Introduction
The innate immune system can detect the presence of
pathogens, such as viruses and bacteria, and activate immune
responses to eliminate the infections. These pathogens can be
recognized by pattern-recognition receptors (PRRs) and trigger
activation of innate immunity (1,2).
The family of Toll-like receptors (TLRs) is a class of PRRs in
mammals (3). TLR4 is an important
receptor recognizing lipopolysaccharide (LPS), which is a component
of the outer membrane of Gram-negative bacteria (4,5).
By contrast, the wall components of Gram-positive bacteria, such as
peptidoglycan (PGN) and lipoteichoic acid (LTA), are recognized by
TLR2 (6-8). PGN and LTA can induce septic shock
and multiple organ failure (9).
TLR2 expression can be detected in various types of
human immune cells, including monocytes, macrophages, dendritic
cells and polymorphonuclear leukocytes (also termed granulocytes
and include neutrophils, basophils and eosinophils) (10). In peripheral blood, neutrophils
are the most abundant type of granulocytes and the first r immune
cells to respond to infections. When human neutrophils are exposed
to LTA, cell migration, degranulation, secretion of
pro-inflammatory factors [including interleukin (IL)-8, tumor
necrosis factor-α (TNF-α) and granulocyte colony-stimulating factor
(G-CSF)], increased production of reactive oxygen species (ROS) and
antimicrobial activity, and activation of TLR2 and NF-κB-mediated
signaling pathways have been reported (11-14).
MicroRNA (miRNA) is a group of small non-coding RNAs
with ~22 nucleotides. Emerging evidence suggests that miRNAs are
involved in regulation of gene expression and immune responses
(15,16). For example, miR-155, miR-146a,
miR-UL112-3p and miR-344b-1-3p have been demonstrated to interact
with TLR2 in pathological conditions (17-21). However, the interaction of miRNA
and LTA-mediated immune activation has not been extensively
investigated in a specific type of immune cells. Thus, the present
study aimed to investigate the expression mRNA and miRNA in
Staphylococcus aureus LTA-stimulated human neutrophils via
next-generation sequencing. To understand the LTA-mediated effect
in healthy immune cells, neutrophils were obtained from the
peripheral blood of a healthy donor.
Materials and methods
Neutrophil isolation and LTA
treatment
The present study was approved by the Institutional
Review Board of Kaohsiung Medical University Hospital (IRB no.
KMUH-IRB-20120287). A total of 10 ml venous blood was obtained from
a healthy donor. The participant agreed to the use of their sample
in research and signed informed consent during the period Jan 2013
to Jan 2014. Human neutrophils were separated from whole blood
using CD66abce microbeads (Miltenyi Biotec GbmH), according to
manufacturer's instruction. Subsequently, 3×107 isolated
neutrophils were cultured in RPMI1640 medium containing 10% fetal
bovine serum, 100 U/ml penicillin G, 100 µg/ml streptomycin
and 0.25 µg/ml amphotericin B (Thermo Fisher Scientific,
Inc.), and 1 µg/ml of LTA (from S. aureus; LTA group;
cat. no. L2515; Sigma-Aldrich; Merck KgaA) or double distilled
water (vehicle group) in 5% CO2 air atmosphere at 37°C
for 16 h. Neutrophils were collected for RNA isolation. The purity
of isolated CD66abce+ cells was evaluated via flow
cytometry. Cells were stained with Alexa Fluor 647-conjugated
anti-human CD66b (1:20; cat. no. 561645; BD Pharmingen), according
to manufacturer's instructions. The cells were then washed and
analyzed using a BD Accuri C6 flow cytometer with BD Accuri C6
software version 1.0.264.21 (BD Biosciences).
RNA isolation
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.) according to the
supplier's protocol. Purified RNA was quantified using a ND-1000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.) and the quality was confirmed using an Agilent 2100
Bioanalyzer and an RNA 6000 Pico LabChip RNA (Agilent Technologies,
Inc.). The RNA integrity number (RIN) resulting from the Agilent
Bioanalyzer was 8.3 for the LTA-stimulated cells and 7.6 for the
vehicle-stimulated cells. The quality report is shown in Fig. S1.
Library preparation, sequencing,
alignment and differential expression analysis
Sequencing for mRNA and miRNA was commercially
performed by Welgene Biotech Co., Ltd. All RNA sample preparation
procedures were carried out according to the official Illumina
protocol (Illumina, Inc.). For mRNA sequencing, Agilent's
SureSelect Strand-Specific RNA Library Preparation kit (Agilent
Technologies, Inc.) was used for library construction, followed by
AMPure XP Beads (Agilent Technologies, Inc.) size selection. The
sequence was directly determined via Illumina's
sequencing-by-synthesis technology. Sequencing data were generated
by Welgene's pipeline based on Illumina's base-calling program
bcl2fastq v2.2.0. For miRNA sequencing, samples were prepared using
the TruSeq™ miRNA Library kit (Illumina, Inc.), following the
supplier's guide. Libraries were sequenced on an Illumina
instrument (75-cycle single-end read; 75SE) and miRNA sequencing
data was processed using the Illumina software BCL2FASTQ v2.20.
Sequence Quality Trimming, performed by Trimmomatic version 0.36
(22). HISAT2 was used for mRNA
alignment (23) and miRDeep2 was
used for miRNA alignment (24).
The expression levels were normalized by calculating fragments per
kilobase of transcript per million mapped reads (FPKM).
Differential expression analysis was performed via Cuffdiff
(Cufflinks 2.2.1) (25). P-value
was calculated by Cuffdiff with non-grouped sample using the
'blind' method, in which all samples are treated as replicates of a
single global 'condition' and used to build one model (25).
Reverse transcription-quantitative PCR
(RT-qPCR)
Isolated cells (5×105) were seeded into
several wells of a 24-well plate and treated with vehicle or 1
µg/ml LTA for 16 h. Total RNA was isolated using TRIzol
reagent (Thermo Fisher Scientific, Inc.). Equal amount of total RNA
was reverse transcribed via the PrimeScript RT reagent kit
(Clontech Laboratories, Inc.). qPCR was performed with SYBR-Green
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.) on
a Real-Time PCR system (QuantStudio 3D Digital PCR System; Thermo
Fisher Scientific, Inc.). The thermocycling conditions were: 20 sec
at 95°C, followed by 40 amplification cycles of 95°C for 3 sec and
60°C for 30 sec. The primers were as follows: Human chemokine (C-C
motif) ligand (CCL) 2, forward 5′-TCTGTGCCTGCTGCTCATAG-3′ and
reverse 5′-TGGAATCCTGAACCCACTTC-3′; human CCL7, forward
5′-ACCACCAGTAGCCACTGTCC-3′ and reverse 5′-TTGGGTTTTCTTGTCCAGGT-3′;
human C-X-C motif chemokine ligand 5 (CXCL5), forward
5′-TGTTTACAGACCACGCAAGG-3′ and reverse 5′-GGGGCTTCTGGATCAAGAC-3′;
and human GAPDH, forward 5′-GAGTCAACGGATTTGGTCGT-3′ and reverse
5′-TTGATTTTGGAGGGATCTCG-3′. The relative mRNA expression levels
were normalized to the GAPDH expression and calculated using the
2−∆∆Cq method (26).
Gene ontology (GO) analysis of genes and
miRNAs
The criteria of differential mRNA expression were
set at fold change ≥2.0, FPKM >0.8 and P-value <0.05. For
determining the function of LTA-affected genes, the biological
process of GO (GOTERM_BP_ALL) analysis and Kyoto encyclopedia of
genes and genomes (KEGG) pathway analysis were performed via DAVID
Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/home.jsp) (27,28). In addition, gene set enrichment
analysis (GSEA; http://www.broad.mit.edu/gsea/) (29,30) was performed using the GO
biological processes database c5.bp.v6.2. The criteria of
differential miRNA expression were set at fold change ≥2.0 and
reads per million (RPM) >1. GO analysis of miRNA was performed
via the GSEA method of miRNA Enrichment Analysis and Annotation
Tool (miEAA; https://ccb-compute2.cs.uni-saar-land.de/mieaa_tool/)
(31).
Interaction between miRNA and mRNA
To predict the miRNA-targeted mRNAs, the Funrich
software version 3.1.3 (32) and
miRDB 6.0 (miRNAs with Target Score >90 were selected) were used
(33). The miRNA target genes
were determined using two databases: TargetScan 7.2 (http://www.targetscan.org/vert_72/) (34) and miRTarBase 7.0 (http://mirtarbase.mbc.nctu.edu.tw/php/index.php)
(35). The network was drawn by
using stringApp 1.4.1 plugin in Cytoscape software 3.7.1 (36,37).
Statistical analysis
The Venn diagram was drawn via the website
http://bioinformatics.psb.ugent.be/webtools/Venn/,
accessed on 22 January 2019. The statistical analysis associated
with the Venn diagram was performed via the website http://nemates.org/MA/progs/overlap_stats.html. The
number of genes in the genome was set to 1,917 miRNAs according to
the latest information of miRBase. For GSEA, P-values <0.01 and
false discovery rate (FDR) <25% were considered significant. For
GSEA of miRNAs P-values <0.05 were considered significant. All
other graphs were produced in GraphPad Prism 8 software (GraphPad
Software, Inc.). The Student's t-test was used for analysis of
differences between the vehicle and LTA-treated groups, using
GraphPad Prism 8. P<0.05 was considered to indicate a
statistically significant difference.
Results
Distribution of mRNA expression in human
neutrophils following LTA stimulation
Previous publications have reported that stimulation
with 0.1-10 µg/ml of S. aureus LTA induced the
release of cytokines, such as IL-8 and TNF-α, in human monocytes
within 1-6 h (38,39). In addition, the gene expression
profile of the human monocyte cell line THP-1 following stimulation
with 25 µg/ml LTA for 6 h was detected via microarray
analysis (40). The results
indicated that genes involved in inflammatory responses, cell
adhesion, cytokines and chemokines were upregulated following S.
aureus LTA stimulation (40).
In the present study, to investigate the mRNA and miRNA expression
changes in human neutrophils, human CD66abce+ cells were
enriched from peripheral blood obtained from a healthy donor. In
peripheral blood, the majority of the enriched CD66abce+
cells are considered neutrophils (41,42). The purity of the
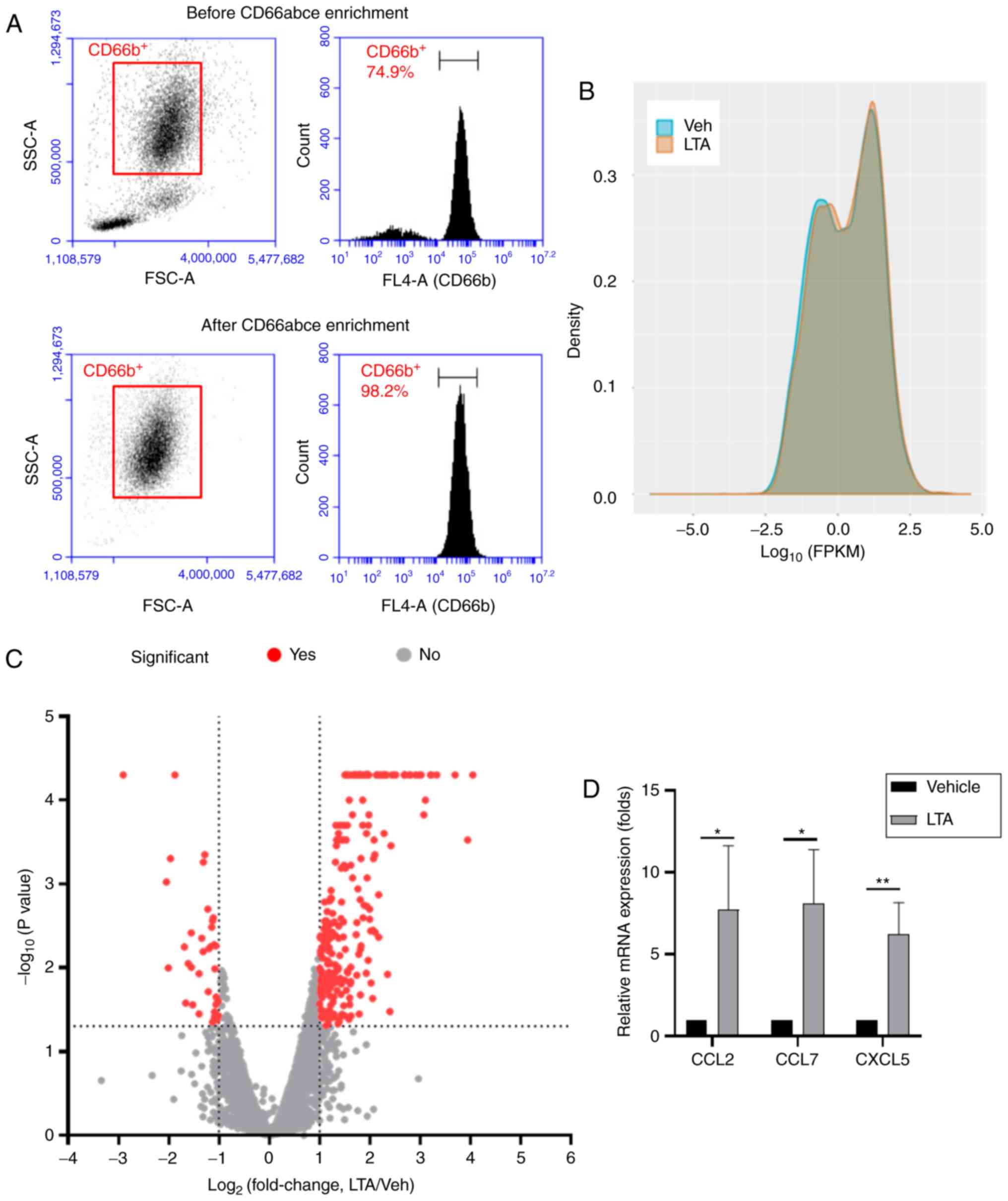
CD66acbe+ cells following enrichment was >98%, as
evidenced by flow cytometry analysis (Fig. 1A). Because 1 µg/ml of LTA
stimulation has been shown to be sufficient to activate signaling
downstream of TLR2 in immune cells (43), and the gene expression profile of
LTA-stimulated neutrophils has not been investigated, the isolated
human CD66abce+ cells were stimulated with 1
µg/ml LTA from S. aureus (LTA group) or vehicle
control (ddH2O; Veh group) for 16 h and then the total RNA was
extracted. Quality assessment of the RNA sequencing analysis is
shown in Figs. S2 and S3, reporting high scores in per-base
sequence quality and per-sequence quality in both groups. The
mapped reads for both RNA and small RNA sequencing are listed in
Table SI. The expression was
normalized in FPKM mapped reads. The distribution of the FPKM
values of the two samples was presented in a density plot (Fig. 1B). The results suggested that the
FPKM distribution was similar in the two samples. To further
investigate the differential gene expression in response to LTA
stimulation, the distribution of differentially expressed genes
between the two samples was plotted in a volcano plot (Fig. 1C). Genes with fold change ≥2.0
(log2 fold change >1 or <-1), FPKM >0.8 and
P-value <0.05 (-log10 P-value >1.3) were
considered as significant. According to these criteria, 290
significant differentially expressed genes were selected for
subsequent analysis (the full gene list is presented in Table SII). Furthermore, the mRNA
expression changes of CCL2, CCL7 and CXCL5 were validated by
RT-qPCR (Fig. 1D), confirming
that these genes were demonstrate to be significantly upregulated
following LTA stimulation by both the RT-qPCR and the RNA
sequencing analyses.
Evaluating the function of LTA-affected
genes
Previous studies have suggested that LTA stimulation
results in induction of inflammatory cytokines and chemokines, cell
migration and antimicrobial responses in immune cells. To further
investigate the function of the differential gene expression in
response to LTA stimulation, the 290 selected genes were subjected
to GO analysis for biological processes and KEGG pathway analysis
via the DAVID gene functional classification tool. Results with
P-values <0.001 and FDR <25% were considered as significantly
enriched biological processes and KEGG pathways. The results
revealed that >200 biological processes and 4 KEGG pathways were
enriched. The gene list in each enriched biological process and
KEGG pathway is present in Tables
SIII and SIV. The top 20
most significant enriched biological processes are presented in
Table I. These biological
processes, including positive regulation of cell migration and
motility, positive regulation of cellular component movement,
response to external stimulus, defense responses, inflammatory
responses and cell surface receptor signaling pathway, were similar
with the LTA-induced responses of neutrophils in previous studies
(11-14). The results of KEGG pathway
analysis are presented Table
II.
 | Table IGO analysis for biological processes
via DAVID gene functional classification tool. |
Table I
GO analysis for biological processes
via DAVID gene functional classification tool.
| GO ID | GO term | Enrichment | P-value | FDR |
|---|
| GO:0030335 | Positive regulation
of cell migration | 6.237193 |
6.81×10−19 |
1.29×10−15 |
| GO:0051272 | Positive regulation
of cellular component movement | 6.020298 |
7.20×10−19 |
1.36×10−15 |
| GO:2000147 | Positive regulation
of cell motility | 6.023695 |
2.17×10−18 |
4.11×10−15 |
| GO:0030334 | Regulation of cell
migration | 4.543111 |
2.72×10−18 |
5.16×10−15 |
| GO:0040017 | Positive regulation
of locomotion | 5.838131 |
6.11×10−18 |
1.16×10−14 |
| GO:2000145 | Regulation of cell
motility | 4.316392 |
8.70×10−18 |
1.65×10−14 |
| GO:0006954 | Inflammatory
response | 4.580108 |
2.92×10−17 |
5.54×10−14 |
| GO:0040012 | Regulation of
locomotion | 4.136543 |
4.70×10−17 |
8.90×10−14 |
| GO:0006952 | Defense
response | 2.989709 |
4.85×10−17 |
9.19×10−14 |
| GO:0051270 | Regulation of
cellular component movement | 4.042019 |
5.25×10−17 |
9.93×10−14 |
| GO:0016477 | Cell migration | 3.285506 |
3.73×10−16 |
6.33×10−13 |
| GO:0070887 | Cellular response
to chemical stimulus | 2.301508 |
9.52×10−16 |
1.89×10−12 |
| GO:0006950 | Response to
stress | 2.010083 |
2.23×10−15 |
4.21×10−12 |
| GO:0009605 | Response to
external stimulus | 2.502035 |
4.33×10−15 |
8.19×10−12 |
| GO:0051674 | Localization of
cell | 3.022336 |
4.56×10−15 |
8.62×10−12 |
| GO:0048870 | Cell motility | 3.022336 |
4.56×10−15 |
8.62×10−12 |
| GO:0007166 | Cell surface
receptor signaling pathway | 2.234817 |
1.38×10−14 |
2.61×10−11 |
| GO:0032879 | Regulation of
localization | 2.280856 |
3.45×10−14 |
6.54×10−11 |
| GO:0010033 | Response to organic
substance | 2.159601 |
4.98×10−14 |
9.42×10−11 |
| GO:0048583 | Regulation of
response to stimulus | 1.961424 |
6.01×10−14 |
1.14×10−10 |
| Table IIKyoto encyclopedia of genes and
genomes pathway analysis via DAVID gene functional classification
tool. |
Table II
Kyoto encyclopedia of genes and
genomes pathway analysis via DAVID gene functional classification
tool.
| Path ID | Path name | Enrichment | P-value | FDR |
|---|
| hsa04060 | Cytokine-cytokine
receptor interaction | 4.2657 |
2.88×10−8 |
3.62×10−5 |
| hsa05205 | Proteoglycans in
cancer | 3.5337 |
7.46×10−5 | 0.0938 |
| hsa04015 | Rap1 signaling
pathway | 3.3655 |
1.26×10−4 | 0.1585 |
| hsa04062 | Chemokine signaling
pathway | 3.5464 |
1.38×10−4 | 0.1738 |
The functions of the 290 genes were also analyzed by
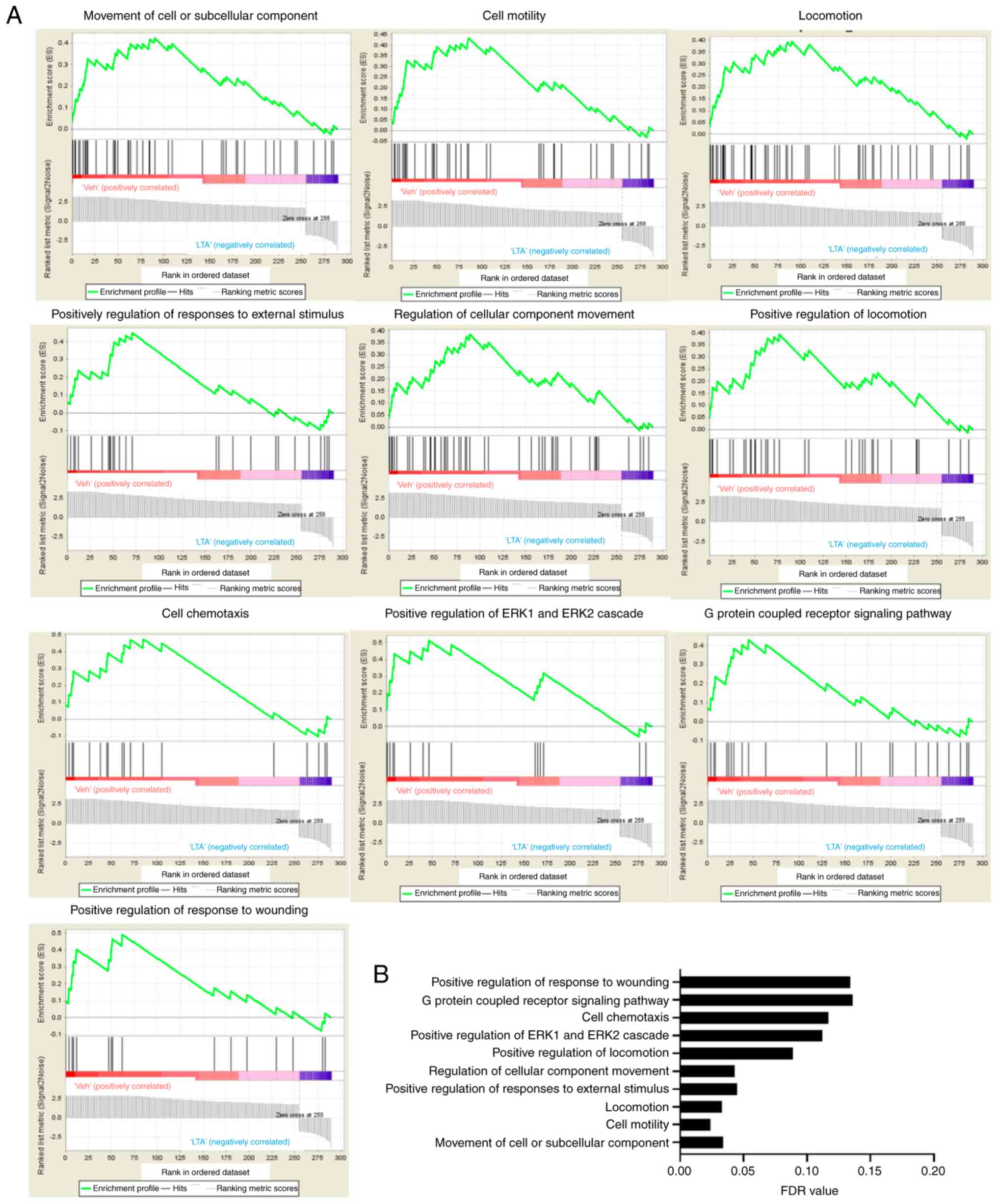
GSEA. The results revealed that 53 gene sets were significant at
FDR <25% and 16 gene sets were significant at nominal P-value
<1%. According to the FDR value, the top 10 most significant
gene sets are presented in Fig.
2. The gene sets identified by GSEA analysis were similar with
those from GO analysis, including cell motility, locomotion,
cellular component movement, G protein-coupled receptor signaling
and positive regulation of ERK1 and ERK2 cascade, were shown.
Upregulation of cell migration and granule degranulation in
LTA-stimulated neutrophils has been reported in previous studies
(11-14). By contrast, upregulation of
cytokines, such as IL-8, IL-6, TNF-α and G-CSF, was not observed in
the present results; the expression changes of those four genes
were <2-fold in the present RNA sequencing results (data not
shown).
Evaluating the function of LTA-affected
miRNAs
The miRNA expression was also determined via miRNA
sequencing in the present study. miRNA expression was considered
significantly changed based on fold change ≥2.0 and reads per
million (RPM) >1. Compared to vehicle-stimulated neutrophils, 38
miRNAs, including 36 downregulated miRNAs and 2 upregulated miRNAs,
were identified as significantly differentially expressed in
LTA-stimulated neutrophils. The list of the 38 significant
differentially expressed miRNAs is presented in Table SV. The miRNA enrichment analysis
was determined via Funrich software according to biological
process. The Funrich analysis suggested that these miRNAs were
signifi-cantly involved in signal transduction and cell
communication (P<0.05; Fig.
3A). In addition, the GSEA-like method of gene ontology
analysis was performed via the miEAA website, which is a miRNA
enrichment analysis and annotation web-based application. Pathways
such as nucleotide binding, signal transduction, cell cortex,
protein autophosphorylation, transcription corepressor and energy
reserve metabolic process were enriched in the LTA-stimulated
neutrophils (Fig. 3B and Table III). Pathways such as
microtubule-based process, cytoskeleton-dependent intracellular
transport, protein polymerization and ubiquitin binding were
suppressed (Fig. 3B and Table III). Signal transduction was the
only enriched biological process observed with both the analysis
methods.
| Table IIIGO analysis of miRNA via miEAA
website. |
Table III
GO analysis of miRNA via miEAA
website.
| GO ID | Path name | Enrichment | P-value | miRNA |
|---|
| GO0000166 | Nucleotide
binding | Enriched | 0.0057 | hsa-miR-10a-3p;
hsa-miR-193a-3p; hsa-miR-22-5p; hsa-miR-331-5p; hsa-miR-34a-5p;
hsa-miR-34c-5p; hsa-miR-362-5p; hsa-miR-378a-5p; hsa-miR-940 |
| GO0007017 | Microtubule based
process | Depleted | 0.0085 | hsa-miR-708-5p;
hsa-miR-940 |
| GO0030705 | Cytoskeleton
dependent | Depleted | 0.0085 | hsa-miR-708-5p;
hsa-miR-940 |
| intracellular
transport | | | |
| GO0051258 | Protein
polymerization | Depleted | 0.0085 | hsa-miR-708-5p;
hsa-miR-940 |
| GO0043130 | Ubiquitin
binding | Depleted | 0.0110 | hsa-miR-34a-5p;
hsa-miR-708-5p; hsa-miR-708-3p; hsa-miR-940 |
| GO0007165 | Signal
transduction | Enriched | 0.0132 | hsa-miR-10a-3p;
hsa-miR-1271-5p; hsa-miR-22-5p; hsa-miR-31-3p; hsa-miR-331-5p;
hsa-miR-337-3p; hsa-miR-34a-5p; hsa-miR-34c-5p; hsa-miR-378a-5p;
hsa-miR-3928-3p; hsa-miR-625-5p; hsa-miR-708-5p |
| GO0005938 | Cell cortex | Enriched | 0.0149 | hsa-miR-193a-3p;
hsa-miR-31-3p; hsa-miR-34a-5p; hsa-miR-34c-5p |
| GO0046777 | Protein
autophosphorylation | Enriched | 0.0149 | hsa-miR-193a-3p;
hsa-miR-31-3p; hsa-miR-34a-5p; hsa-miR-34c-5p |
| GO0003714 | Transcription
corepressor activity | Enriched | 0.0176 | hsa-miR-193a-3p;
hsa-miR-34a-5p; hsa-miR-34c-5p; hsa-miR-362-3p;
hsa-miR-378a-5p |
| GO0006112 | Energy reserve
metabolic process | Enriched | 0.0176 | hsa-miR-10a-3p;
hsa-miR-337-3p; hsa-miR-34a-5p; hsa-miR-34c-5p;
hsa-miR-378a-5p |
Evaluating the potential interaction
between LTA-affected miRNA and genes
To further identify whether the 38 miRNAs may
interact with the 290 LTA-affected genes, the Funrich software and
the miRDB website were used. The analysis results of the Funrich
software and the miRDB website, respectively, indicated 264 miRNAs
and 350 miRNAs might target to 342 genes. Seven and 15 shared
miRNAs were respectively identified between the 38 miRNAs and 264
Funrich-predicted miRNAs (Fig.
4A), and 38 miRNAs and 350 miRDB-predicted miRNAs (Fig. 4B). The results further revealed
that 5 miRNAs, hsa-miR-1271-5p, hsa-miR-708-5p, hsa-miR-362-3p,
hsa-miR-34c-5p and hsa-miR-34a-5p, were observed in both analyses.
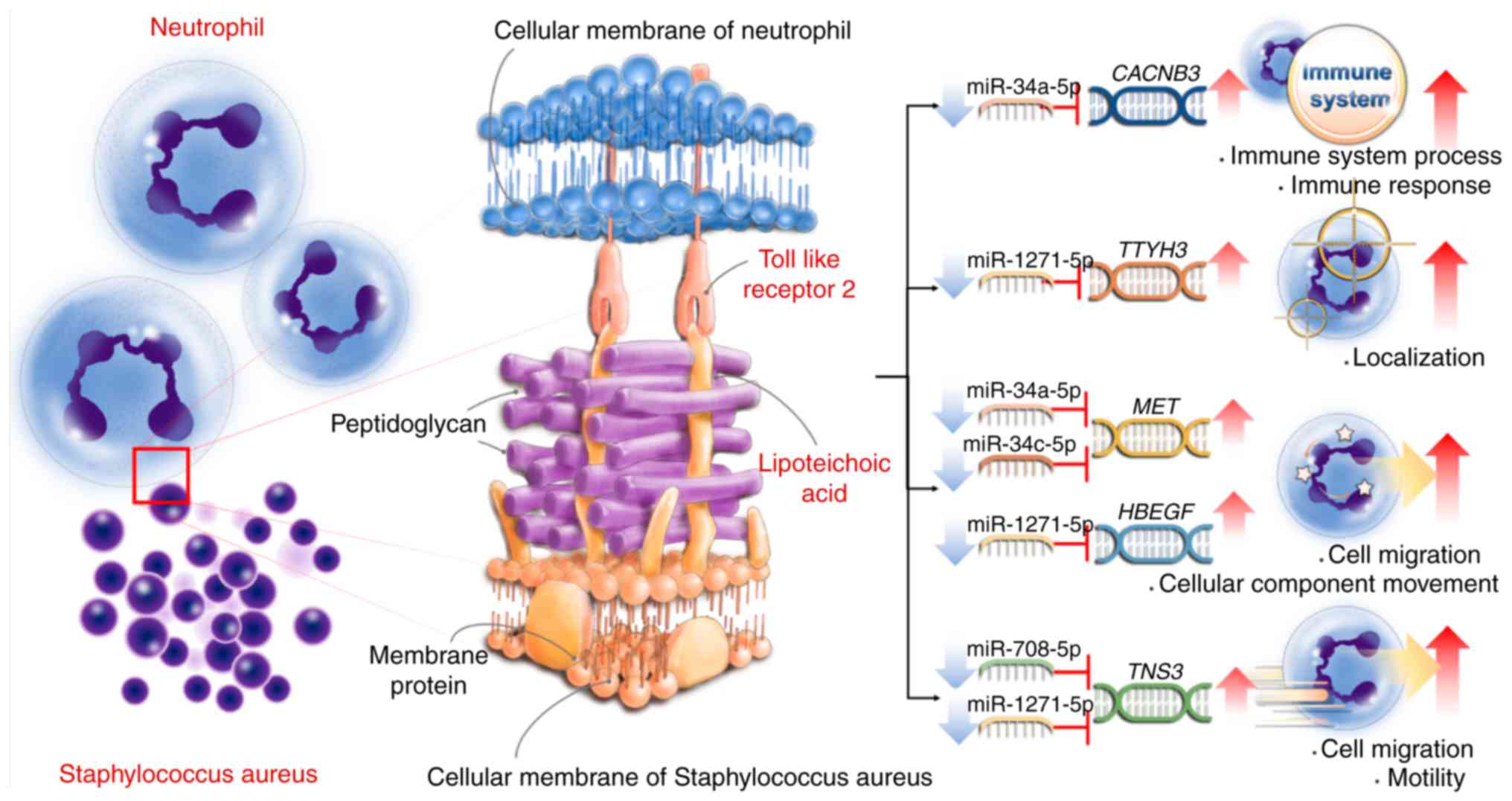
The potential interaction between 5 miRNAs and 290 genes was
further validated by Targetscan and miRTarBase database analyses.
The interaction between the 4 miRNAs and the 5 genes is presented
in Table IV. These genes were
involved in various biological processes. For example, MET
proto-oncogene (MET) and heparin binding EGF like growth factor
(HBEGF) were involved in positive regulation of cell migration and
cellular component movement, calcium voltage-gated channel
auxiliary subunit β3 (CACNB3) was involved in immune system process
and immune response, tensin 3 (TNS3) was involved in cell migration
and motility, and tweety family member 3 (TTYH3) involved in
localization (Table SII). The
interactions between hsa-miR-34a-5p, hsa-miR-34c-5p and MET,
hsa-miR-34a-5p and CACNB3, and their biological function have been
validated in other publications (44-52).
| Table IVTarget genes of miRNAs. |
Table IV
Target genes of miRNAs.
| miRNA | Fold change of
miRNAa | Gene symbol | Fold change of
mRNAa | TargetScan | miRTarBase | (Refs.) |
|---|
| hsa-miR-34a-5p | −2.18 | MET | 2.23 | Yes | Yes | (40,46-48) |
| hsa-miR-34a-5p | −2.18 | CACNB3 | 0.49 | Yes | Yes | (45) |
| hsa-miR-34c-5p | −2.11 | MET | 2.23 | Yes | Yes | (40-44) |
| hsa-miR-708-5p | −2.07 | TNS3 | 6.41 | Yes | No | |
|
hsa-miR-1271-5p | −2.98 | TTYH3 | 2.70 | Yes | No | |
|
hsa-miR-1271-5p | −2.98 | TNS3 | 6.41 | Yes | No | |
|
hsa-miR-1271-5p | −2.98 | HBEGF | 3.78 | Yes | No | |
Although interactions were predicted between the 4
miRNAs and the 5 genes, potential interactions between most of
genes and miRNAs identified in the present study were not
identified. Because LTA stimulation induces defense responses,
further analysis focused on the genes and miRNAs involved in the
biological processes of positive regulation of cell migration and
motility and cellular component movement (Fig. 5). The results revealed that
various genes with >2-fold changes may also interact with miRNAs
with <2-fold changes. Further experimental evidence will be
necessary to confirm whether miRNA-mRNA interactions may be
important for regulation of LTA-induced signaling pathways.
Discussion
LTA is a cell wall polymer in Gram-positive bacteria
and a risk factor for sepsis. Based on their chemical structures,
LTAs can be grouped into different types. Type I LTA is present in
bacteria including S. aureus, Listeria monocytogenes
and Bacillus subtilis (53). Prior publications have reported
that LTAs from S. aureus and Streptococcus pneumoniae
(type IV) can induce secretion of IL-8, IL-6, IL-1β and TNF-α in
monocytes and macrophages (54,55). Although the half-life of
circulating neutrophils is only 6-8 h (56), a previous study demonstrated that
the production of IL-1β, IL-8 and TNF-α was significantly induced
when human neutrophils were incubated with 10 µg/ml S.
aureus LTA for 16 and 24 h (14). In addition, Hattar et al
(14) demonstrated that the
protein levels of IL-8 were induced by stimulation with 1, 5 and 10
µg/ml LTA after 16 h of incubation. In the present study,
the results of RNA sequencing provided a whole molecular picture of
LTA-induced gene expression, including a trend for increased
expression of IL-8, IL-6, TNF-α and G-CSF (although <2-fold),
and 290 genes with significantly differential expression in human
neutrophils following 16 h stimulation with 1 µg/ml LTA.
LTA-affected genes have been reported in several
types of cells in previous studies. Two previously published
datasets in the Gene Expression Omnibus (GEO) database, GSE15512
and GSE21188, include results from microarray analysis determining
the gene expression of an LTA-treated monocyte cell line (25
µg/ml LTA was used to stimulate THP-1 cells for 6 h) and
peripheral blood mononuclear cells (PBMCs; 10 µg/ml LTA was
used to stimulate PBMCs for 7 h), respectively. Although the dose
and duration of the LTA stimulations were not identical, the
expression of inflammatory genes in the present study was compared
with that in both datasets. In general, the upregulated expression
of genes including IL-1β, IL-6, CXCL8, CCL2 and CCL20 were similar
in the present study and both datasets. Notably, LTA stimulation in
THP-1 cells induced more significant changes in gene expression
compared with LTA stimulation in PBMCs and in the present study.
Based on 20 shared inflammatory-associated genes among the present
study and the two public databases, the gene expression profile in
the present study was moderately positively correlated with that in
the public databases. These findings might suggest that genes can
be affected differently by the various doses of LTA in a short (6-7
h) and long (16 h) incubation period.
The results of mRNA sequencing revealed various
biological processes and signaling pathways that were enriched
following LTA stimulation. In Table
I, positively regulation of cell migration, cell motility and
locomotion, as well as defense responses and inflammatory
responses, were observed. Additionally, KEGG pathway analysis
revealed that 4 pathways were enriched, including cytokine-cytokine
receptor interaction and chemokine signaling pathway (Table II). A previous study demonstrated
that B. subtilis LTA increases the secretion of CCL2 and
CXCL10 in odontoblasts (57). To
the best of our knowledge, the induction of chemokines is not fully
elucidated in neutrophils. In odontoblasts, fibroblasts and pulpal
cells, activation of TLR2, TLR3 and TLR4 pathways induces the
production of several chemokines, such as CCL2, CCL7, IL-8 (CXCL8)
and CXCL10 (58,59). In human lymphatic endothelium
cells, LTA stimulation induces the expression of CCL2, CCL5, CXCL1,
CXCL3, CXCL5, CXCL6 and IL-8 (CXCL8) through a TLR2-depedent
mechanism (60). The present
study revealed upregulation of CCL2, CCL7 and CXCL5 in
LTA-stimulated neutrophils (Fig.
5). In addition, the expression of TLR2 was also upregulated
(by 2.02-fold) following LTA stimulation. Therefore, it is supposed
that TLR2 might be also essential for chemokine signaling pathway
in human neutrophils. The role of TLR2 in the regulation of the
chemokine signaling pathway, the function of proteoglycans, and the
role of the Rap1 signaling pathway in neutrophils, will be further
investigated in subsequent studies.
The effect of LTA stimulation on the miRNA
expression remained unclear. In a mouse model, Staphylococcus
epidermidis LTA induced the expression of miR-143 via TLR2
signaling (61). When mice were
exposed to LTA from B. subtilis, S. faecalis and
S. aureus, the expression of miR-451, miR-668, miR-1902 and
miR-1904 was induced in whole blood and serum (62). The present study found 38 miRNAs
with >2-fold change in expression following LTA stimulation; the
majority of these 38 miRNAs were novel and not reported in previous
publications. However, miR-143, miR-451, miR-668, miR-1902 and
miR-1904 were not significantly altered in the human LTA-stimulated
neutrophils in the present study. Because a miRNA can target many
genes (63), GO analysis of the
38 miRNAs was performed (Table
III). However, the function of these miRNAs and the regulatory
mechanism of these enriched biological processes and LTA-mediated
responses remained unknown. Therefore, the miRNA-target gene
interactions were further investigated via multiple bioin-formatic
tools. The results revealed potential novel interactions between
hsa-miR-34a-5p, hsa-miR-34c-5p, hsa-miR-708-5p hsa-miR-1271-5p and
MET, HBEGF, CACNB3, TNS3 and TTYH3, and that these interactions may
regulate cell migration and motility, cellular component movement,
immune system process and immune response. Although these findings
were interesting, there are some limitations in the current study.
Firstly, only one sample from one donor was analyzed in the present
study. Furthermore, the interactions between miRNA and mRNA were
not experimentally confirmed. The proposed interactions require
further validation through functional experiments in the future.
The summary of the present findings is presented in Fig. 6.
To the best of our knowledge, the present study is
the first to provide comprehensive information about transcriptome
analysis of LTA-stimulated human neutrophils. A total of 290 mRNAs
and 38 miRNAs which were significantly altered by 16 h-stimulation
of S. aureus LTA in human neutrophils were identified.
Furthermore, bioinformatic analysis proposed novel interactions
between 4 miRNAs and 5 target genes. These findings may provide new
insights of the LTA-mediated effect on peripheral neutrophils and
the innate immune responses in a healthy person.
Supplementary Data
Acknowledgments
The authors thank the Center for Research Resources
and Development of Kaohsiung Medical University.
Funding
This study was supported by grants from the Ministry
of Science and Technology (grant nos. MOST 108-2314-B-037-097-MY3,
107-2320-B-037-011-MY3 and 106-2320-B-037-029-MY3), the Kaohsiung
Medical University Hospital (grant nos. KMUH107-7M36, KMUH107-7R81,
KMUHS10701 and KMUHS10712), the Kaohsiung Medical University
Research Center Grant (grant no. KMU-TC108A04) and the Kaohsiung
Medical University (grant nos. KMU-DK108003 and KMU-Q108005).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MY and MT conceived and designed the experiments.
IY, KL and SJ prepared the materials and performed the experiments.
MY, IY, CL, MT and PK analyzed the data. MY wrote the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Institutional Review
Board of Kaohsiung Medical University Hospital (IRB no.
KMUH-IRB-20120287). Signed informed consent was obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Brubaker SW, Bonham KS, Zanoni I and Kagan
JC: Innate immune pattern recognition: A cell biological
perspective. Annu Rev Immunol. 33:257–290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mogensen TH: Pathogen recognition and
inflammatory signaling in innate immune defenses. Clin Microbiol
Rev. 22:240–273, Table of Contents, 2009. PubMed/NCBI
|
|
3
|
Kawasaki T and Kawai T: Toll-like receptor
signaling pathways. Front Immunol. 5:4612014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu YC, Yeh WC and Ohashi PS: LPS/TLR4
signal transduction pathway. Cytokine. 42:145–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park BS and Lee JO: Recognition of
lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med.
45:e662013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seo HS, Michalek SM and Nahm MH:
Lipoteichoic acid is important in innate immune responses to
gram-positive bacteria. Infect Immun. 76:206–213. 2008. View Article : Google Scholar :
|
|
7
|
Schwandner R, Dziarski R, Wesche H, Rothe
M and Kirschning CJ: Peptidoglycan- and lipoteichoic acid-induced
cell activation is mediated by toll-like receptor 2. J Biol Chem.
274:17406–17409. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oliveira-Nascimento L, Massari P and
Wetzler LM: The role of TLR2 in infection and immunity. Front
Immunol. 3:792012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kengatharan KM, De Kimpe S, Robson C,
Foster SJ and Thiemermann C: Mechanism of gram-positive shock:
Identification of peptidoglycan and lipoteichoic acid moieties
essential in the induction of nitric oxide synthase, shock, and
multiple organ failure. J Exp Med. 188:305–315. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kurt-Jones EA, Mandell L, Whitney C,
Padgett A, Gosselin K, Newburger PE and Finberg RW: Role of
toll-like receptor 2 (TLR2) in neutrophil activation: GM-CSF
enhances TLR2 expression and TLR2-mediated interleukin 8 responses
in neutrophils. Blood. 100:1860–1868. 2002.PubMed/NCBI
|
|
11
|
Lotz S, Aga E, Wilde I, van Zandbergen G,
Hartung T, Solbach W and Laskay T: Highly purified lipoteichoic
acid activates neutrophil granulocytes and delays their spontaneous
apoptosis via CD14 and TLR2. J Leukoc Biol. 75:467–477. 2004.
View Article : Google Scholar
|
|
12
|
Ginsburg I: Role of lipoteichoic acid in
infection and inflammation. Lancet Infect Dis. 2:171–179. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nathan C: Neutrophils and immunity:
Challenges and opportunities. Nat Rev Immunol. 6:173–182. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hattar K, Grandel U, Moeller A, Fink L,
Iglhaut J, Hartung T, Morath S, Seeger W, Grimminger F and Sibelius
U: Lipoteichoic acid (LTA) from Staphylococcus aureus stimulates
human neutrophil cytokine release by a CD14-dependent,
Toll-like-receptor-independent mechanism: Autocrine role of tumor
necrosis factor-[alpha] in mediating LTA-induced interleukin-8
generation. Crit Care Med. 34:835–841. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Drury RE, O'Connor D and Pollard AJ: The
clinical application of MicroRNAs in infectious disease. Front
Immunol. 8:11822017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Lei C, He Q, Pan Z, Xiao D and Tao
Y: Nuclear functions of mammalian MicroRNAs in gene regulation,
immunity and cancer. Mol Cancer. 17:642018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X
and Xiong S: Autoantibody induction by DNA-containing immune
complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J
Immunol. 190:5411–5422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao H, Zhang H, Lan K, Wang H, Su Y, Li D,
Song Z, Cui F, Yin Y and Zhang X: Purified Streptococcus pneumoniae
endo-peptidase O (PepO) enhances particle uptake by macrophages in
a toll-like receptor 2- and miR-155-dependent manner. Infect Immun.
85:e01012–e01016. 2017. View Article : Google Scholar :
|
|
19
|
Xu H, Wu Y, Li L, Yuan W, Zhang D, Yan Q,
Guo Z and Huang W: MiR-344b1-3p targets TLR2 and negatively
regulates TLR2 signaling pathway. Int J Chron Obstruct Pulmon Dis.
12:627–638. 2017. View Article : Google Scholar :
|
|
20
|
Landais I, Pelton C, Streblow D,
DeFilippis V, McWeeney S and Nelson JA: Human cytomegalovirus
miR-UL112-3p targets TLR2 and modulates the TLR2/IRAK1/NFκB
signaling pathway. PLoS Pathog. 11:e10048812015. View Article : Google Scholar
|
|
21
|
Quinn EM, Wang JH, O'Callaghan G and
Redmond HP: MicroRNA-146a is upregulated by and negatively
regulates TLR2 signaling. PLoS One. 8:e622322013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for Illumina sequence data.
Bioinformatics. 30:2114–2120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim D, Langmead B and Salzberg SL: HISAT:
A fast spliced aligner with low memory requirements. Nat Methods.
12:357–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Friedlander MR, Mackowiak SD, Li N, Chen W
and Rajewsky N: miRDeep2 accurately identifies known and hundreds
of novel microRNA genes in seven animal clades. Nucleic Acids Res.
40:37–52. 2012. View Article : Google Scholar :
|
|
25
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar
|
|
29
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mootha VK, Lindgren CM, Eriksson KF,
Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E,
Ridderstråle M, Laurila E, et al: PGC-1alpha-responsive genes
involved in oxidative phosphorylation are coordinately
downregulated in human diabetes. Nat Genet. 34:267–273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Backes C, Khaleeq QT, Meese E and Keller
A: miEAA: microRNA enrichment analysis and annotation. Nucleic
Acids Res. 44:W110–W116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu W and Wang X: Prediction of functional
microRNA targets by integrative modeling of microRNA binding and
target expression data. Genome Biol. 20:182019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garcia DM, Baek D, Shin C, Bell GW,
Grimson A and Bartel DP: Weak seed-pairing stability and high
target-site abundance decrease the proficiency of lsy-6 and other
microRNAs. Nat Struct Mol Biol. 18:1139–1146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46:D296–D302.
2018. View Article : Google Scholar :
|
|
36
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Doncheva NT, Morris JH, Gorodkin J and
Jensen LJ: Cytoscape stringApp: Network analysis and visualization
of proteomics data. J Proteome Res. 18:623–632. 2019. View Article : Google Scholar
|
|
38
|
Finney SJ, Leaver SK, Evans TW and
Burke-Gaffney A: Differences in lipopolysaccharide- and
lipoteichoic acid-induced cytokine/chemokine expression. Intensive
Care Med. 38:324–332. 2012. View Article : Google Scholar :
|
|
39
|
Schröder NW, Morath S, Alexander C, Hamann
L, Hartung T, Zähringer U, Göbel UB, Weber JR and Schumann RR:
Lipoteichoic acid (LTA) of Streptococcus pneumoniae and
Staphylococcus aureus activates immune cells via Toll-like receptor
(TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14,
whereas TLR-4 and MD-2 are not involved. J Biol Chem.
278:15587–15594. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zeng RZ, Kim HG, Kim NR, Gim MG, Ko MY,
Lee SY, Kim CM and Chung DK: Differential gene expression profiles
in human THP-1 monocytes treated with Lactobacillus plantarum or
Staphylococcus aureus lipoteichoic acid. J Korean Soc Appl Bi.
54:763–770. 2011. View Article : Google Scholar
|
|
41
|
Sharma S, Davis RE, Srivastva S, Nylen S,
Sundar S and Wilson ME: A subset of neutrophils expressing markers
of antigen-presenting cells in human visceral leishmaniasis. J
Infect Dis. 214:1531–1538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen X, Li SJ, Ojcius DM, Sun AH, Hu WL,
Lin X and Yan J: Mononuclear-macrophages but not neutrophils act as
major infiltrating anti-leptospiral phagocytes during
leptospirosis. PLoS One. 12:e01810142017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Long EM, Millen B, Kubes P and Robbins SM:
Lipoteichoic acid induces unique inflammatory responses when
compared to other toll-like receptor 2 ligands. PLoS One.
4:e56012009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar
|
|
45
|
Cai KM, Bao XL, Kong XH, Jinag W, Mao MR,
Chu JS, Huang YJ and Zhao XJ: Hsa-miR-34c suppresses growth and
invasion of human laryngeal carcinoma cells via targeting c-Met.
Int J Mol Med. 25:565–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dong F and Lou D: MicroRNA-34b/c
suppresses uveal melanoma cell proliferation and migration through
multiple targets. Mol Vis. 18:537–546. 2012.PubMed/NCBI
|
|
47
|
Hagman Z, Haflidadottir BS, Ansari M,
Persson M, Bjartell A, Edsjö A and Ceder Y: The tumour suppressor
miR-34c targets MET in prostate cancer cells. Br J Cancer.
109:1271–1278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang F, Lu J, Peng X, Wang J, Liu X, Chen
X, Jiang Y, Li X and Zhang B: Integrated analysis of microRNA
regulatory network in nasopharyngeal carcinoma with deep
sequencing. J Exp Clin Cancer Res. 35:172016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bavamian S, Mellios N, Lalonde J, Fass DM,
Wang J, Sheridan SD, Madison JM, Zhou F, Rueckert EH, Barker D, et
al: Dysregulation of miR-34a links neuronal development to genetic
risk factors for bipolar disorder. Mol Psychiatry. 20:573–584.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yan D, Zhou X, Chen X, Hu DN, Dong XD,
Wang J, Lu F, Tu L and Qu J: MicroRNA-34a inhibits uveal melanoma
cell proliferation and migration through downregulation of c-Met.
Invest Ophthalmol Vis Sci. 50:1559–1565. 2009. View Article : Google Scholar
|
|
51
|
Guessous Li Y, Zhang F, Dipierro Y, Kefas
C, Johnson B, Marcinkiewicz E, Jiang L, Yang J, Schmittgen YTD, et
al: MicroRNA-34a inhibits glioblastoma growth by targeting multiple
oncogenes. Cancer Res. 69:7569–7576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yan K, Gao J, Yang T, Ma Q, Qiu X, Fan Q
and Ma B: MicroRNA-34a inhibits the proliferation and metastasis of
osteosarcoma cells both in vitro and in vivo. PLoS One.
7:e337782012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Percy MG and Grundling A: Lipoteichoic
acid synthesis and function in gram-positive bacteria. Annu Rev
Microbiol. 68:81–100. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Standiford TJ, Arenberg DA, Danforth JM,
Kunkel SL, VanOtteren GM and Strieter RM: Lipoteichoic acid induces
secretion of interleukin-8 from human blood monocytes: A cellular
and molecular analysis. Infect Immun. 62:119–125. 1994.PubMed/NCBI
|
|
55
|
Mattsson E, Verhage L, Rollof J, Fleer A,
Verhoef J and van Dijk H: Peptidoglycan and teichoic acid from
Staphylococcus epidermidis stimulate human monocytes to release
tumour necrosis factor-alpha, interleukin-1 beta and interleukin-6.
FEMS Immunol Med Microbiol. 7:281–287. 1993.PubMed/NCBI
|
|
56
|
Summers C, Rankin SM, Condliffe AM, Singh
N, Peters AM and Chilvers ER: Neutrophil kinetics in health and
disease. Trends Immunol. 31:318–324. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Durand SH, Flacher V, Roméas A, Carrouel
F, Colomb E, Vincent C, Magloire H, Couble ML, Bleicher F, Staquet
MJ, et al: Lipoteichoic acid increases TLR and functional chemokine
expression while reducing dentin formation in in vitro
differentiated human odontoblasts. J Immunol. 176:2880–2887. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Park C, Lee SY, Kim HJ, Park K, Kim JS and
Lee SJ: Synergy of TLR2 and H1R on Cox-2 activation in pulpal
cells. J Dent Res. 89:180–185. 2010. View Article : Google Scholar
|
|
59
|
Staquet MJ, Durand SH, Colomb E, Roméas A,
Vincent C, Bleicher F, Lebecque S and Farges JC: Different roles of
odonto-blasts and fibroblasts in immunity. J Dent Res. 87:256–261.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sawa Y, Tsuruga E, Iwasawa K, Ishikawa H
and Yoshida S: Leukocyte adhesion molecule and chemokine production
through lipoteichoic acid recognition by toll-like receptor 2 in
cultured human lymphatic endothelium. Cell Tissue Res. 333:237–252.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xia X, Li Z, Liu K, Wu Y, Jiang D and Lai
Y: Staphylococcal LTA-induced miR-143 inhibits propionibacterium
acnes-mediated inflammatory response in skin. J Invest Dermatol.
136:621–630. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hsieh CH, Yang JC, Jeng JC, Chen YC, Lu
TH, Tzeng SL, Wu YC, Wu CJ and Rau CS: Circulating microRNA
signatures in mice exposed to lipoteichoic acid. J Biomed Sci.
20:22013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|