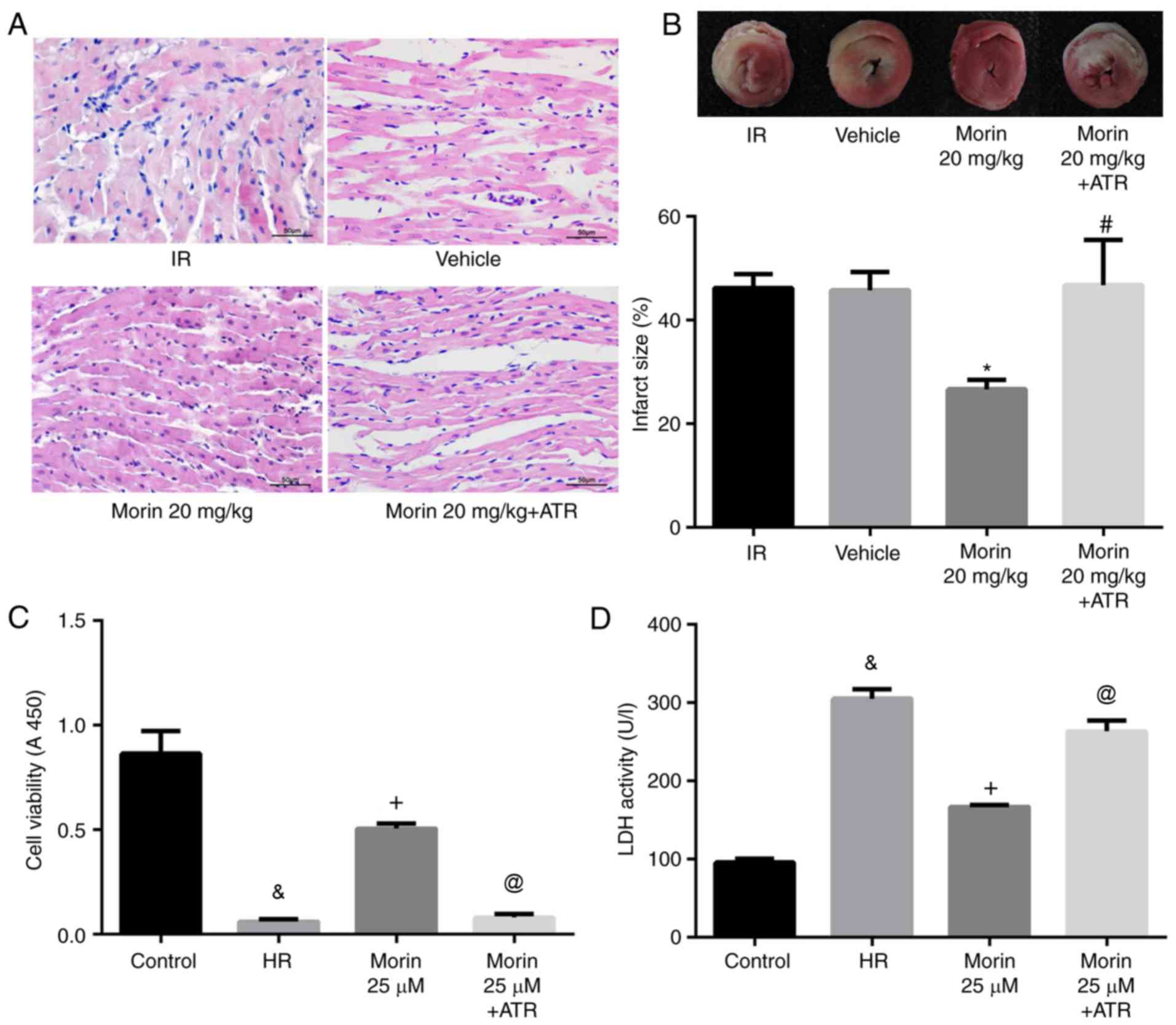
Effect of morin on myocardial damage
The results of the HE staining indicated that
myocardial damage was ameliorated following treatment with morin at
different doses, as evidenced by less vacuolation in cytoplasm and
a decreased number of cells exhibiting nuclear condensation,
dissolution and fragmentation compared with the IR group. Compared
with the morin10 group, fewer instances of severe myocardial damage
were observed in the morin20 group (Fig. 2A). In addition, the myocardial
infarct sizes following treatment with morin at different doses
were significantly decreased compared with the IR group (Fig. 2B). Although there was no
significant difference in myocardial infarct size between morin20
and morin10 groups, it was identified visually that the extent of
myocardial damage indicated in morin20 group was less compared with
that in morin10 group through the result of HE staining. Therefore,
the dose of 20 mg/kg was selected for the additional animal
experiments.
The damage of cardiomyocytes caused by
hypoxia/reoxygenation was evaluated by determining cell viability
and LDH activity. The results demonstrated that cell viability was
significantly increased and LDH activity was markedly decreased in
different morin treatment groups (morin12.5, morin25 and morin50)
compared with those in the HR group. However, there was no
difference in cell viability and LDH activity among groups treated
with different doses of morin (Fig.
2C and D). Although different doses of morin treatment groups
exhibited no statistical difference in cell viability, the cell
viability in morin 25 µM group was increased by almost 10%
compared with that in morin 12.5 µM group (0.449±0.052 vs.
0.409±0.017; P=0.213). Considering that the sample size was
relatively small (n=3), we hypothesized that the statistical
difference between these two groups may be observed if the sample
size became larger. Therefore, the dose of 25 µM was
selected for additional cell experimentation.
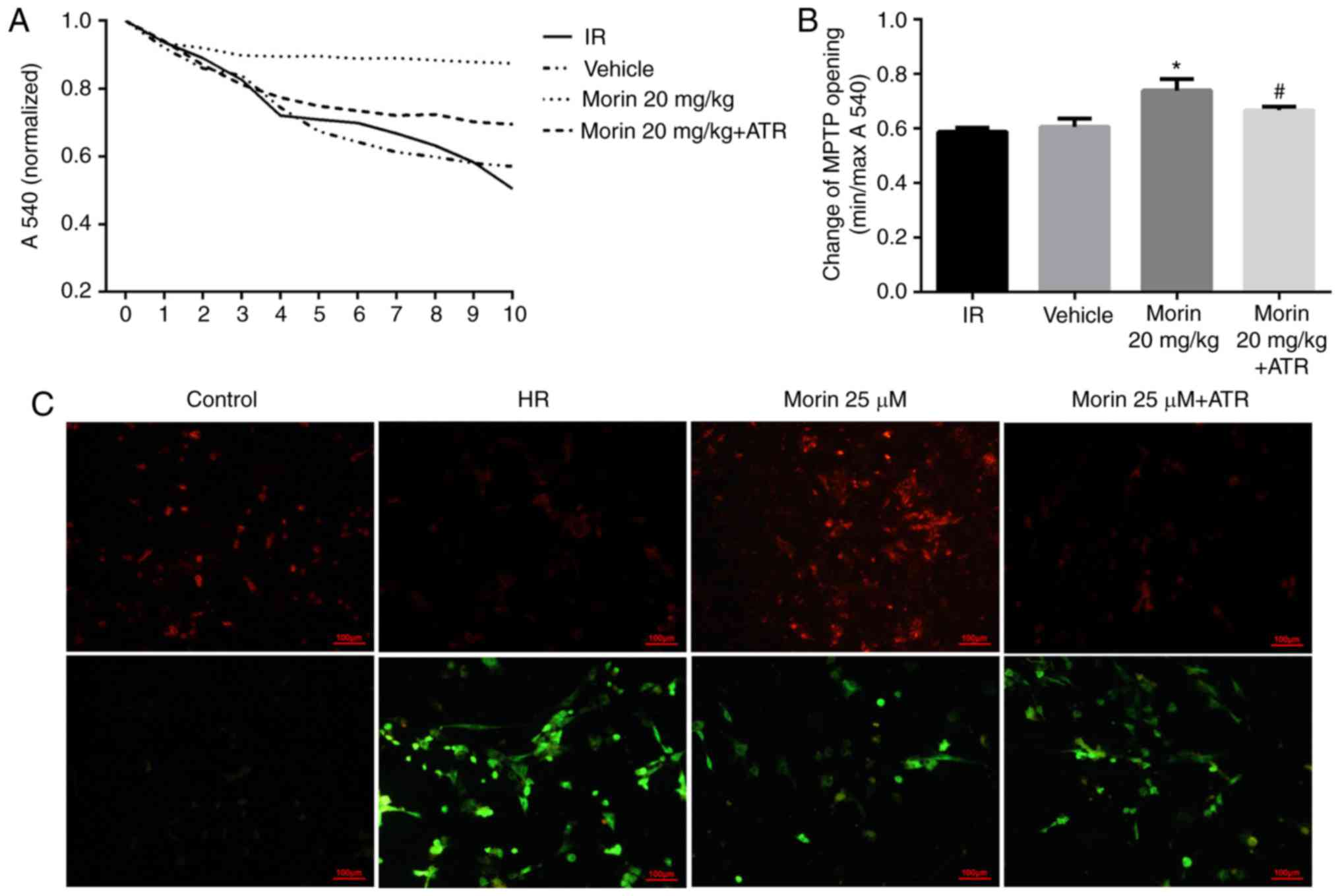
Effect of morin on the alterations in
MPTP opening and ΔΨm
Calcium may induce MPTP opening (16). The sensitivity of mitochondria to
calcium is a marker for MPTP opening (27). The results indicated that the
sensitivity of mitochondria to calcium was significantly attenuated
in the morin treatment group compared with the IR group, suggesting
that morin inhibited the extent of MPTP opening (Fig. 3A and B). In addition, the results
of ΔΨm assay indicated that in the cardiomyocytes, the red
fluorescence intensity was increased and green fluorescence
intensity was decreased in the morin treatment group compared with
the HR group, indicating that morin significantly inhibited the
decrease of the ΔΨm induced by hypoxia/reoxygenation (Fig. 3C).
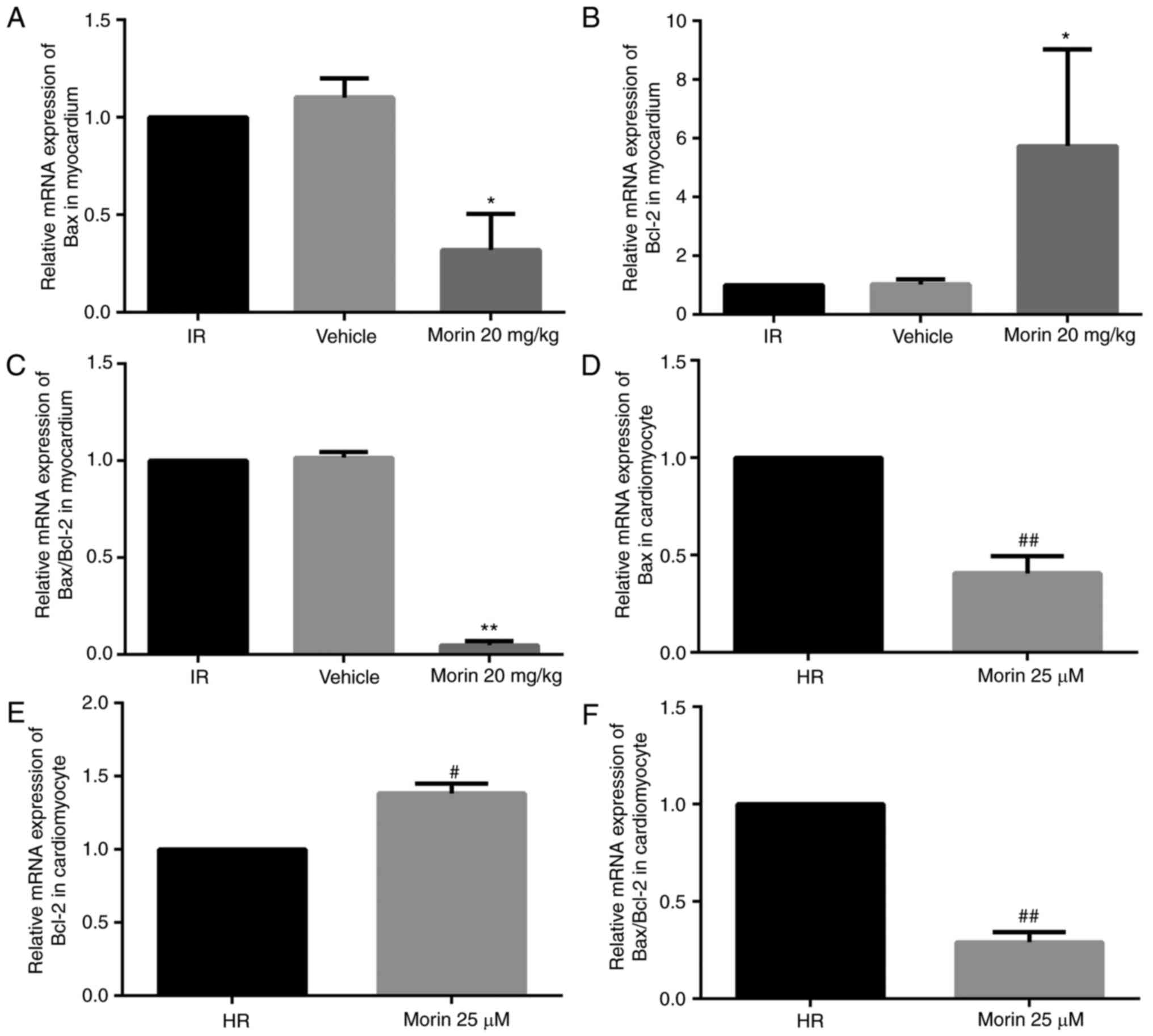
Effect of morin on the expression levels
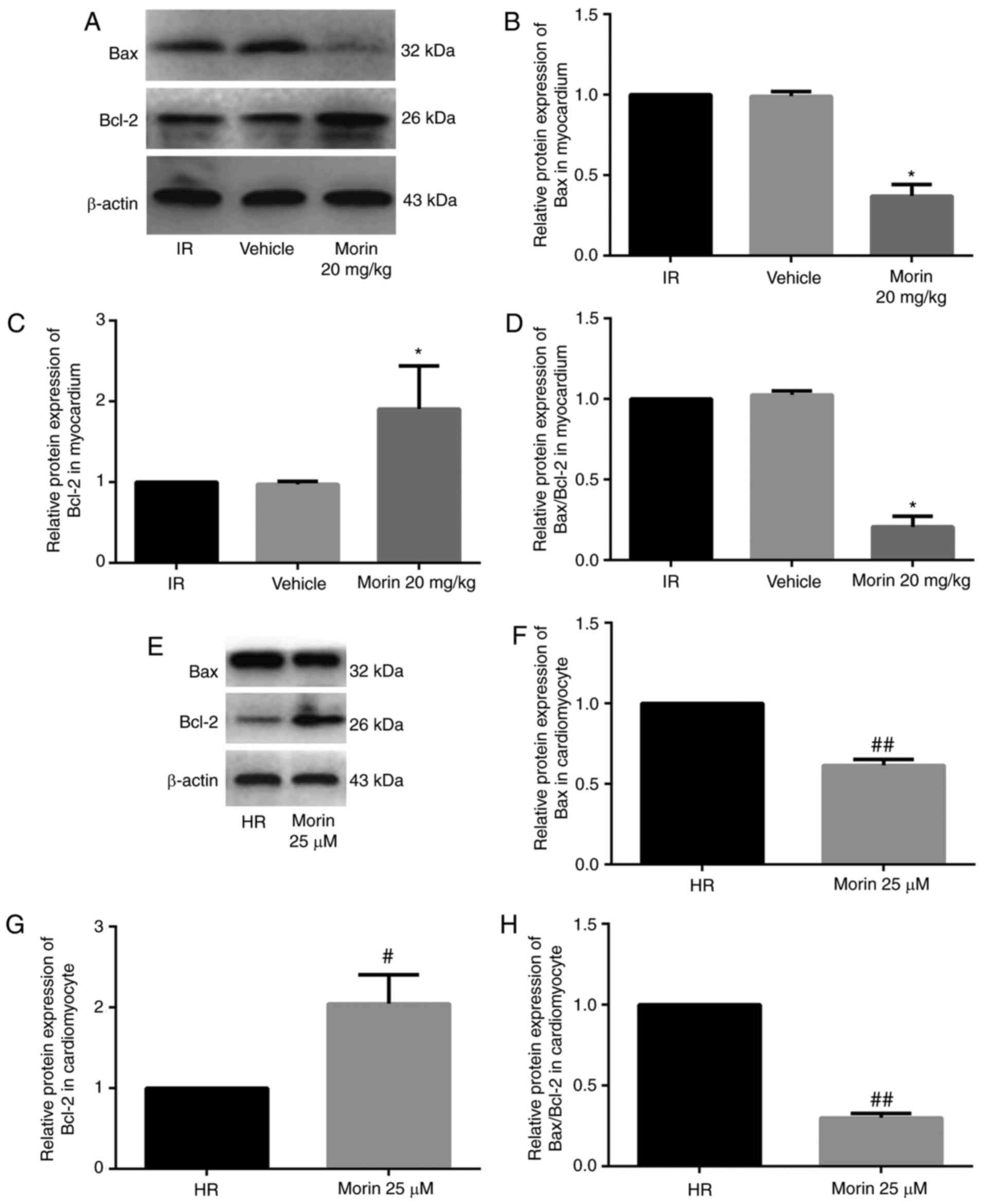
of Bax and Bcl-2
Morin treatment significantly downregulated the mRNA
expression of Bax, upregulated the mRNA expression of Bcl-2 and
decreased the Bax/Bcl-2 ratio in the myocardium and in
cardiomyocytes (Fig. 4).
Consistent with the changes in mRNA expression levels, morin
treatment also markedly downregulated the protein expression of
Bax, upregulated the protein expression of Bcl-2 and decreased the
Bax/Bcl-2 ratio in the myocardium and in cardiomyocytes (Fig. 5).
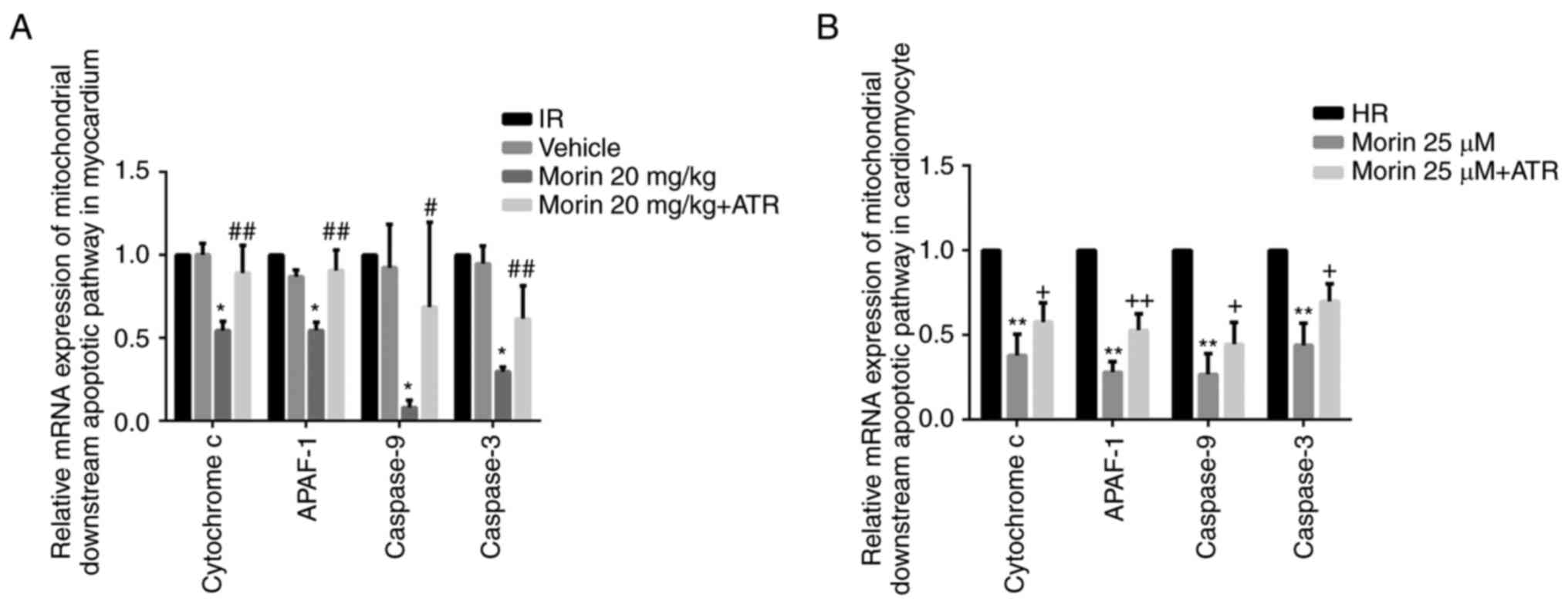
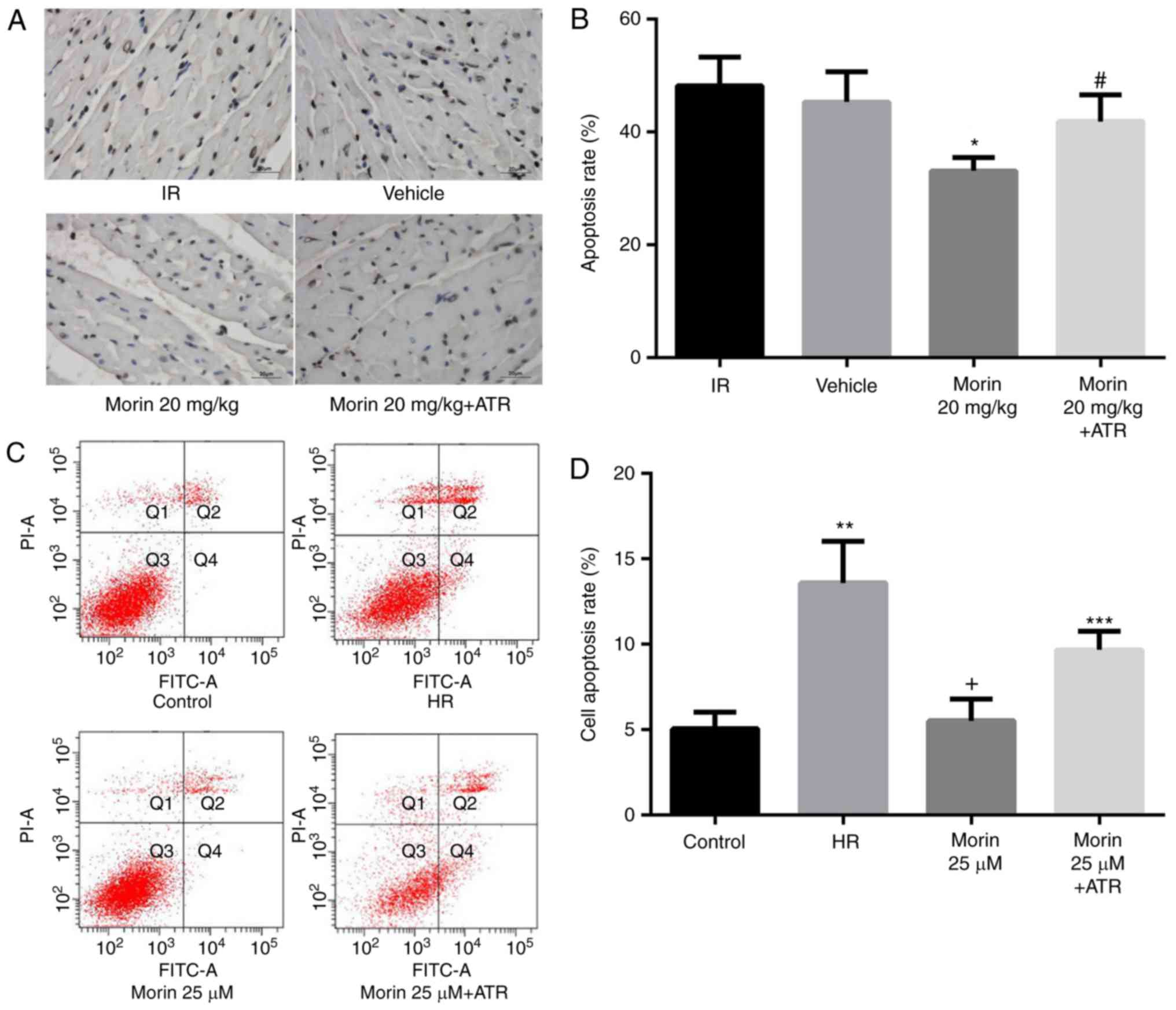
Effect of morin on the mitochondrial
apoptotic pathway and apoptosis
MPTP opening may activate the mitochondrial
apoptotic pathway and induce apoptosis (15). Therefore, the present study
explored the effect of morin on the apoptotic rate and on the
expression of mitochondrial apoptosis-associated proteins. The
results indicated that morin significantly decreased the mRNA and
protein expression of cytochrome c, APAF-1, caspase-9 and
caspase-3 in the myocardium and in cardiomyocytes (Figs. 6 and 7). In addition, morin also significantly
decreased the apoptotic rate of treated cells compared with the HR
cells (Fig. 8).
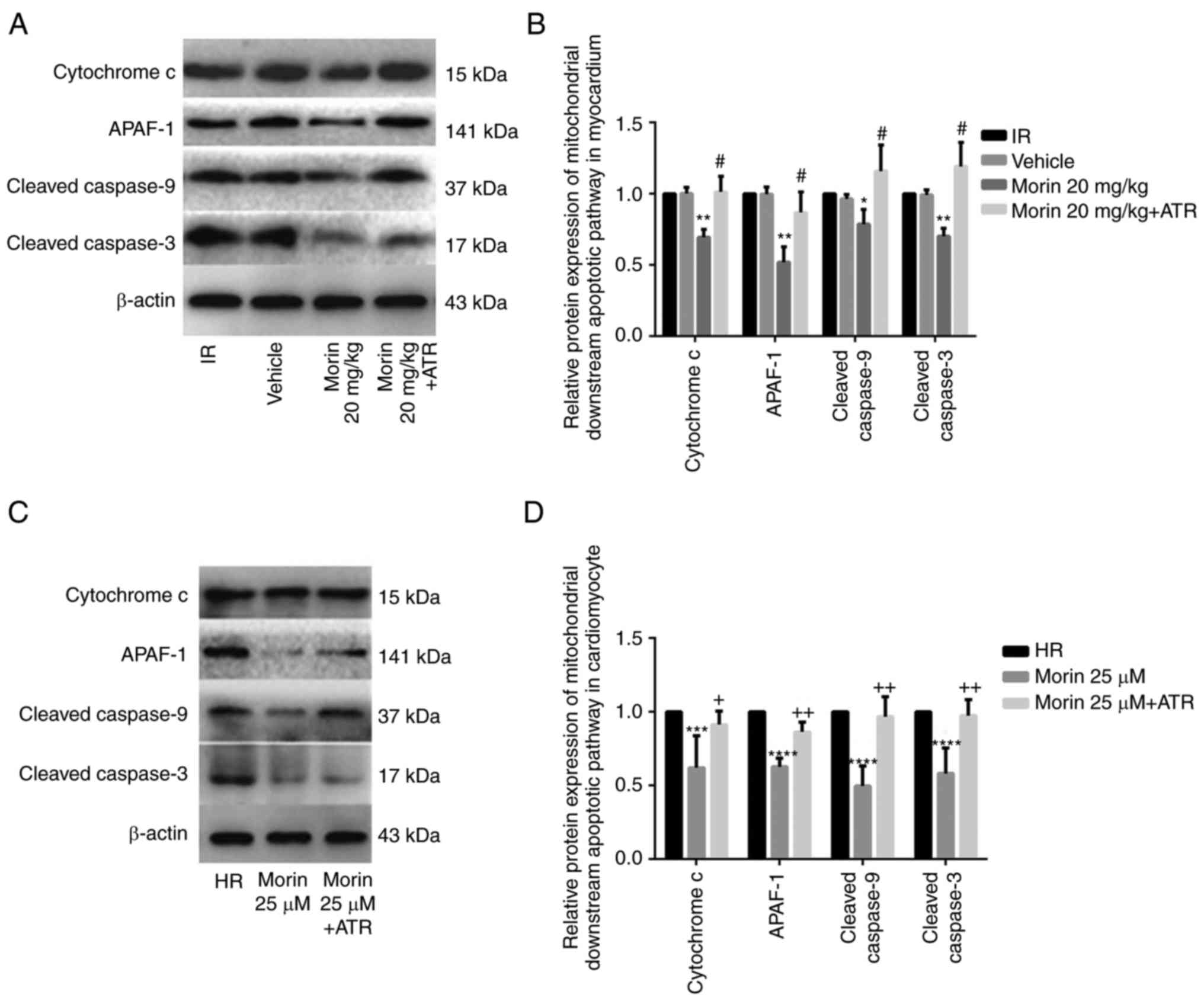 | Figure 7Relative protein expression levels of
downstream genes of the mitochondrial apoptotic pathway. (A)
Protein expression levels of cytochrome c, APAF-1, cleaved
caspase-9 and cleaved caspase-3 in the heart tissue detected by
western blot analysis. (B) Relative protein expression levels of
cytochrome c, APAF-1, cleaved caspase-9 and cleaved
caspase-3 in the heart tissue. (C) Protein expression levels of
cytochrome c, APAF-1, cleaved caspase-9 and cleaved
caspase-3 in cardiomyocytes detected by western blot analysis. (D)
Relative protein expression levels of cytochrome c, APAF-1,
cleaved caspase-9 and cleaved caspase-3 in cardiomyocytes. Values
are presented as the mean ± standard deviation; n=3-5;
*P<0.05 vs. the IR group; **P<0.01 vs.
the IR group; #P<0.01 vs. the morin20 group;
***P<0.05 vs. the HR group; ****P<0.01
vs. the HR group; +P<0.05 vs. the morin25 group;
++P<0.01 vs. the morin25 group. APAF-1, apoptotic
protease activating factor-1; IR, ischemia-reperfusion; HR,
hypoxia/reoxygenation. |
ATR abrogated the protective effects of
morin
To additionally confirm whether morin prevented MIRI
through inhibition of MPTP opening, the effect of ATR, a known MPTP
opener, on the protective effect of morin was examined. As
expected, the inhibitory effects of morin on MPTP opening (Fig. 3A and B), the decrease of ΔΨm
(Fig. 3C), the expression of
mitochondrial apoptosis-associated proteins (Figs. 6 and 7) and apoptosis were reversed by ATR
(Fig. 8). In addition, it was
observed that myocardial infarct size was increased, cell viability
was decreased and LDH activity was increased in the morin combined
with ATR pretreatment groups as compared with the morin
pretreatment groups (Fig. 9).
Considering that previous studies had demonstrated that ATR at a
dose of 5 mg/kg does not aggravate myocardial IR injury (28,29), these results suggest that the
protective effects of morin were abrogated by induction of MPTP
opening using ATR.
Discussion
In the present study, it was identified that morin
treatment prior to ischemia markedly alleviated the extent of MIRI,
as evidenced by the increased cell viability, decreased LDH
activity and cell apoptosis, the improved recovery of cardiac
function and the reduction of myocardial infarct size observed.
Although Al-Numair et al (30) previously demonstrated that morin
exhibited an antioxidative activity in an isoproterenol-induced
model of myocardial infarction, they did not provide direct
evidence of the protective effect of morin on myocardial ischemia
injury. To the best of our knowledge, this is the first study to
describe the protective effect of morin on MIRI in detail.
Therefore, morin seems to be a promising compound that may be used
for preventing MIRI. A number of drugs and chemical compounds,
including rosuvastatin (31),
tauroursodeoxycholic acid (32),
tilianin (33) and
phosphodiesterases (34), have
been demonstrated to prevent MIRI by inhibition of MPTP opening.
Woodman et al (35)
confirmed that 3′,4′-dihydroxyflavonol, a flavonoid compound, may
inhibit MPTP opening and preserve mitochondrial function. In the
present study, the results indicated that morin decreased the
sensitivity of MPTP to calcium, which is a marker of MPTP opening,
therefore suggesting that morin inhibited the opening of MPTP. To
the best of the authors’ knowledge, this is a novel result within
this field of study. The irreversible opening of MPTP causes the
loss of ΔΨm (36), release of
cytochrome c to the cytoplasm and activation of
mitochondrial apoptosis-associated proteins, including APAF-1,
caspase-9 and caspase-3, subsequently resulting in apoptosis
induction (37,38). As expected, the present study also
identified that morin prevented the loss and preserved the
stability of ΔΨm. In addition, the expression levels of cytochrome
c, APAF-1, cleaved caspase-9 and cleaved caspase-3 were
significantly decreased by morin treatment. Similar to the results
of the present study, Chen et al (22) identified that morin decreased the
expression of caspase3 in focal cerebral ischemic rats. In
addition, the present study demonstrated that the beneficial
effects of morin on MIRI were reversed by an MPTP opener (ATR),
therefore providing additional evidence of morin-mediated
inhibition of MPTP opening (39).
Taken together, we hypothesize that morin may prevent MIRI by
inhibiting MPTP opening.
MPTP opening may be regulated by Bax and Bcl-2
(40). The pro-apoptotic protein
Bax may mediate MPTP opening through binding of adenine nucleotide
translocase (ANT) or voltage-dependent anion channel (VDAC), while
the anti-apoptotic protein Bcl-2 may directly inhibit the
interaction between Bax and ANT/VDAC to inhibit MPTP opening by
competing with ANT for the Bax binding site (41). The Bax/Bcl-2 balance is critical
for maintaining cell homeostasis (42). Overexpression of Bcl-2 inhibits
mitochondrial ROS production and the release of cytochrome
c, and apoptosis-inducing factor. It may also increase
mitochondrial calcium tolerance load, prevent caspase cascade
activation and exert anti-apoptotic effects (43,44). In the present study, the results
demonstrated that morin treatment may significantly downregulate
the expression of Bax, upregulate the expression of Bcl-2 and
decrease the ratio of Bax to Bcl-2. In addition, the Bax and Bcl-2
expression levels following ATR rescue were also examined, but the
results indicated that ATR did not affect the expressions of Bax
and Bcl-2 (data not shown). The primary explanation may be that ATR
is an inducer of MPTP opening through the inhibition of adenine
nucleotide translocator (45,46). It may induce MPTP opening and
activate mitochondrial downstream apoptotic pathway proteins, but
cannot affect the expressions of Bax and Bcl-2. Therefore, we
hypothesized that morin may regulate the MPTP opening by
controlling the expression of Bax and Bcl-2.
Due to the absence of neural and humoral regulation
in the isolated rat heart models of the present study, the
pathophysiological changes normally observed during MIRI were not
able to be simulated completely, which is a limitation of the
present study. Therefore, the protective effect of morin on MIRI
requires additional verification in vivo.
In conclusion, to the best of our knowledge, this is
the first study to demonstrate morin-mediated prevention of MIRI.
The protective effect of morin on MIRI may be associated with
inhibition of MPTP opening. Therefore, morin may be a promising
compound for MIRI prevention in the future.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81670320) and the
Natural Science Foundation of Liaoning Province (grant no.
201602826).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
DJ and NW conceived and designed the experiments. SL
conducted the experiments. JM, ZH and XL participated in the
completion of the experiments. SL, PJ and YG analyzed the data. SL
wrote the paper. NW revised the manuscript. All the authors read
and approved the final paper.
Ethics approval and consent to
participate
The experimental protocol was approved by the
Institutional Ethics Committee of China Medical University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Writing Group Members; Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, Fullerton HJ, et al: Heart disease and
stroke statistics-2016 update: A report from the American Heart
Association. Circulation. 133:e38–360. 2016. View Article : Google Scholar
|
|
2
|
Guo X, Li Z, Vittinghoff E, Sun Y and
Pletcher MJ: Trends in rate of acute myocardial infarction among
patients aged <30 years. Nat Rev Cardiol. 15:1192018. View Article : Google Scholar
|
|
3
|
Plakht Y, Gilutz H and Shiyovich A: Excess
long-term mortality among hospital survivors of acute myocardial
infarction. Soroka Acute Myocardial Infarction (SAMI) project.
Public Health. 143:25–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bainey KR and Armstrong PW: Clinical
perspectives on reperfusion injury in acute myocardial infarction.
Am Heart J. 167:637–645. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gerczuk PZ and Kloner RA: An update on
cardioprotection: A review of the latest adjunctive therapies to
limit myocardial infarction size in clinical trials. J Am Coll
Cardiol. 59:969–978. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neri M, Riezzo I, Pascale N, Pomara C and
Turillazzi E: Ischemia/reperfusion injury following acute
myocardial infarction: A critical issue for clinicians and forensic
pathologists. Mediators Inflamm. 2017:70183932017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Murry CE, Jennings RB and Reimer KA:
Preconditioning with ischemia: A delay of lethal cell injury in
ischemic myocardium. Circulation. 74:1124–1136. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F,
Wang NP, Guyton RA and Vinten-Johansen J: Inhibition of myocardial
injury by ischemic postconditioning during reperfusion: Comparison
with ischemic preconditioning. Am J Physiol Heart Circ Physiol.
285:H579–H588. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donato M, Evelson P and Gelpi RJ:
Protecting the heart from ischemia/reperfusion injury: An update on
remote ischemic preconditioning and postconditioning. Curr Opin
Cardiol. 32:784–790. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zou R, Shi W, Tao J, Li H, Lin X, Yang S
and Hua P: SIRT5 and post-translational protein modifications: A
potential therapeutic target for myocardial ischemia-reperfusion
injury with regard to mitochondrial dynamics and oxidative
metabolism. Eur J Pharmacol. 818:410–418. 2018. View Article : Google Scholar
|
|
12
|
Liu J, Wang H and Li J: Inflammation and
inflammatory cells in myocardial infarction and reperfusion injury:
A double-edged sword. Clin Med Insights Cardiol. 10:79–84. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Badalzadeh R, Mokhtari B and Yavari R:
Contribution of apoptosis in myocardial reperfusion injury and loss
of cardioprotection in diabetes mellitus. J Physiol Sci.
65:201–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morciano G, Bonora M, Campo G, Aquila G,
Rizzo P, Giorgi C, Wieckowski MR and Pinton P: Mechanistic role of
mPTP in ischemia-reperfusion injury. Adv Exp Med Biol. 982:169–189.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ong SB, Samangouei P, Kalkhoran SB and
Hausenloy DJ: The mitochondrial permeability transition pore and
its role in myocardial ischemia reperfusion injury. J Mol Cell
Cardiol. 78:23–34. 2015. View Article : Google Scholar
|
|
16
|
Hurst S, Hoek J and Sheu SS: Mitochondrial
Ca2+ and regulation of the permeability transition pore.
J Bioenerg Biomembr. 49:27–47. 2017. View Article : Google Scholar
|
|
17
|
Kwong JQ and Molkentin JD: Physiological
and pathological roles of the mitochondrial permeability transition
pore in the heart. Cell Metab. 21:206–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Šileikytė J and Forte M: Shutting down the
pore: The search for small molecule inhibitors of the mitochondrial
permeability transition. Biochim Biophys Acta. 1857.1197–1202.
2016.
|
|
19
|
Lim WY, Messow CM and Berry C: Cyclosporin
variably and inconsistently reduces infarct size in experimental
models of reperfused myocardial infarction: A systematic review and
meta-analysis. Br J Pharmacol. 165:2034–2043. 2012. View Article : Google Scholar :
|
|
20
|
Piot C, Croisille P, Staat P, Thibault H,
Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant
D, et al: Effect of cyclosporine on reperfusion injury in acute
myocardial infarction. N Engl J Med. 359:473–481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rizvi F, Mathur A and Kakkar P: Morin
mitigates acetaminophen-induced liver injury by potentiating Nrf2
regulated survival mechanism through molecular intervention in
PHLPP2-Akt-Gsk3β axis. Apoptosis. 20:1296–1306. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Y, Li Y, Xu H, Li G, Ma Y and Pang
YJ: Morin mitigates oxidative stress, apoptosis and inflammation in
cerebral ischemic rats. Afr J Tradit Complement Altern Med.
14:348–355. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang R, Kang KA, Piao MJ, Maeng YH, Lee
KH, Chang WY, You HJ, Kim JS, Kang SS and Hyun JW: Cellular
protection of morin against the oxidative stress induced by
hydrogen peroxide. Chem Biol Interact. 177:21–27. 2009. View Article : Google Scholar
|
|
24
|
Kastenmayer RJ, Moore RM, Bright AL,
Torres-Cruz R and Elkins WR: Select agent and toxin regulations:
Beyond the eighth edition of the Guide for the Care and Use of
Laboratory Animals. J Am Assoc Lab Anim Sci. 51:333–338.
2012.PubMed/NCBI
|
|
25
|
Wu N, Li W, Shu W and Jia D: Protective
effect of picroside II on myocardial ischemia reperfusion injury in
rats. Drug Des Devel Ther. 8:545–554. 2014.PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
27
|
Strutyns’ka NA, Dorofeieva NO, Vavilova HL
and Sahach VF: Hydrogen sulfide inhibits Ca2+-induced
mitochondrial permeability transition pore opening in spontaneously
hypertensive rats. Fiziol Zh. 59:3–10. 2013.In Ukrainian.
View Article : Google Scholar
|
|
28
|
Nazari A, Sadr SS, Faghihi M, Azizi Y,
Hosseini MJ, Mobarra N, Tavakoli A and Imani A: Vasopressin
attenuates ischemia-reperfusion injury via reduction of oxidative
stress and inhibition of mitochondrial permeability transition pore
opening in rat hearts. Eur J Pharmacol. 760:96–102. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Alizadeh AM, Faghihi M, Khori V, Sohanaki
H, Pourkhalili K, Mohammadghasemi F and Mohsenikia M: Oxytocin
protects cardiomyocytes from apoptosis induced by
ischemia-reperfusion in rat heart: Role of mitochondrial
ATP-dependent potassium channel and permeability transition pore.
Peptides. 36:71–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Al-Numair KS, Chandramohan G, Alsaif MA,
Veeramani C and El Newehy AS: Morin, a flavonoid, on lipid
peroxidation and antioxidant status in experimental myocardial
ischemic rats. Afr J Tradit Complement Altern Med. 11:14–20. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu CW, Yang F, Cheng SZ, Liu Y, Wan LH
and Cong HL: Rosuvastatin postconditioning protects isolated hearts
against ischemia-reperfusion injury: The role of radical oxygen
species, PI3K-Akt-GSK-3β pathway, and mitochondrial permeability
transition pore. Cardiovasc Ther. 35:3–9. 2017. View Article : Google Scholar
|
|
32
|
Xie Y, He Y, Cai Z, Cai J, Xi M, Zhang Y
and Xi J: Tauroursodeoxycholic acid inhibits endoplasmic reticulum
stress, blocks mitochondrial permeability transition pore opening,
and suppresses reperfusion injury through GSK-3β in cardiac H9c2
cells. Am J Transl Res. 8:4586–4597. 2016.
|
|
33
|
Wang Y, Yuan Y, Wang X, Wang Y, Cheng J,
Tian L, Guo X, Qin D and Cao W: Tilianin post-conditioning
attenuates myocardial ischemia/reperfusion injury via mitochondrial
protection and inhibition of apoptosis. Med Sci Monit.
23:4490–4499. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chanoit G, Zhou J, Lee S, McIntosh R, Shen
X, Zvara DA and Xu Z: Inhibition of phosphodiesterases leads to
prevention of the mitochondrial permeability transition pore
opening and reperfusion injury in cardiac H9c2 cells. Cardiovasc
Drugs Ther. 25:299–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Woodman OL, Long R, Pons S, Eychenne N,
Berdeaux A and Morin D: The cardioprotectant
3′,4′-dihydroxyflavonol inhibits opening of the mitochondrial
permeability transition pore after myocardial ischemia and
reperfusion in rats. Pharmacol Res. 81:26–33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsujimoto Y and Shimizu S: Role of the
mitochondrial membrane permeability transition in cell death.
Apoptosis. 12:835–840. 2007. View Article : Google Scholar
|
|
37
|
Hüttemann M, Helling S, Sanderson TH,
Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J,
Ramzan R, et al: Regulation of mitochondrial respiration and
apoptosis through cell signaling: Cytochrome c oxidase and
cytochrome c in ischemia/reperfusion injury and inflammation.
Biochim Biophys Acta. 1817.598–609. 2012.
|
|
38
|
Kim JS, He L and Lemasters JJ:
Mitochondrial permeability transition: A common pathway to necrosis
and apoptosis. Biochem Biophys Res Commun. 304:463–470. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang S, Li H, Tang L, Ge G, Ma J, Qiao Z,
Liu H and Fang W: Apelin-13 protects the heart against
ischemia-reperfusion injury through the RISK-GSK-3β-mPTP pathway.
Arch Med Sci. 11:1065–1073. 2015.PubMed/NCBI
|
|
40
|
Chen Z, Chua CC, Ho YS, Hamdy RC and Chua
BH: Overexpression of Bcl2 attenuates apoptosis and protects
against myocardial I/R injury in transgenic mice. Am J Physiol
Heart Circ Physiol. 280:H2313–H2320. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dejean LM, Martinez-Caballero S, Guo L,
Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B,
Jonas EA and Kinnally KW: Oligomeric Bax is a component of the
putative cytochrome c release channel MAC, mitochondrial
apoptosis-induced channel. Mol Biol Cell. 16:2424–2432. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong JW, Zhu HF, Zhu WZ, Ding HL, Ma TM
and Zhou ZN: Intermittent hypoxia attenuates ischemia/reperfusion
induced apoptosis in cardiac myocytes via regulating Bcl2/Bax
expression. Cell Res. 13:385–391. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Imahashi K, Schneider MD, Steenbergen C
and Murphy E: Transgenic expression of Bcl2 modulates energy
metabolism, prevents cytosolic acidification during ischemia, and
reduces ischemia/reperfusion injury. Circ Res. 95:734–741. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hochhauser E, Kivity S, Offen D, Maulik N,
Otani H, Barhum Y, Pannet H, Shneyvays V, Shainberg A, Goldshtaub
V, et al: Bax ablation protects against myocardial
ischemia-reperfusion injury in transgenic mice. Am J Physiol Heart
Circ Physiol. 284:H2351–H2359. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Halestrap AP, Clarke SJ and Javadov SA:
Mitochondrial permeability transition pore opening during
myocardial reperfusion-a target for cardioprotection. Cardiovasc
Res. 61:372–385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Javadov S, Karmazyn M and Escobales N:
Mitochondrial permeability transition pore opening as a promising
therapeutic target in cardiac diseases. J Pharmacol Exp Ther.
330:670–678. 2009. View Article : Google Scholar : PubMed/NCBI
|