Introduction
Intermittent hypoxia/reoxygenation (IHR) has been
reported to contribute to the pathogenic process of obstructive
sleep apnea (OSA); it also appears to be involved in promoting
resistance to radiotherapy and chemotherapy in solid tumors, and is
encountered in a wide range of respiratory and cardiac disorders,
including chronic obstructive pulmonary disease and congestive
cardiac failure (1-5). During these disease processes, IHR
is always associated with increased reactive oxygen species (ROS)
production, oxidative stress, and inflammation (6,7).
Emerging evidence indicates that chronic kidney disease is a
prevalent complication of untreated OSA with symptoms of polyuria
and proteinuria (8,9). However, the precise mechanisms
between these two diseases remain to be fully elucidated.
In contrast to the adaptation effect in response to
continuous hypoxia (CH) through hypoxia-inducible factor 1 (HIF-1)
signaling, the body undergoes tissue damage and cellular
inflammation, mainly caused by nuclear factor-κB (NF-κB) signaling,
following IHR challenge (10).
NF-κB is considered the main pro-inflammatory family of
transcription factors (11,12), NF-κB is released consequently and
translocates to the nucleus where it activates the expression of
NF-κB-related inflammatory genes (13). In this process, transforming
growth factor-β-activated kinase-1 (TAK1) has been reported to be
crucial through its kinase activity to phosphorylate and activate
inhibitor of NF-κB (IκB) kinase (IKK) β, facilitating IKKβ-mediated
IκBα degradation and downstream activation of NF-κB (14). However, the role of TAK1 in the
IHR-induced activation of NF-κB has not been reported.
Deubiquitination is catalyzed by deubiquitinating
(DUB) enzymes, which are proteases that cleave ubiquitin or
ubiquitin-like proteins from target proteins (15). Ubiquitin-specific peptidase 8
(USP8; also known as Ubpy) is a cysteine protease and member of the
ubiquitin-specific protease family of DUB enzymes capable of
catalyzing the complete breakdown of K48- and K63-linked
polyubiquitin into its component monomers (16,17). USP8 has an important physiological
function in cell growth, and the deletion of USP8 causes embryonic
death in mice (18,19). USP8 has been reported to
negatively regulate the ubiquitination of epidermal growth factor
receptor, neuregulin receptor degradation protein-1, and β-site
amyloid precursor protein-cleaving enzyme to regulate the process
of cell proliferation, neuroinflammation and neurological disorder
(20-23). However, the effect of USP8 on
IHR-induced inflammation and the underlying targets in the
IHR-induced activation of NF-κB remain to be fully elucidated. The
present study demonstrated for the first time, to the best of our
knowledge, that the expression of USP8 was decreased in IHR
conditions but not in normoxic or CH conditions in renal tubular
epithelial cells. Furthermore, the overexpression of USP8
significantly suppressed IHR-induced pro-inflammatory cytokine
expression and inhibited the activation of NF-κB. It was also
observed that USP8 interacted with TAK1 and deubiquitinated the
K63-linked ubiquitination of TAK1, leading to the inactivation of
TAK1 and NF-κB. In conclusion, the present study revealed an
anti-inflammatory role of USP8 in IHR-induced inflammation and
suggested USP8 as a potential target for the treatment of
IHR-related diseases.
Materials and methods
Cell culture and hypoxia treatment
The HK-2 (human renal tubular epithelial cells) and
TCMK-1 (murine renal tubular epithelial cells) cells were purchased
from the American Type Culture Collection (Manassas, VA, USA).
These cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
that was supplemented with 10% heat-inactivated fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). The hypoxia treatment
was performed as described previously (24). Briefly, for the normoxic cultures,
the cells were cultured in a humidified incubator (37°C, 21%
O2, 74% N2 and 5% CO2) for 48 h.
For the CH cultures, the cells were grown in a sealed hypoxic
incubator for 48 h (37°C, 1% O2, 94% N2 and
5% CO2). For the IHR cultures, the cells were subjected
to a specified number of hypoxia (37°C, 1% O2, 94%
N2 and 5% CO2) and reoxygenation (37°C, 21%
O2, 74% N2 and 5% CO2) cycles;
each hypoxia and reoxygenation cycle included 3 h of hypoxic
incubation followed by 3 h of normoxic culture. When reoxygenated,
the cell culture medium was replaced, and the culture dishes were
placed into a normoxic chamber.
Plasmid and short hairpin (sh)RNA
The human USP8 and murine USP8 expression plasmids
were purchased from Vigene Biosciences, Inc. (Jinan, China). Human
USP8 and murine USP8 shRNA plasmids (cat. nos. 76975 and 76976)
were purchased from Santa Cruz Biotechnology, Inc., (Dallas, TX,
USA). For the transfection of plasmids into HK-2 and TCMK-1 cells,
X-treme Gene 9 reagents were used according to the standard
protocol (Roche Diagnostics, Basel, Switzerland). Following
transfection, the cells were incubated at 37°C for 36 or 48 h to
allow for maximal overexpression or knockdown of the target gene,
and then exposed to eight cycles of IHR, followed by the
examination of protein expression or inflammatory cytokine
detection.
Dual-luciferase reporter gene assays
The experiment was performed as described previously
(25). The NF-κB promoter plasmid
and phRL-TK plasmid were obtained from Dr Guo Litao from Affiliated
Hospital of Chengde Medical University (Chengde, China). The
reporter plasmid was transfected into HK-2 cells using the
Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, Inc.),
and the phRL-TK plasmid was cotransfected as internal control. A
total of 36 h later, the cells were exposed to IHR treatment and
the luciferase activities were measured on a SpectraMax M5 reader
(Molecular Devices LLC, Sunnyvale, CA, USA) and normalized to
Renilla luciferase activity using the Dual Luciferase
Reporter Assay System (Promega Corporation, Madison, WI, USA).
RNA analysis
The cells were collected and TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used to extract
total RNA according to the manufacturer's protocol. The cDNA
Reverse Transcription kit (Thermo Fisher Scientific, Inc.) was used
for using 1 µg of total RNA from each sample according to the
manufacturer's protocol. The temperature protocol was as follows:
RNA and kit reagents were mixed and incubated for 2 min at 37°C in
a water bath. Then the other components were added and incubated
for 10 min at 25°C followed by 15 min at 50°C. The reaction was
terminated by heating at 85°C for 5 min.
A LightCycler (ABI PRISM 7000; Applied Biosystems;
Thermo Fisher Scientific, Inc.) and a SYBR RT-PCR kit (Takara
Biotechnology Co., Ltd., Dalian, China) were used for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis. Reaction conditions for SYBR Green quantitative PCR were:
Pre-denaturation at 95°C for 10 min; followed by 40 cycles of
denaturation at 95°C for 15 sec, annealing at 60°C for 20 sec, and
extension at 72°C for 20 sec. GAPDH was used as the internal
control, and the 2-ΔΔCq method was used to evaluate the
relative quantities of each amplified product in the samples
(26). The primer sequences used
in RT-qPCR analysis are presented in Table I.
 | Table IList of primers used in the present
study. |
Table I
List of primers used in the present
study.
| Number | Gene | Sequence (5′-3′) |
|---|
| 1 | USP8 | F AAA GAC AGG CAG GAG
GAA GC |
| (Homo
sapiens) | R CCA ATG TGC CAC CAT
CCT CT |
| 2 | USP8 | F AAC CAG CAC CAA GAA
TAC CAA |
| (Mus
musculus) | R CCG AAC TTC AGC CTC
TTC GT |
| 3 | TNF-α | F CTA AGA GGG AGA GAA
GCA |
| (Homo
sapiens) | R AGA GGC TGA GGA ACA
AGC |
| 4 | TNF-α | F GCC ACC ACG CTC TTC
TGT CT |
| (Mus
musculus) | R TGA GGG TCT GGG CCA
TAG AAC |
| 5 | IL-6 | F CCT TCG GTC CAG TTG
CCT TCT |
| (Homo
sapiens) | R CCA GTG CCT CTT TGC
TGC TTT |
| 6 | IL-6 | F ACA ACC ACG GCC
TTC CC TAC |
| (Mus
musculus) | R CAT TTC CAC GAT
TTC CCA GA |
| 7 | IL-1β | F AGC TAC GAA TCT
CCG ACC |
| (Homo
sapiens) | R TGG CCA CAA CAA
CTG ACG |
| 8 | IL-1β | F ACC TTC CAG GAT
GAG GAC ATG |
| (Mus
musculus) | R AAC GTC ACA CAC
CAG CAG GTT |
| 9 | IL-8 | F TTG CCA AGG AGT
GCT AAA GAA |
| (Homo
sapiens) | R GCC CTC TTC AAA
AAC TTC TCC |
| 10 | CXCL15 | F CAA GGC TGG TCC
ATG CTC C |
| (Mus
musculus) | R TGC TAT CAC TTC
CTT TCT GTT GC |
| 11 | GAPDH | F AAT GAC CCC TTC
ATT GAC |
| (Universal
primer) | R TCC ACG ACG TAC
TCA GCG |
Western blot analysis and
immunoprecipitation (IP)
The cells were lysed using RIPA buffer (Thermo
Fisher Scientific, Inc.), and total protein in the supernatants was
quantified using a BCA protein assay kit (Thermo Fisher Scientific,
Inc.). IP and western blot analysis were performed as described
previously (27). Equal amounts
of protein (50 µg protein per lane) were loaded on 10%
SDS-PAGE for separating them and then transferred to polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). Following
blocking with 5% non-fat milk for 1 h, the membranes were probed
with primary antibodies at 4°C overnight. The membranes were washed
three times with TBS and 0.1% Tween 20 (TBST) the next day, and
then incubated with HRP-conjugated anti-rabbit secondary antibodies
(1:5,000; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. Following washing with TBST, the immunoreactive
protein bands were visualized by using Western Blotting Luminol
Reagent (cat no. sc-2048; Santa Cruz Biotechnology, Inc.) and
detected with Quantity One software version 4.6.2 (Bio-Rad
Laboratories, Inc.). The antibodies targeting USP8 (1:1,000; cat.
no. 8728), p65 (1:1,000; cat. no. 8242), phosphorylated (p-)p65
(1:1,000; cat. no. 3033), IKKβ (1:1,000; cat. no. 8943), p-IKKβ
(1:1,000; cat. no. 2697), Flag (1:1,000; cat. no. 14793), Myc
(1:1,000; cat. no.2276), ubiquitin (linkage-specific K63; 1:500;
cat. no. 5621) and β-actin (1:2,000; cat. no. 3700) were all
purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA).
Statistical analysis
All data are presented as the mean ± standard
deviation of three experiments. Statistical significance was
determined with (two-tailed) Student's t-test, P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were conducted using SPSS software (SPSS
program, version 13.0, SPSS, Inc., Chicago, IL, USA).
Results
Expression of USP8 is decreased in
IHR-treated renal tubular epithelial cells
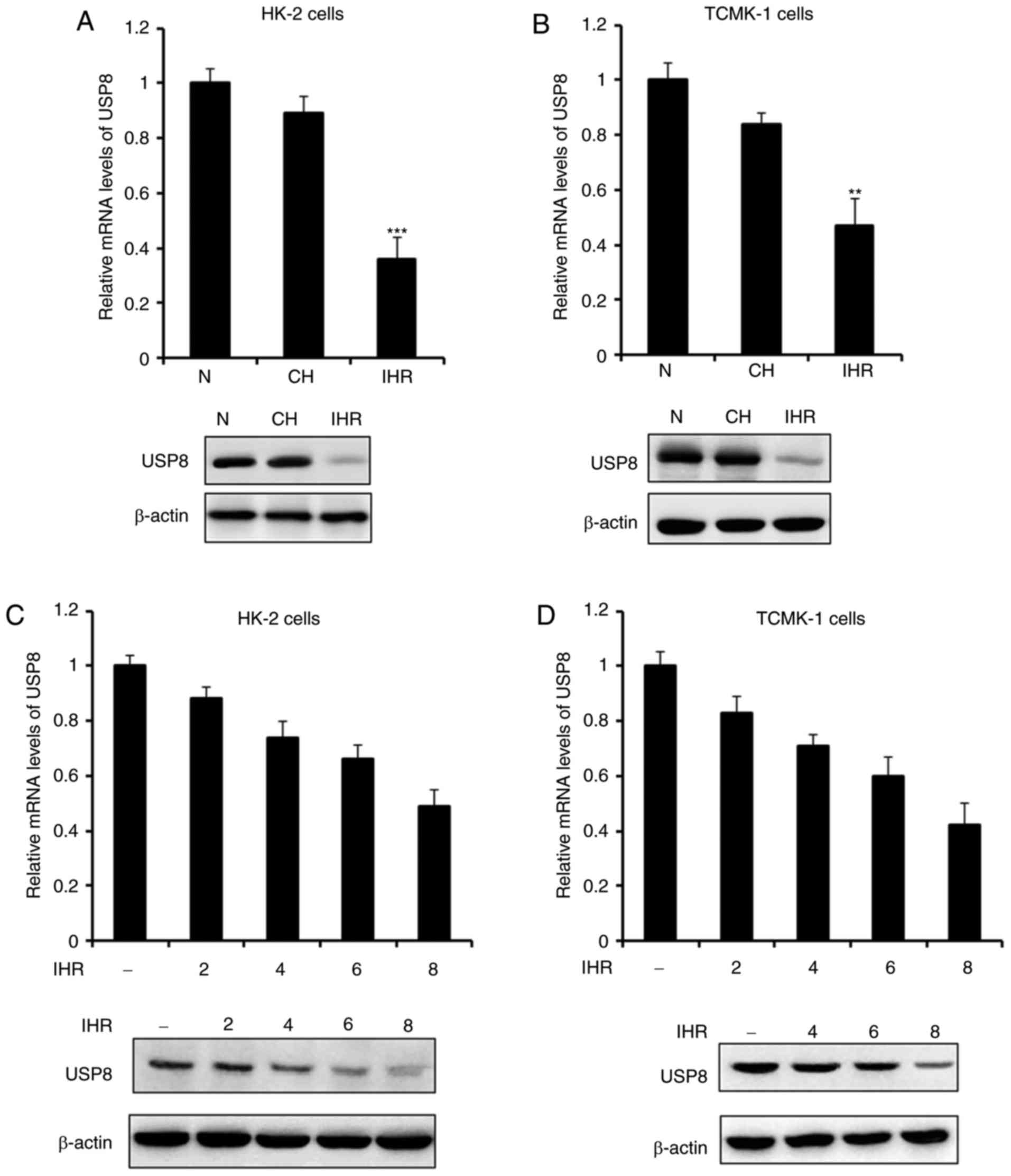
The present study first examined the expression of
USP8 in normoxia, IHR or CH. As shown in Fig. 1A and B, the expression of USP8 was
not affected by the CH conditions, however, it was significantly
decreased in the IHR-treated HK-2 and TCMK-1 cells. The association
between the expression of USP8 and IHR cycles was then examined. It
was demonstrated that the mRNA and protein levels of USP8 were
attenuated in a time-dependent manner (Fig. 1C and D), which indicated that, as
the number of IHR cycles increased, the expression of USP8 was
decreased.
Overexpression of USP8 alleviates
IHR-induced inflammation
It has been reported that USP8 is essential in
downregulating neuroinflammation (23); the present study investigate
whether USP8 regulates the inflammation induced by IHR. First, USP8
was overexpressed in HK-2 and TCMK-1 cells, and the overexpression
of USP8 was confirmed as shown in Fig. 2A and B. The mRNA and protein
levels of pro-inflammatory cytokines were detected, and it was
demonstrated that these cytokines, including tumor necrosis factor
(TNF)-α, interleukin (IL)-6, IL-8 and IL-1β, were all downregulated
in the USP8-overexpressing HK-2 cells following IHR challenge
(Fig. 2C and D). Consistently,
similar results were observed in the TCMK-1 cells (Fig. 2E and F).
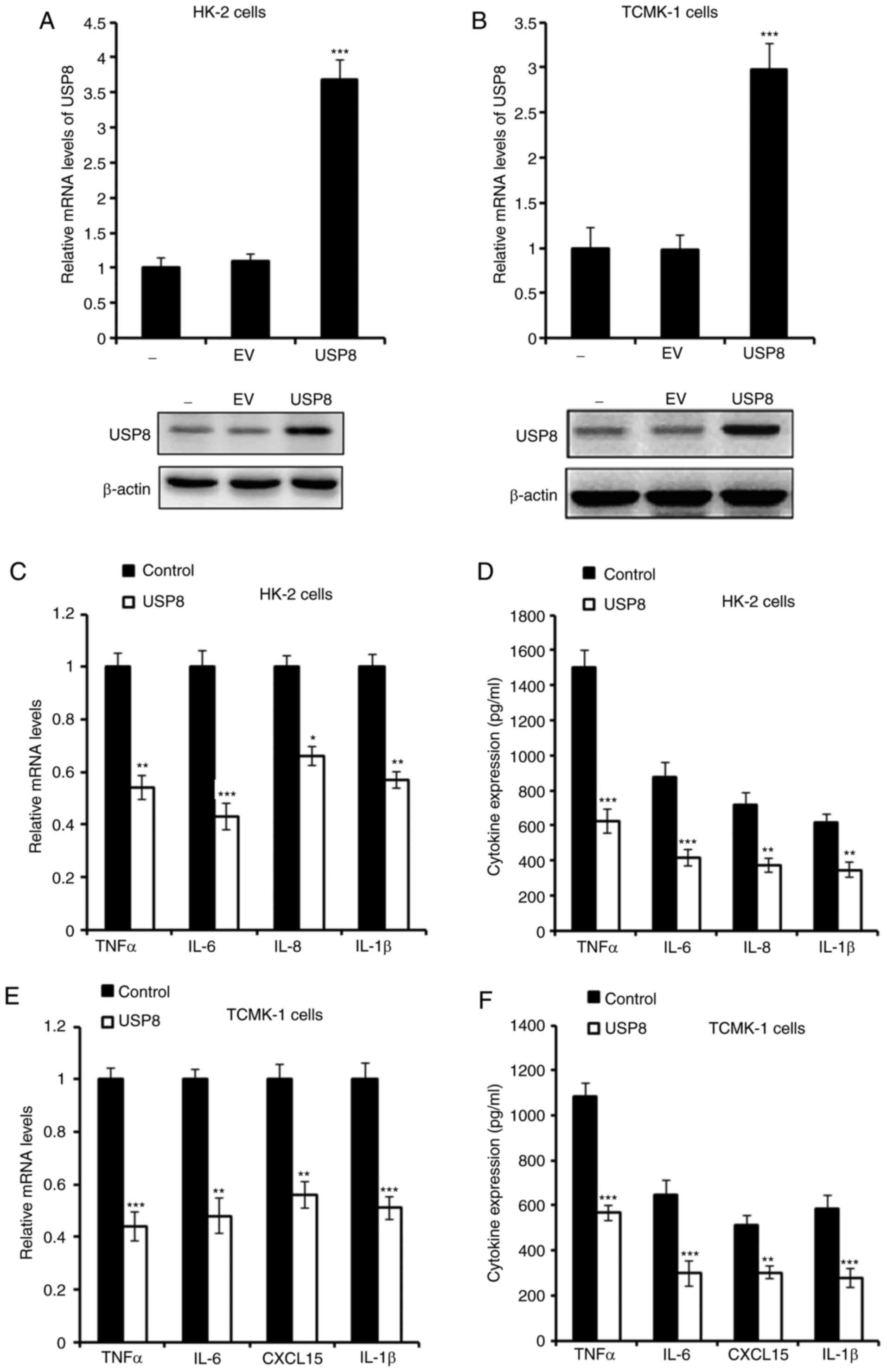 | Figure 2Overexpression of USP8 alleviates
IHR-induced inflammation. (A) mRNA and protein levels of USP8 in
USP8-overexpressing HK-2 cells. (B) mRNA and protein levels of USP8
in USP8-overexpressing TCMK-1 cells. (C) mRNA and (D) protein
levels of TNF-α, IL-6, IL-8 and IL-1β in USP8-overexpressing HK-2
cells in IHR conditions. (E) mRNA and (F) protein levels of TNF-α,
IL-6, CXCL15 and IL-1β in USP8-overexpressing TCMK-1 cells in IHR
conditions. Data are representative of three independent
experiments (mean ± standard deviation). *P<0.05;
**P<0.01; ***P<0.001. USP8,
ubiquitin-specific peptidase 8; IHR, intermittent
hypoxia/reoxygenation; EV, empty vector; TNF-α, tumor necrosis
factor-α; IL, interleukin; CXCL15, chemokine (C-X-C motif) ligand
15. |
Silencing of USP8 aggravates IHR-induced
inflammation
An shRNA plasmid of USP8 was purchased to knock down
the expression of USP8, and the effect of shRNAs was detected
(Fig. 3A and B). As presented in
Fig. 3C and D, the silencing of
USP8 enhanced the IHR-induced production of TNF-α, IL-6, IL-8 and
IL-1β in the HK-2 cells. Consistent with these overexpression
results, it was found that the expression of pro-inflammatory
cytokines in TCMK-1 cells was also increased following USP8 shRNA
treatment in IHR conditions.
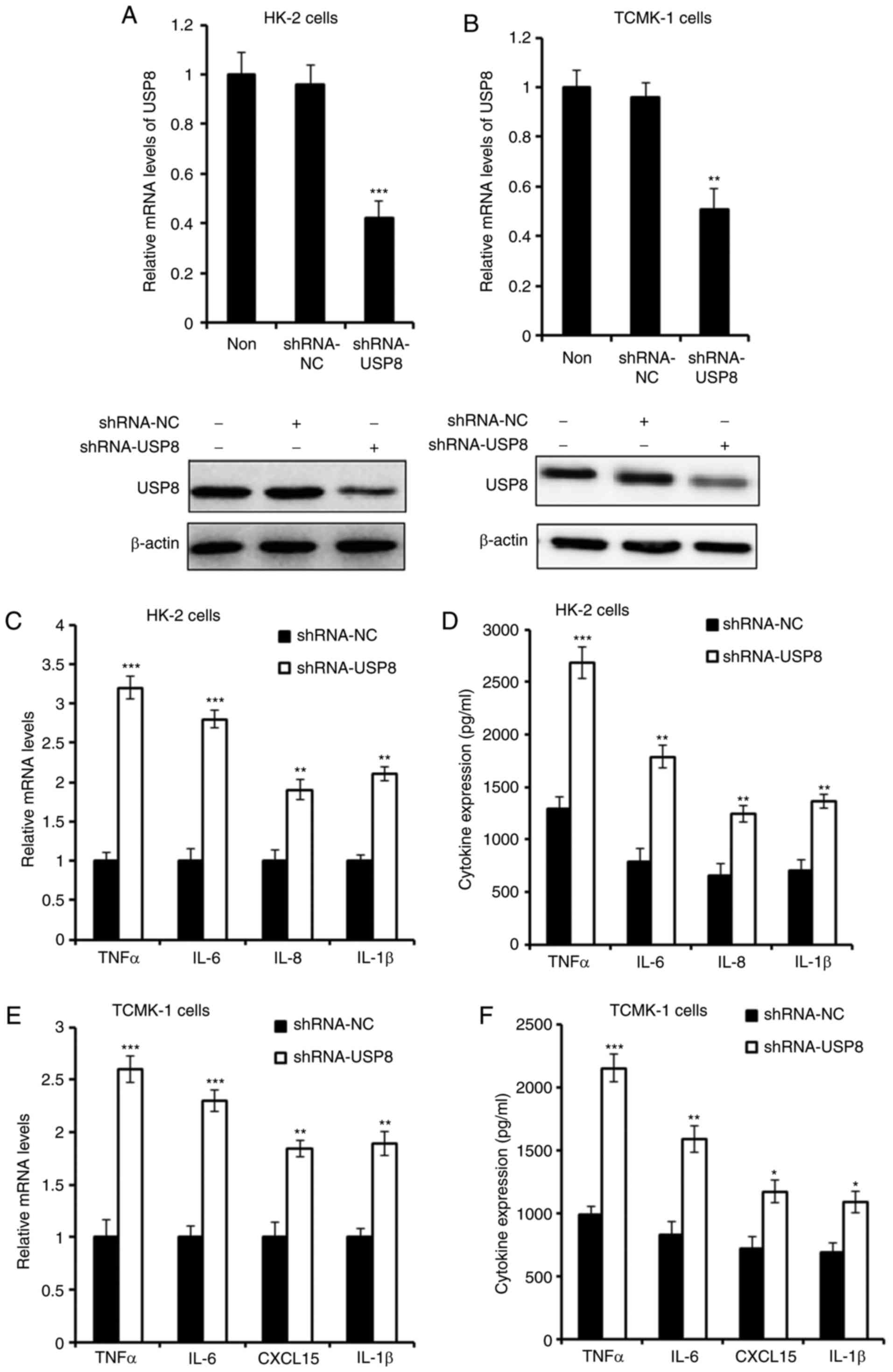 | Figure 3Silencing of USP8 aggravates
IHR-induced inflammation. (A) mRNA and protein levels of USP8 in
USP8-silenced HK-2 cells. (B) mRNA and protein levels of USP8 in
USP8-silenced TCMK-1 cells. (C) mRNA and (D) protein levels of
TNF-α, IL-6, IL-8 and IL-1β in USP8-silenced HK-2 cells in IHR
conditions. (E) mRNA and (F) protein levels of TNF-α, IL-6, CXCL15
and IL-1β in USP8-silenced TCMK-1 cells in IHR conditions. Data are
representative of three independent experiments (mean ± standard
deviation). *P<0.05; **P<0.01;
***P<0.001. USP8, ubiquitin-specific peptidase 8;
IHR, intermittent hypoxia/reoxygenation; TNF-α, tumor necrosis
factor-α; IL, interleukin; CXCL15, chemokine (C-X-C motif) ligand
15; shRNA, short hairpin RNA; NC, negative control. |
USP8 inhibits IHR-induced activation of
NF-κB
It has been reported that IHR-induced inflammation
occurs mainly through NF-κB signalling (10). The present study investigated
whether USP8 also regulates IHR-induced pro-inflammatory cytokine
production through the NF-κB pathway. As presented in Fig. 4A, a Dual-Luciferase Reporter Assay
was used to examine the activation of NF-κB signaling, and it was
found that the overexpression of USP8 significantly suppressed the
activation of NF-κB in the IHR-treated HK-2 cells. Consistently, it
was demonstrated that silencing of USP8 enhanced the IHR-induced
activation of NF-κB (Fig. 4B).
Subsequently, the phosphorylation of p65 and IKKβ was examined, and
it was demonstrated that the overexpression of USP8 inhibited the
IHR-induced phosphorylation of p65 and IKKβ (Fig. 4C and D).
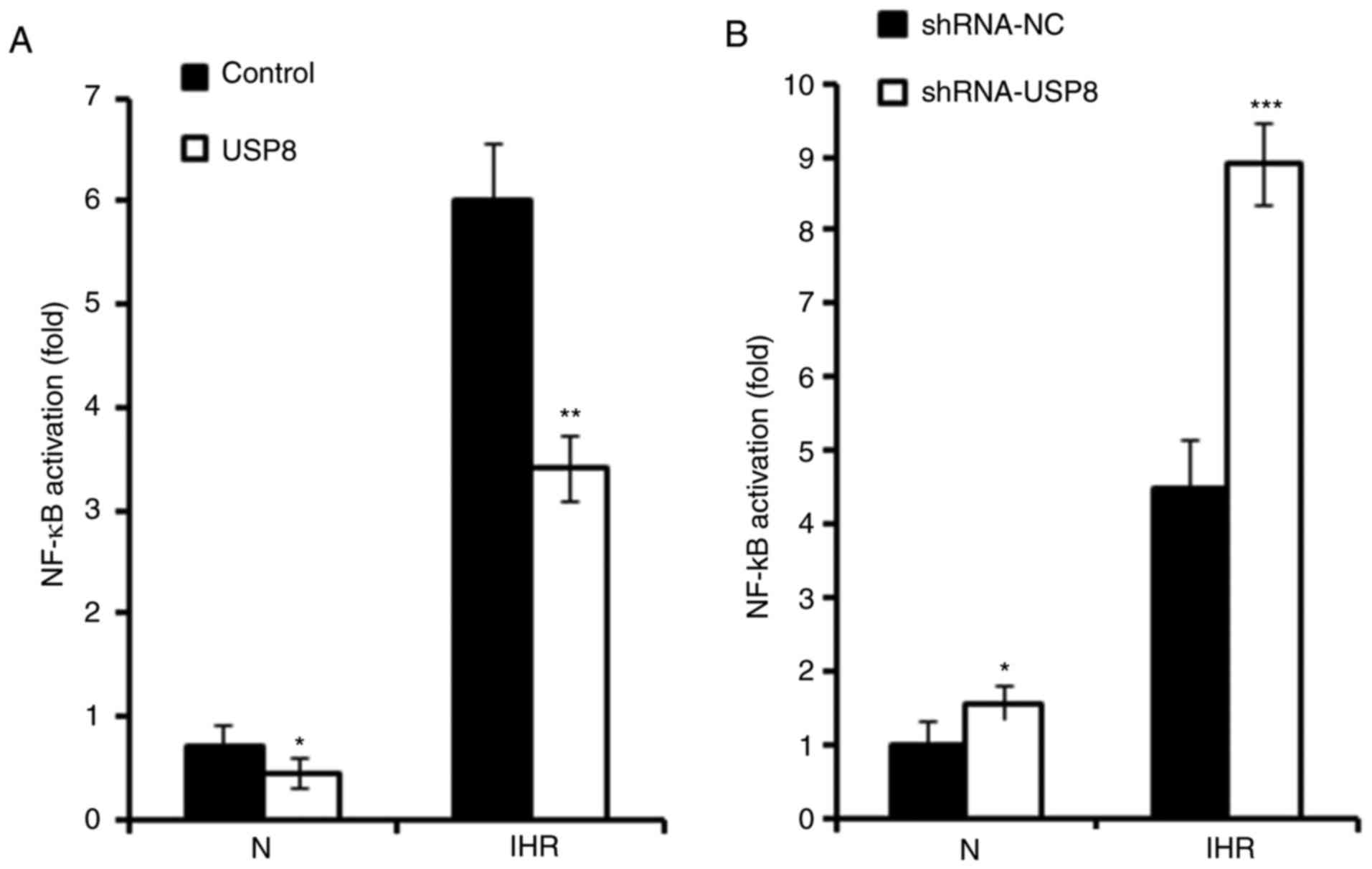 | Figure 4USP8 inhibits IHR-induced NF-κB
activation. (A) HK-2 cells were transfected with control or USP8
expression plasmid, followed by transfection with the NF-κB
reporter plasmid together with the phRL-TK plasmid (internal
control); 36 h later the cells were treated with eight cycles of
IHR, the activation of NF-κB promoter was measured using a
Dual-Luciferase reporter gene assay. (B) HK-2 cells were
transfected with the shRNA-NC or shRNA-USP8 plasmid, followed by
transfection with the NF-κB reporter plasmid together with the
phRL-TK plasmid (internal control); 36 h later the cells were
treated with eight cycles of IHR, the activation of NF-κB promoter
was measured using a Dual-Luciferase reporter gene assay. (C)
Phosphorylation of p65 and IKKβ were examined by a western blot
assay in USP8-overexpressing HK-2 cells treated with eight cycles
of IHR and (D) protein levels protein levels of p-p65 and p-IKKβ
were quantified. Data are representative of three independent
experiments (mean ± standard deviation). *P<0.05;
**P<0.01; ***P<0.001. USP8,
ubiquitin-specific peptidase 8; shRNA, short hairpin RNA; NC,
negative control; N, normoxia; IHR, intermittent
hypoxia/reoxygenation; NF-κB, nuclear factor-κB; IKKβ, inhibitor of
NF-κB kinase β; p-, phosphorylated. |
USP8 interacts with TAK1 and
deubiquitinates the K63-linked ubiquitination of TAK1
TAK1 has been demonstrated to be essential in
regulating the activation of NF-κB (14). The present study examined whether
USP8 regulates TAK1 and thus affects the NF-κB pathway. As
presented in Fig. 5A, it was
demonstrated that, following the overexpression of USP8, the
TAK1-induced activation of NF-κB was suppressed, whereas
IKKβ-induced NF-κB activation was not affected. IP experiments were
performed to examine the interaction between TAK1 and USP8, and it
was found that USP8 interacted with TAK1 (Fig. 5B). On examining whether the
expression of TAK1 was regulated by USP8, however, it was observed
that the expression of TAK1 was not affected by the overexpression
of USP8 (Fig. 5C). USP8 is
reported to be a deubiquitinating enzyme, and it has been reported
to deubiquitinate the K63-linked ubiquitination of diverse targets
(16,17), therefore, the present study
examined the K63-linked ubiquitination level of TAK1. As shown in
Fig. 5D, following IHR treatment,
the overexpression of USP8 downregulated the K63-linked
ubiquitination of TAK1, indicating that USP8 suppressed the
activation of TAK1 through regulating TAK1 K63-linked
ubiquitination.
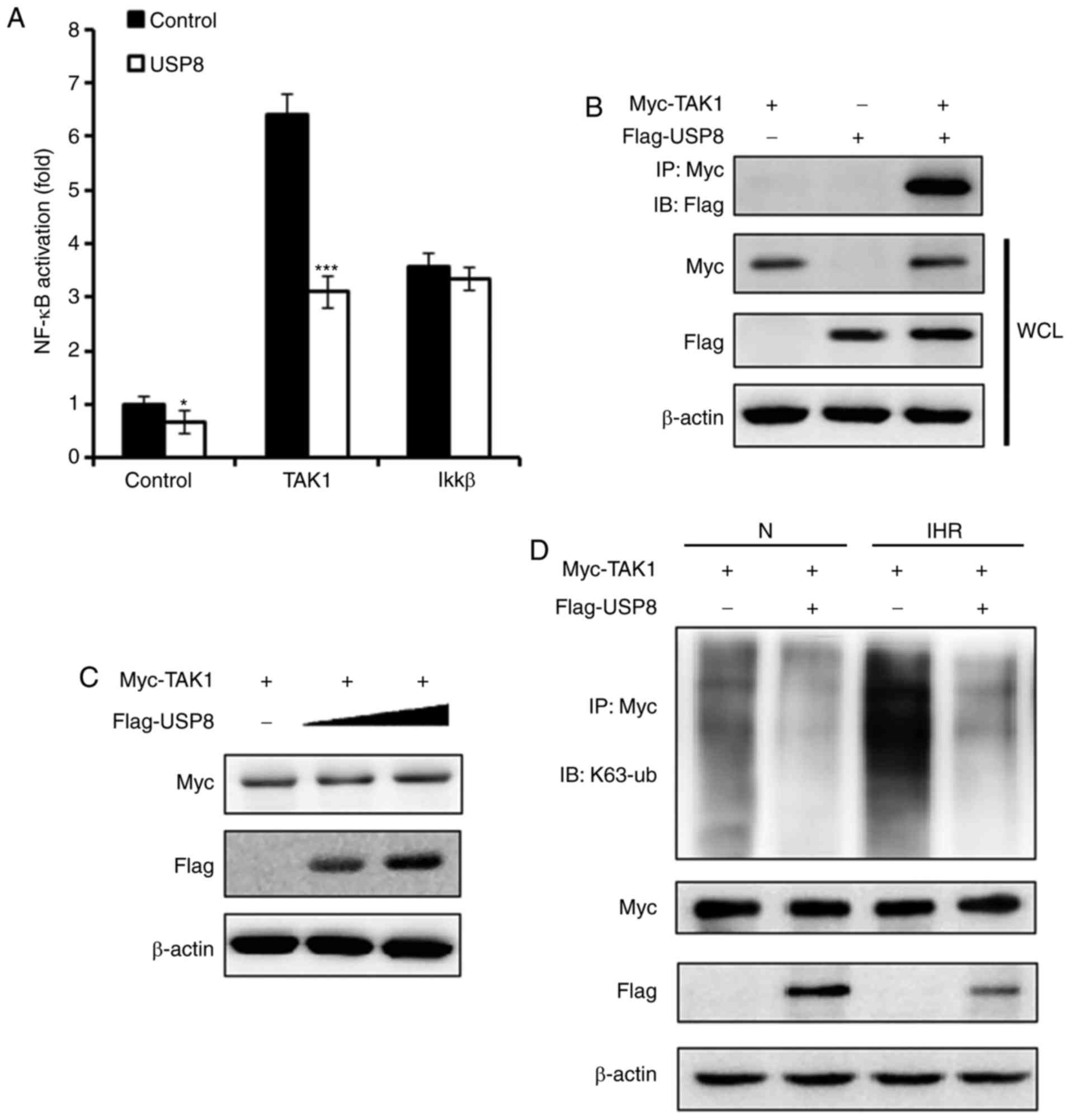 | Figure 5USP8 interacts with TAK1 and
deubiquitinates the K63-linked ubiquitination of TAK1. (A) HK-2
cells were treated with control or USP8 expression plasmid,
followed by transfection with the NF-κB reporter plasmid, phRL-TK
plasmid, together with the control, TAK1 or IKKβ expression
plasmid; 36 h later, the cells were treated with eight cycles of
IHR and activation of the NF-κB promoter was measured using a
Dual-Luciferase reporter gene assay. (B) HK-2 cells were
transfected with Myc-TAK1 and Flag-USP8 expression plasmids, and
the interaction between TAK1 and USP8 was examined by
immunoprecipitation and western blot experiments. (C) HK-2 cells
were transfected with Myc-TAK1, and co-transfected with increasing
quantities of Flag-USP8 plasmid; the expression of TAK1 was
examined by western blot analysis. (D) Western blot analysis of the
K63-linked ubiquitination level of TAK1 in control- or USP8
expression plasmid-treated HK-2 cells in normoxia or IHR
conditions. Data are representative of three independent
experiments (mean ± standard deviation). *P<0.05;
***P<0.001. IHR, intermittent hypoxia/reoxygenation;
N, normoxia; NF-κB, nuclear factor-κB; IKKβ, inhibitor of NF-κB
kinase β; TAK1, transforming growth factor-β-activated kinase-1;
WCL, whole cell lysis. |
Discussion
To the best of our knowledge, the present study is
the first to demonstrate the essential role of USP8 in IHR-induced
inflammation in renal tubular epithelial cells. It is also the
first report to reveal that TAK1 is one of the targets of USP8 and
provide evidence to illustrate that USP8 negatively regulates the
activity of TAK1 through deubiquitinating the K63-linked
ubiquitination of TAK1.
During the progression of acute kidney injury, renal
tubular epithelial cells undergo ischemia and hypoxia, which
include a series of events that cause pathological damage (28,29). Patients with severe OSA also
exhibit a high prevalence of chronic kidney disease and it has been
found to accelerate the loss of kidney function (30). Therefore, it is of significance to
examine the underlying pathogenesis of the disease and identify
other appropriate drug targets. In the present study, it was found
that, in cultured human and murine renal tubular epithelial cells,
the expression of USP8 was significantly decreased in IHR
conditions, however, the expression of USP8 was not affected in CH
conditions. It was hypothesized that this may relate to the
oxidative stress and production of ROS induced by IHR, and future
investigations aim to investigate this to confirm this
hypothesis.
It has been reported that NF-κB-dependent
inflammatory gene expression is induced by IHR (10). Consistent with previous studies,
the present study found in renal tubular epithelial cells that the
activation of NF-κB and pro-inflammatory cytokine production was
increased in IHR conditions. It was also observed that USP8
markedly suppressed the IHR-induced activation of NF-κB and
cytokine production, indicating that USP8 has an anti-inflammatory
effect on IHR-induced inflammation. For the first time, to the best
of our knowledge, the present study revealed that TAK1 is a target
of USP8. TAK1 directly interacted with USP8, and the overexpression
of USP8 significantly suppressed the TAK1-induced activation of
NF-κB. These findings also indicated that TAK1 is the key regulator
of IHR-induced pro-inflammatory cytokine production, which improves
current understanding of TAK1-related diseases.
Ubiquitin has seven lysines (K6, K11, K27, K29, K33,
K48 and K63), all of which can be conjugated to another ubiquitin
to form a polyubiquitin chain through different lysine linkages to
serve distinct functions in the cells (31). The K63-linked polyubiquitination
of TAK1 has been reported to be crucial in the activation of TAK1
and NF-κB (32,33). In the present study, it was found
that E3 ligase USP8 directly interacted with TAK1, but the
overexpression of USP8 did not affect the expression of TAK1,
indicating that the K48-linked polyu-biquitination of TAK1 was not
regulated by USP8. Finally, it was observed that USP8 cleaved
K63-linked polyubiquitin chains from TAK1 to inhibit TAK1 kinase
activity, leading to the attenuated activation of NF-κB and
IHR-induced inflammation. This provides novel ideas for drug
development for IHR-related diseases, including OSA.
In conclusion, the present study found that the
expression of USP8 was decreased in renal tubular epithelial cells
in IHR conditions, and USP8 suppressed the IHR-induced activation
of NF-κB and pro-inflammatory cytokine production, mainly through
deubiquitinating the K63-linked ubiquitination of TAK1. The results
demonstrated the role of USP8 in IHR-induced inflammation and
suggested USP8 as a potential and specific therapeutic target for
IHR-related diseases.
Acknowledgements
The authors would like to thank Dr. Guo Litao from
the Affiliated Hospital of Chengde Medical University for providing
plasmids.
Funding
The present study was supported by grants from
Natural Science Foundation of China (grant nos. 30700890 and
81100059).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YWZ and YQL performed the experiments and drafted
the manuscript. YW and HL assisted with the cell culture and
western blot. YY and QW conceived and designed the study, analyzed
and interpreted the data, and critically revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors confirm that they have no competing
interests.
References
|
1
|
Passali D, Corallo G, Yaremchuk S, Longini
M, Proietti F, Passali GC and Bellussi L: Oxidative stress in
patients with obstructive sleep apnoea syndrome. Acta
Otorhinolaryngol Ital. 35:420–425. 2015.
|
|
2
|
Toffoli S and Michiels C: Intermittent
hypoxia is a key regulator of cancer cell and endothelial cell
interplay in tumours. FEBS J. 275:2991–3002. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prabhakar NR: Oxygen sensing during
intermittent hypoxia: Cellular and molecular mechanisms. J Appl
Physiol. 90:1986–1994. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Agani F and Jiang BH: Oxygen-independent
regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in
cancer. Curr Cancer Drug Targets. 13:245–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Z, Wu S, Liao J, Zhong L, Xing T, Fan
J and Peng Z: Interleukin-6 and rs1800796 locus single nucleotide
polymorphisms in response to hypoxia/reoxygenation in hepatocytes.
Int J Mol Med. 38:192–200. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dyugovskaya L, Polyakov A, Lavie P and
Lavie L: Delayed neutrophil apoptosis in patients with sleep apnea.
Am J Respir Crit Care Med. 177:544–554. 2008. View Article : Google Scholar
|
|
7
|
Lavie L: Oxidative stress in obstructive
sleep apnea and intermittent hypoxia-revisited-the bad ugly and
good: Implications to the heart and brain. Sleep Med Rev. 20:27–45.
2015. View Article : Google Scholar
|
|
8
|
Nicholl DD, Ahmed SB, Loewen AH,
Hemmelgarn BR, Sola DY, Beecroft JM, Turin TC and Hanly PJ:
Clinical presentation of obstructive sleep apnea in patients with
chronic kidney disease. J Clin Sleep Med. 8:381–387.
2012.PubMed/NCBI
|
|
9
|
Kimmel PL, Miller G and Mendelson WB:
Sleep apnea syndrome in chronic renal disease. Am J Med.
86:308–314. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryan S, McNicholas WT and Taylor CT: A
critical role for p38 map kinase in NF-kappaB signaling during
intermittent hypoxia/reoxy-genation. Biochem Biophys Res Commun.
355:728–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Wang C, Li N and Zhang L: Propofol
selectively inhibits nuclear factor-kappaB activity by suppressing
p38 mitogen-activated protein kinase signaling in human EAhy926
endothelial cells during intermittent hypoxia/reoxygenation. Mol
Med Rep. 9:1460–1466. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar
|
|
13
|
Tian Y, Zhang Y, Zhong B, Wang YY, Diao
FC, Wang RP, Zhang M, Chen DY, Zhai ZH and Shu HB: RBCK1 negatively
regulates tumor necrosis factor- and interleukin-1-triggered
NF-kappaB activation by targeting TAB2/3 for degradation. J Biol
Chem. 282:16776–16782. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shuto T, Xu H, Wang B, Han J, Kai H, Gu
XX, Murphy TF, Lim DJ and Li JD: Activation of NF-kappa B by
nontypeable Hemophilus influenzae is mediated by toll-like receptor
2-TAK1-dependent NIK-IKK alpha/beta-I kappa B alpha and MKK3/6-p38
MAP kinase signaling pathways in epithelial cells. Proc Natl Acad
Sci USA. 98:8774–8779. 2001. View Article : Google Scholar
|
|
15
|
Nijman SM, Luna-Vargas MP, Velds A,
Brummelkamp TR, Dirac AM, Sixma TK and Bernards R: A genomic and
functional inventory of deubiquitinating enzymes. Cell.
123:773–786. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
MacDonald E, Urbe S and Clague MJ: USP8
controls the trafficking and sorting of lysosomal enzymes. Traffic.
15:879–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jeong M, Lee EW, Seong D, Seo J, Kim JH,
Grootjans S, Kim SY, Vandenabeele P and Song J: USP8 suppresses
death receptor-mediated apoptosis by enhancing FLIPL stability.
Oncogene. 36:458–470. 2017. View Article : Google Scholar
|
|
18
|
Naviglio S, Mattecucci C, Matoskova B,
Nagase T, Nomura N, Di Fiore PP and Draetta GF: UBPY: A
growth-regulated human ubiquitin isopeptidase. EMBO J.
17:3241–3250. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Niendorf S, Oksche A, Kisser A, Löhler J,
Prinz M, Schorle H, Feller S, Lewitzky M, Horak I and Knobeloch KP:
Essential role of ubiquitin-specific protease 8 for receptor
tyrosine kinase stability and endocytic trafficking in vivo. Mol
Cell Biol. 27:5029–5039. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Alwan HA and van Leeuwen JE: UBPY-mediated
epidermal growth factor receptor (EGFR) de-ubiquitination promotes
EGFR degradation. J Biol Chem. 282:1658–1669. 2007. View Article : Google Scholar
|
|
21
|
Wu X, Yen L, Irwin L, Sweeney C and
Carraway KL III: Stabilization of the E3 ubiquitin ligase Nrdp1 by
the deubiquiti-nating enzyme USP8. Mol Cell Biol. 24:7748–7757.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yeates EF and Tesco G: The
endosome-associated deubiquiti-nating enzyme USP8 regulates bACE1
enzyme ubiquitination and degradation. J Biol Chem.
291:15753–15766. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu L, Bi W and Lu D, Zhang C, Shu X, Wang
H, Qi R, Shi Q and Lu D: Regulation of ubiquitin-specific
processing protease 8 suppresses neuroinflammation. Mol Cell
Neurosci. 64:74–83. 2015. View Article : Google Scholar
|
|
24
|
Litao L, Wenlan L, Lili W, Ting Z, Jianhua
Z and Ni X: Hypoxiainducible factor 1 mediates intermittent
hypoxiainduced migration of human breast cancer MDAMB231 cells.
Oncol Lett. 14:7715–7722. 2017.
|
|
25
|
Guo L, Dong W, Fu X, Lin J, Dong Z, Tan X
and Zhang T: Tripartite Motif 8 (TRIM8) positively regulates
pro-inflammatory responses in Pseudomonas aeruginosa-Induced
keratitis through promoting K63-Linked polyubiquitination of tAK1
protein. Inflammation. 40:454–463. 2017. View Article : Google Scholar
|
|
26
|
Kenneth J and Livak TD: Analysis of
relative gene expression data using rea l-time quantitative PCR a
nd the 2(-Delta Delta C(T)) Method. Method. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Lin Y and Luo Z: NLRP6 facilitates the
interaction between TAB2/3 and TRIM38 in rheumatoid arthritis
fibroblast-like synoviocytes. FEBS Lett. 591:1141–1149. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bonventre JV and Yang L: Cellular
pathophysiology of ischemic acute kidney injury. J Clin Invest.
121:4210–4221. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cantaluppi V, Quercia AD, Dellepiane S,
Figliolini F, Medica D and De Lena M: New mechanisms and recent
insights in the pathogenesis of acute kidney injury (AKI). G Ital
Nefrol. 29:535–547. 2012.In Italian. PubMed/NCBI
|
|
30
|
Yayan J, Rasche K and Vlachou A:
Obstructive sleep apnea and chronic kidney disease. Adv Exp Med
Biol. 1022:11–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Adhikari A and Chen ZJ: Diversity of
polyubiquitin chains. Dev Cell. 16:485–486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sorrentino A, Thakur N, Grimsby S,
Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH and
Landström M: The type I TGF-beta receptor engages TRAF6 to activate
TAK1 in a receptor kinase-independent manner. Nat Cell Biol.
10:1199–1207. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fan YH, Yu Y, Mao RF, Tan XJ, Xu GF, Zhang
H, Lu XB, Fu SB and Yang J: USP4 targets TAK1 to downregulate
TNFα-induced NF-kappaB activation. Cell Death Differ. 18:1547–1560.
2011. View Article : Google Scholar : PubMed/NCBI
|