Introduction
Pancreatic cancer is a common and highly malignant
tumor of the digestive system. In recent years, the global
morbidity and mortality of pancreatic cancer is increasing. In
Europe, new cases of pancreatic cancer in 2006 were 60,000, and
59,000 cases of death, ranking fifth in cancer mortality in Europe
(1). In the United States, new
cases were 37,170 in 2006, and 33,370 cases of death; in 2010, new
cases were 43140, ranking fourth in cancer mortality in the United
States (2,3). Surgical resection is the only hope to
improve survival rate, but the vast majority of pancreatic cancer
patients are diagnosed at advanced stage, and prognosis is poor,
the average 5-year survival rate is less than 5% (3). In addition to surgery, chemotherapy
is still an important means to treat advanced pancreatic cancer,
prevent post-surgical recurrence, prolong survival time and improve
life quality. Gemcitabine is currently the best first-line
chemotherapy to treat advanced pancreatic cancer (4), but due to acquired or intrinsic drug
resistance of pancreatic cancer cells (5,6),
effect of using gemcitabine to treat pancreatic cancer is still not
ideal (7), which causes patients
treated with gemcitabine to suffer from side effects of
chemotherapy without good treatment. Thus, it is particularly
important to find a drug that can reverse the drug-resistance and
enhance the treatment of gemcitabine. Emodin
(6-methyl-1,3,8-tanthragallol), which is the main active monomer
separated from Rheum, Polygonum, buckthorn and senna, is a tyrosine
kinase II inhibitor, it has effects of anti-microbial activity
(8–11), anti-inflammatory (12,13),
immunosuppression (14),
anti-tumor (15,16), and activity in protection of liver
cells (17). Our team previously
found emodin could promote apoptosis (16), and established the
gemcitabine-resistance cell line SW1990/Gem, we confirmed that
emodin could increase the sensibility of resistance cell line to
gemcitabine through inhibiting the expression of NF-κB (18). However, it is still unclear whether
emodin can reverse the gemcitabine-resistant human pancreatic
cancer cells, thus this study mainly investigates the effect of
emodin reversing the gemcitabine-resistance on SW1990/Gem cell
line, and its possible mechanisms.
Materials and methods
Chemicals and cell lines
The followings were purchased: Emodin (purity
>98%), MTT, DMSO (Sigma), gemcitabine (Eli Lilly and Co.), FBS,
trypsin containing EDTA, Roswell Park Memorial Institute-1640
(RPMI-1640) (Gibco), BCA protein assay kit (Pik-day Institute of
Biotechnology), Rhodamine123 (Rh123), Annexin V-FITC/PI apoptosis
detection kit (Biological Development Co., Ltd. Nanjing KGI), RNA
extraction kit (Life Technologies Co.); cDNAfirst strand synthesis
kit (Fermentas), 2X Taq PCR MasterMix (Tiangen), P-gp, NF-κB,
Bcl-2, Bax, cytochrome-C antibodies (Santa Cruz). Drug preparation:
Emodin was dissolved in DMSO as a 0.2 mM stock solution and stored
at −20°C. DMSO concentration <0.1% (it has no effect on cell
proliferation when concentration was <0.1%). Gemcitabine is
reconstituted as a 0.02 mM stock solution in sterile saline. Cell
culture: The human pancreatic cancer cell line SW1990 was purchased
from the American Type Culture Collection. Cells were cultured in
RPMI-1640 medium, supplemented with 10% fetal calf serum (FCS), in
a humidified atmosphere of 5% CO2 and 95% air at
37°C.
Establishment of gemcitabine-resistant
human pancreatic cancer cell line SW1990/Gem
Gemcitabine-resistant pancreatic cancer cell line
SW1990/Gem was obtained by culture of pancreatic cancer cell line
SW1990 in vitro with intermittently increasing the
concentration of gencitabine in the culture medium for 10 months.
After cultivating SW1990 cells with different concentrations of
gemcitabine for 1 week, we checked the cell death conditions and
chose the concentration of median lethal dose (LD80)
(which could kill 80% cells) as the initial concentration to
cultivate the resistant cell line. Cells were cultivated in this
medium for 48 h, and then incubated in RPMI-1640 medium without
drugs. When cells grew stably and entered the logarithmic growth
phase, they were passaged twice, and exposed to gemcitabine in
double LD80 concentration, after nine concentration
gradients and ~10 months of cultivation, they were finally
incubated in RPMI-1640 medium without drugs for 2 months.
Morphological assay of
gemcitabine-resistant cell line SW1990/Gem
Two lines of logarithmic phase SW1990/Gem and SW1990
cells were incubated in a 6-well plate at a density of 100,000
cells per well for 2 days, and were observed by optical microscope
(Nikon, TS100), and then were collected separately and fixed for
electron microscopic observation of cell ultra-structures.
Sensitivity analysis of SW1990/Gem to
gemcitabine
The logarithmic phase SW1990/Gem and SW1990 cells
were incubated in a 96-well plate at a density of 4,000 cells per
well. Cells were cultured in different concentrations (20, 40, 80
and 160 μM) of gemcitabine for 48 h after they adhered. Each group
had 6-wells. The supernatant was discarded and 20 μl MTT (5 mg/ml)
was added with 180 μl medium to each well, 4 h later the culture
medium was removed and 150 μl DMSO was added to each well. The
plate was shaken by microplate shaker for 10 min and the absorbance
(A) of samples was measured at 490 nm by automatic enzyme-linked
immunosorbent assay. The experiment was repeated three times. The
drug inhibition of cells was calculated by the following formula:
Inhibition = 1-dosing group A/control group A × 100%. Data was
graphed on a semi-logarithmic curve with drug concentrations
plotted on the x-axis and cell inhibitions on the y-axis. SPSS
software was used to calculate the 50% inhibitory inhibition
(IC50) (19) and the
resistance index (RI). RI = IC50 of resistance cell
line/IC50 of the sensitive cell line.
Effect of gemcitabine on SW1990/Gem
proliferation after pretreatment with emodin
SW1990/Gem cells were incubated in a 96-well plate
at a density of 4,000 cells per well overnight. Cells were
pretreated with low emodin (10 μM) for different periods (12, 24,
36, 48 and 60 h) and then incubated with gemcitabine for 48 h.
Emodin was not added to the control group, and it was directly
incubated in gemcitabine for 48 h. The supernatant was discarded
and MTT (5 mg/ml) was added, 4 h later the culture medium was
removed and 150 μl DMSO was added to each well. The plate was
shaken by a microplate shaker for 10 min and absorbance (A) of
samples were measured. Each group had 6-wells. The experiment was
repeated three times, and the cell viability was calculated.
Effect of emodin on SW1990/Gem cell
apoptosis
The logarithmic phase SW1990/Gem cells were
incubated in a 6-well plate (4×105/well), treated with
different concentrations of emodin (10, 20, 40, 80 and 160 μM) and
the control group when cells were 80% confluent. Forty-eight hours
later, cells were collected and centrifuged at 1000 rpm/min for 5
min. Cells were washed with cold PBS 3 times and resuspended with
500 μl binding buffer, then 5 μl Annexin V-FITC was added, mixed,
and cultured in the dark for 5 min, adding 10 μl PI for 5 min. The
fluorescence of cells was measured by flow a cytometer at 488/530
nm. The experiment was repeated three times, and cells were
analyzed with Cell Quest software.
Protein expression of MDR-1 (P-gp),
NF-κB, Bcl-2, Bax, cytochrome-C (cytosol), caspase-9 and -3 were
detected in SW1990/Gem and SW1990 by Western blotting
SW1990/Gem and SW1990 cells were collected, the
cytoplasmic protein was evaluated with the
mitochondrial/cytoplasmic protein isolation kit to detect
cytochrome-C levels according to the instructions. Cells were lysed
with RIPA, centrifuged at 12000 rpm/min, collecting the
supernatant. Protein concentrations were measured with BCA kit,
then the amount of protein was equaled, in 10% SDS-PAGE
electrophoresis, PVDF membrane was transferred, then blocked with
5% skimmed milk powder, incubated with antibodies at 4°C overnight,
then washed with TBST and incubated with secondary antibodies for 2
h, after washing with TBST and coloring with ECL, the membranes
were exposed to X-rays. The experiment was repeated three
times.
Protein expression of P-gp, NF-κB, Bcl-2,
Bax, cytochrome-C, caspase-9 and -3 in SW1990/Gem after treatment
with emodin and/or gemcitabine by Western blotting
SW1990/Gem was treated with emodin (10 μM) and
gemcitabine (20 μM) alone or together for 48 h, cytoplasmic protein
was gathered with the mitochondrial/cytoplasmic protein isolation
kit to detect cytochrome-C levels according to the instructions.
The other steps were as above.
Analysis of MDR-1 (P-gp), NF-κB, Bcl-2,
Bax, cytochrome-C, caspase-9 and -3 in SW1990/Gem after treatment
with emodin and/or gemcitabine by reverse transcription-PCR
After cultivating SW1990/Gem cells treated with
emodin (10 μM) and gemcitabine (20 μM) alone or combined for 48 h,
cells were lysed by TRIzol and RNA was extracted, then the content
of RNA was measured by UV spectrophotometer at 260 nm. cDNA under
the instruction of the first Fermentas cDNA strand synthesis kit
was synthesized (Tiangen 2X Taq PCR MasterMix instruction) the PCR
amplification conditions were: MDR-1, 94°C 30 sec, 57°C 30 sec,
72°C 30 sec, 30 cycles; NF-κB, 94°C 30 sec, 54°C 30 sec, 72°C 20
sec, 30 cycles; Bcl-2, 94°C 20 sec, 58°C 20 sec, 72°C 20 sec, 35
cycles; Bax, 94°C 30 sec, 57°C 30 sec, 72°C 20 sec, 30 cycles;
cytochrome-C, 94°C 30 sec, 60°C 30 sec, 72°C 30 sec, 35 cycles;
caspase-9, 94°C 30 sec, 56°C 30 sec, 72°C 30 sec, 35 cycles;
caspase-3, 94°C 30 sec, 57°C 30 sec, 72°C 30 sec, 35 cycles; GAPDH,
94°C 30 sec, 54°C 30 sec, 72°C 20 sec, 25 cycles. GAPDH was used as
an internal control. Product (5-μl) was added to the 1.5% agarose
gel electrophoresis and images were taken. The RT-PCR sequences of
primers and the size of the sequences are shown in Table I.
 | Table IRT-PCR sequences of primers and the
size of the sequences. |
Table I
RT-PCR sequences of primers and the
size of the sequences.
| Gene | Sense primer | Antisense
primer | PCR product
(bp) |
|---|
| MDR-1 |
GAATCTGGAGGAAGACATGACC |
TCCAATTTTGTCACCAATTCC | 259 |
| NF-κB |
AGCACAGATACCACCAAGACCC |
CCCACGCTGCTCTTCTATAGCAAC | 300 |
| Bcl-2 |
AGCCGGGAGAACAGGGTATG |
ATCCAGGTGTGCATGCCG | 549 |
| Bax |
ATGGCTGGGGAGACACCTGA |
TGGGCGTCCCGAAGTAGGAA | 394 |
| Cyt-c |
GCGTGTCCTTGGACTTAGAG |
GGCGGCTGTGTAAGAGTATC | 241 |
| Caspase-9 |
GGTTCTGGAGGATTTGGTGA |
GACAGCCGTGAGAGAGAATGA | 325 |
| Caspase-3 |
AGCAAACCTCAGGGAAACATT |
GTCTCAATGCCACAGTCCAGT | 309 |
| GAPDH |
AACGGATTTGGTCGTATTGGG |
TCGCTCCTGGAAGATGGTGAT | 216 |
Analysis of P-gp function in SW1990/Gem
by Rhodamine123 (Rh123) efflux experiment (flow cytometric
analysis)
After SW1990/Gem cells were incubated with emodin
and gemcitabine alone or combination for 48 h, they were
resuspended in medium (1×106/ml), then Rhodamine 123
staining solution 10 μg/ml was added and cultured in an incubator
with 37°C 5% CO2 for 30 min, centrifuged and washed with
medium twice, resuspended in medium and incubated in the incubator
for 120 min, centrifuged again and washed with PBS twice, measured
by flow cytometry at 488/530 nm. This experiment was repeated three
times.
Statistical analysis
Data were expressed as the mean ± SD and evaluated
by SPSS16.0. The differences were considered to be statistically
significant at P<0.05.
Results
Biological properties of the
gemcitabine-resistant cell line SW1990/Gem and sensitivity
testing
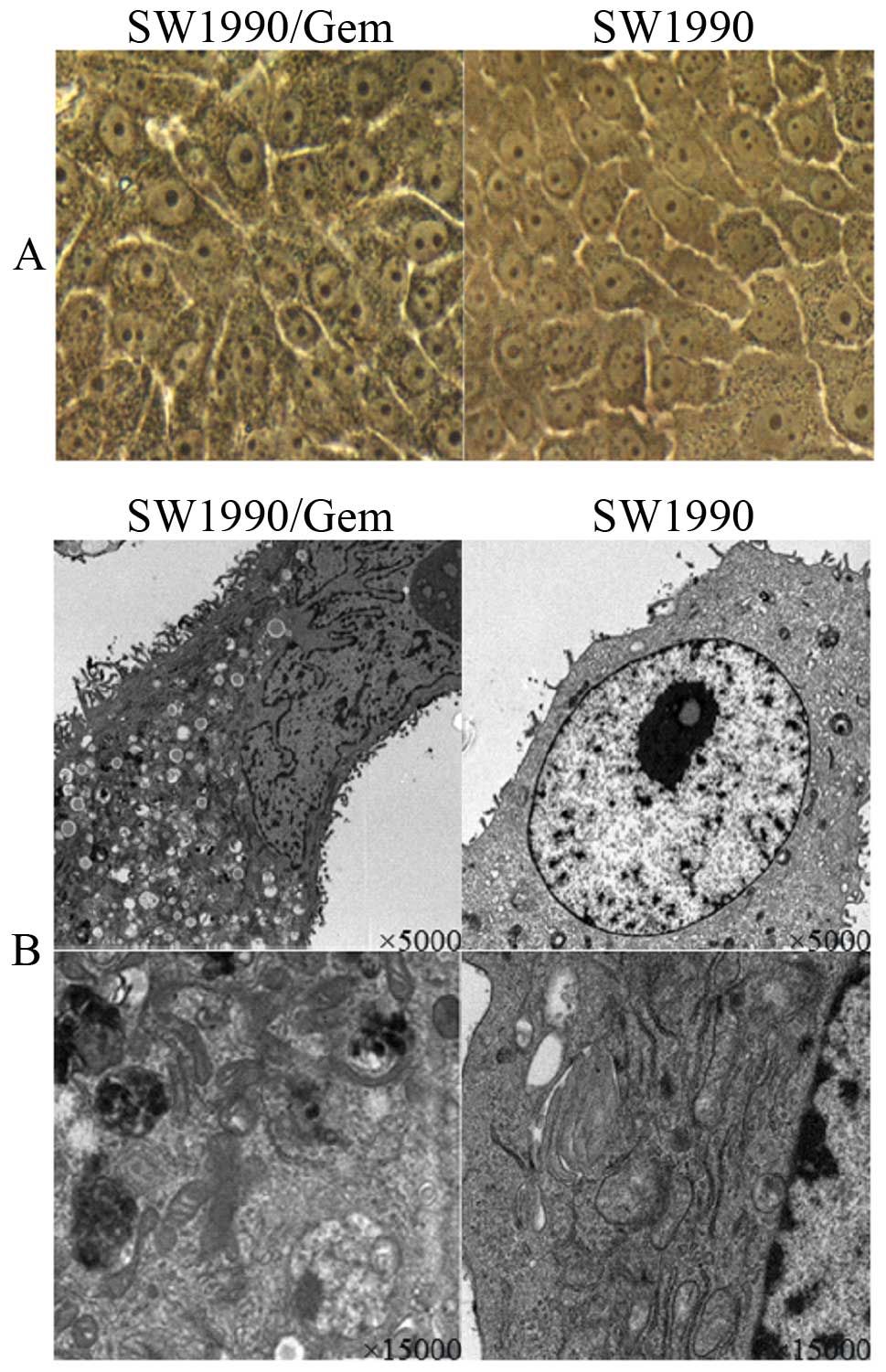
We achieved the stable passage resistance cell line
after 10 months via in vitro culture, after incubation
without drugs for 2 months, compared with parental cell line
SW1990, the SW1990/Gem changed significant in morphology. Under
light microscope (x400) (Fig. 1A),
the volume of SW1990/Gem cells increased, was different in size and
the granular substances increased, SW1990/Gem cells with more
nucleoli increased. Under electron microscopy (×5,000 and ×15,000)
(Fig. 1B), microvilli at the
surface of the SW1990/Gem cell membranes increased, the SW1990/Gem
cell surface area increased, cell organelles in the cytoplasm
increased, mitochondria cristaes were disordered, vacuoles were
also found in the SW1990/Gem cell matrix, endoplasmic reticulum and
the vacuole structures in cytoplasm were increased. The
gemcitabine-resistant cell line SW1990/Gem 50% inhibitory
concentration (IC50) was 1267.53±26.78 μM, the
resistance index was 48.63. The resistant cell line SW1990/Gem
showed significant resistance to gemcitabine.
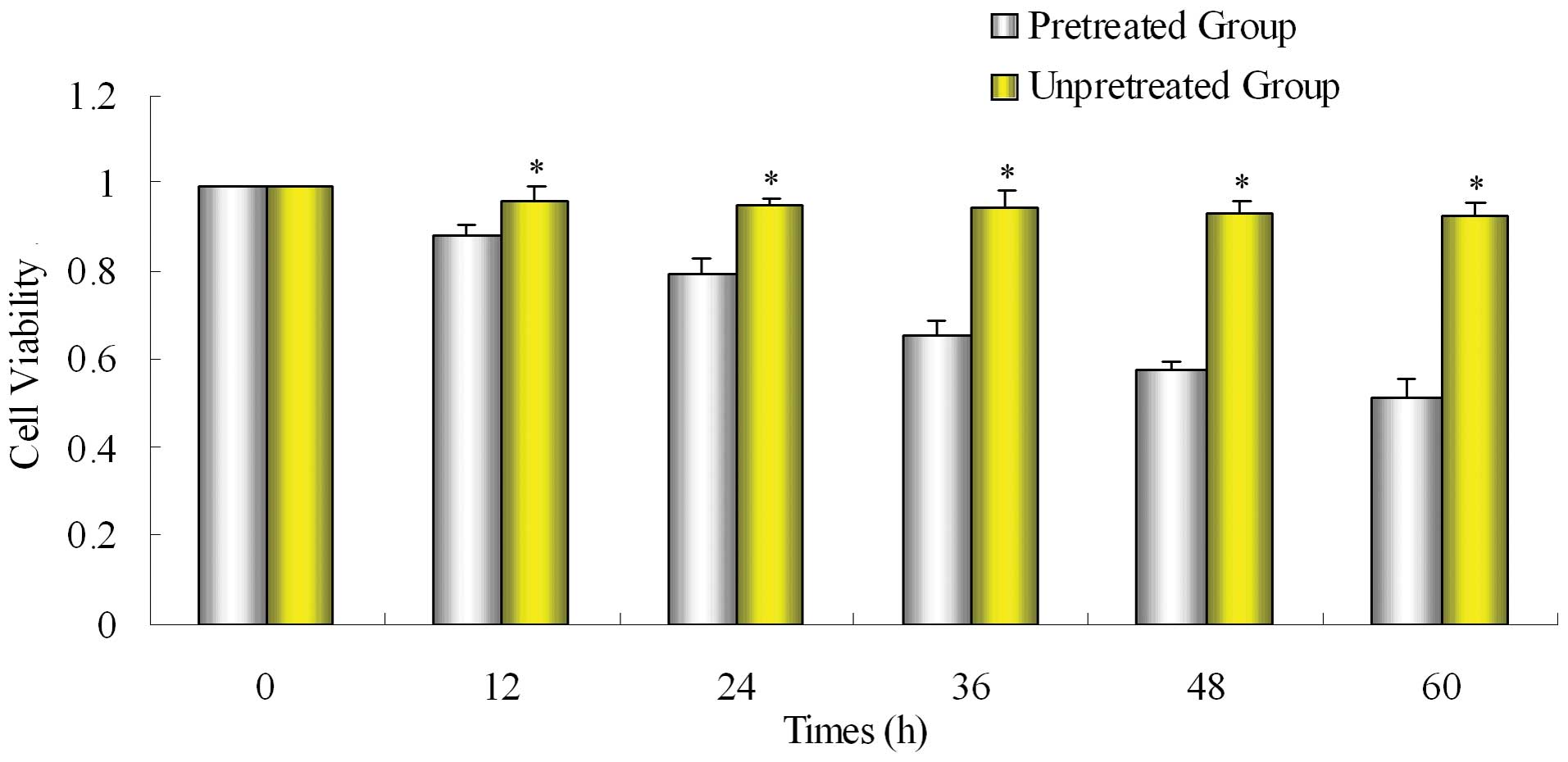
After SW1990/Gem cells were pretreated
with emodin, the sensitivity to gemcitabine was significantly
enhanced
Pretreated group was pretreated with emodin and then
treated with gemcitabine for 48 h, unpretreated group was treated
with only gemcitabine for 48 h. Compared with the unpretreated
group, the inhibiting effect of gemcitabine on proliferation of
gemcitabine-resistant cell line SW1990/Gem was significantly
enhanced in pretreated group (Fig.
2).
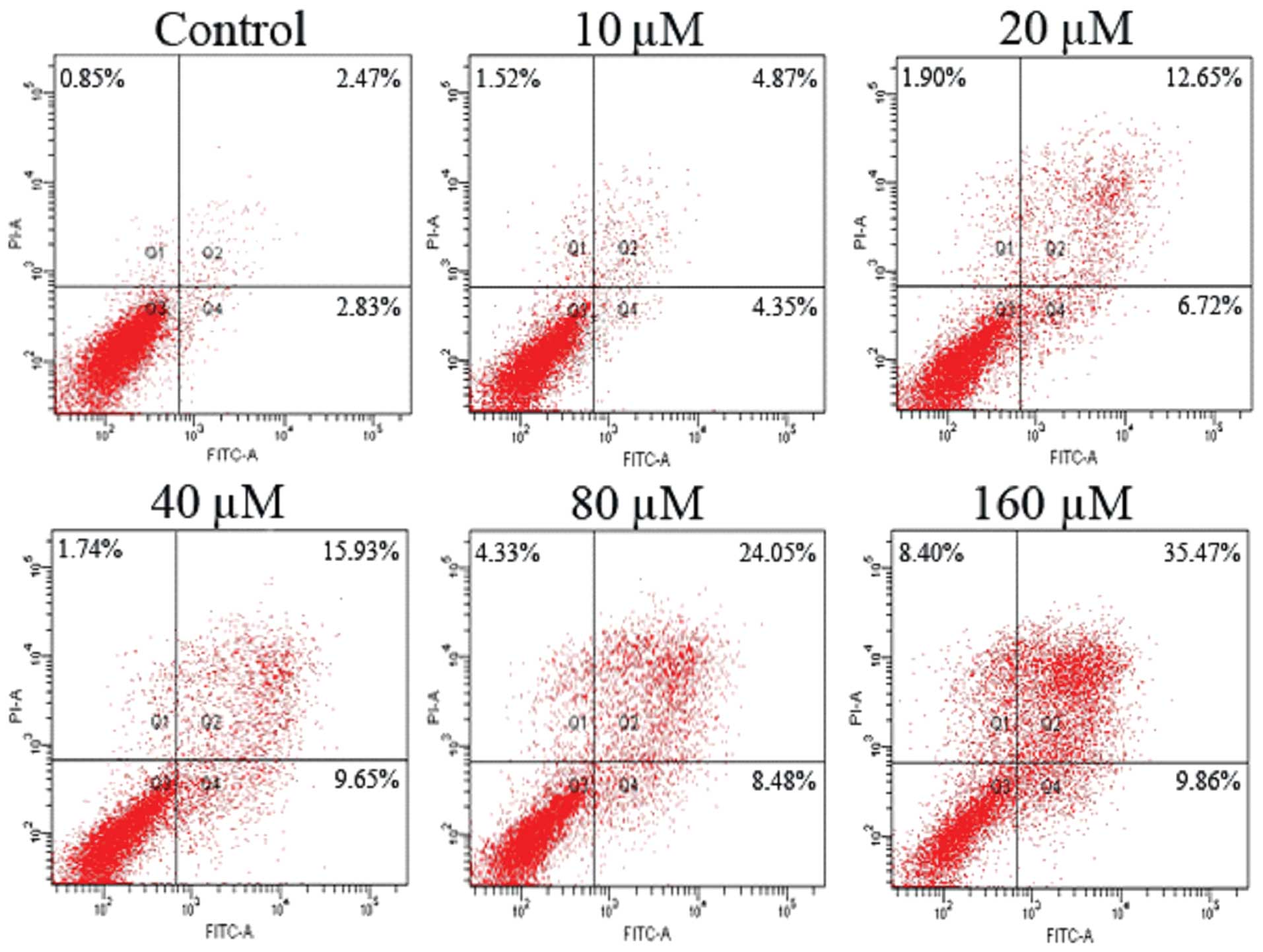
Promoting effect of emodin on SW1990/Gem
cell apoptosis
SW1990/Gem cells were treated with different
concentrations of emodin (10, 20, 40, 80 and 160 μM) for 48 h, FCM
was applied to analyze cell apoptosis. As Fig. 3 showed that emodin promoted cell
apoptosis of gemcitabine-resistant cell line SW1990/Gem in a
dose-dependent manner.
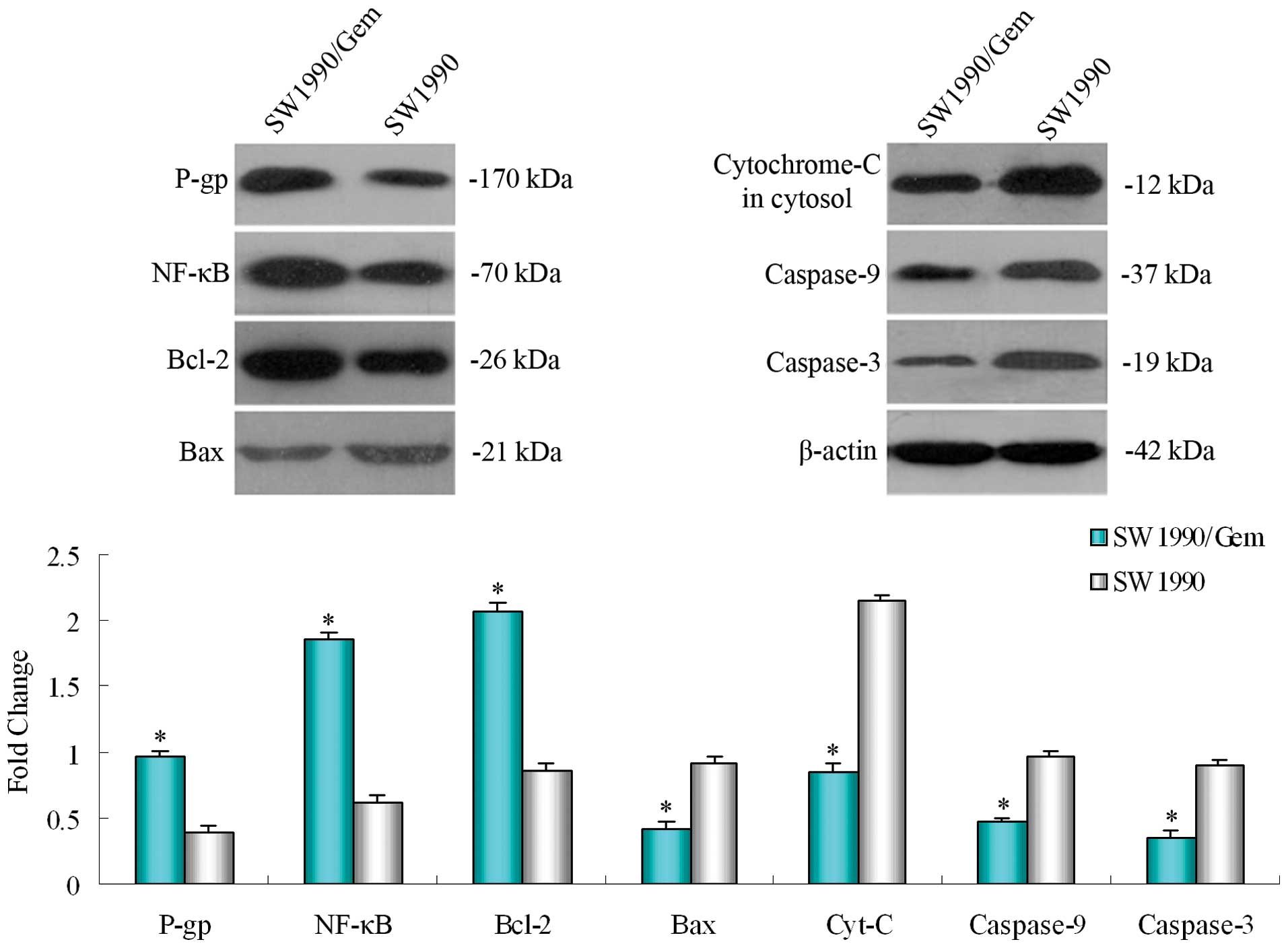
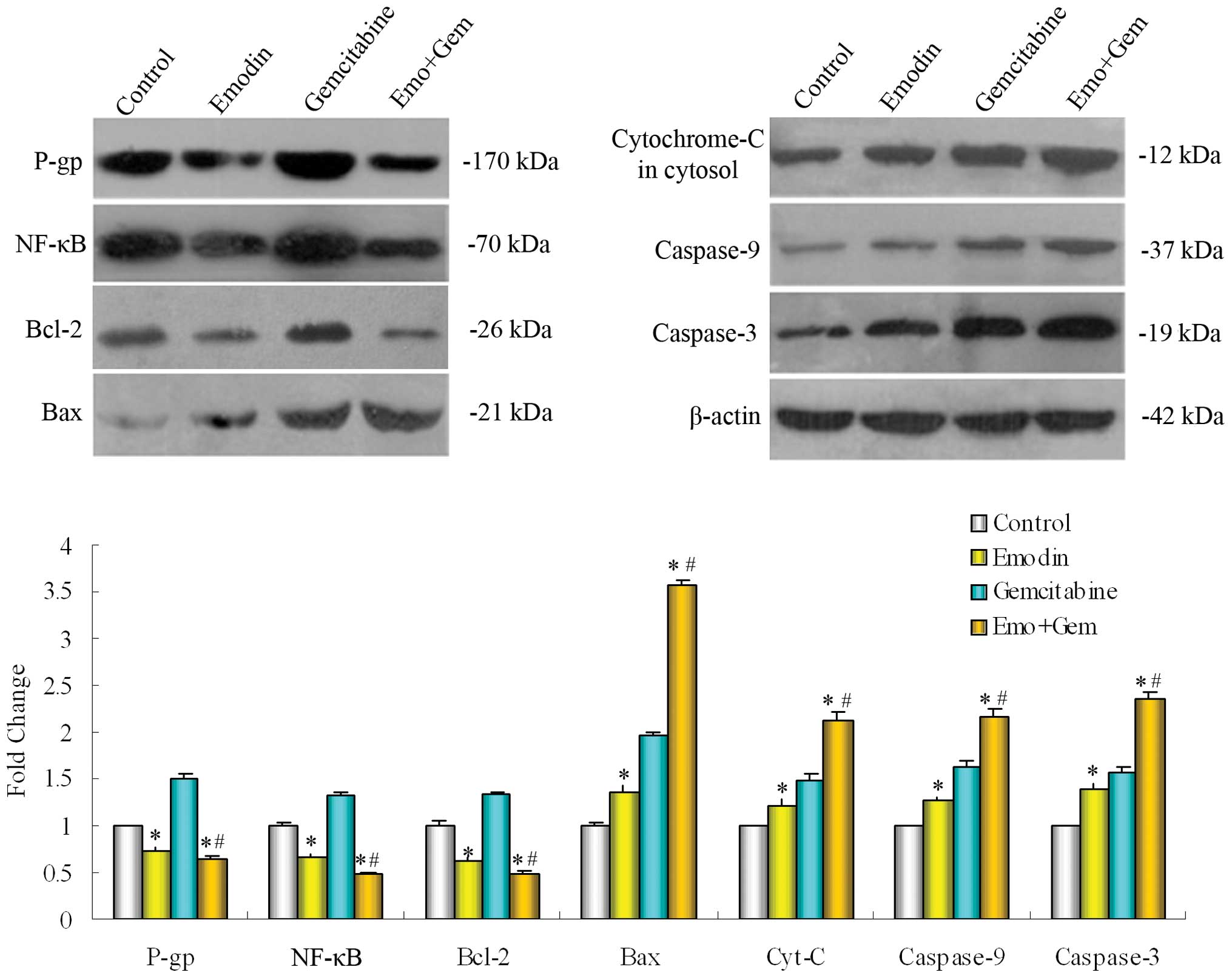
Different protein expression of the P-gp,
NF-κB, Bcl-2, Bax, cytochrome-C (cytosol), caspase-9 and -3 in
SW1990/Gem and SW1990 cells
Determined the basal expression of protein P-gp,
NF-κB, Bcl-2, Bax, cytochrome-C (cytosol), caspase-9 and-3 in
SW1990/Gem cells and SW1990 cells by Western blotting. Compared
with parental cell line SW1990, the expression of
multidrug-resistance gene encoding protein P-gp, apoptosis
regulatory protein in mitochondrial pathway Bcl-2 and the NF-κB
increased in the SW1990/Gem cells, while the expression of Bax
regulated by Bcl-2 in mitochondrial pathway, cytochrome-C
(cytosol), the caspase-9 and -3 of caspase family decreased
obviously (Fig. 4).
Effect of emodin on NF-κB and its related
proteins in SW1990/Gem cells
SW1990/Gem cells were treated with emodin (10 μM)
alone or combined with gemcitabine (20 μM) for 48 h. The expression
of protein was measured by Western blotting. As shown in Fig. 5, emodin alone or combined with
gemcitabine down-reguglated the expression of NF-κB in SW1990/Gem
cells, and then decreased the NF-κB regulated multidrug resistance
protein P-gp and the Bcl-2 in mitochondrial pathway, then increased
the expression of Bax and cytochrome-C (cytosol) regulated by Bcl-2
in mitochondrial pathway. Caspase cascade was triggered followed by
the apoptosis of pancreatic cancer cells, thus, inhibition of
pancreatic cancer growth and promotion of apoptosis occurred.
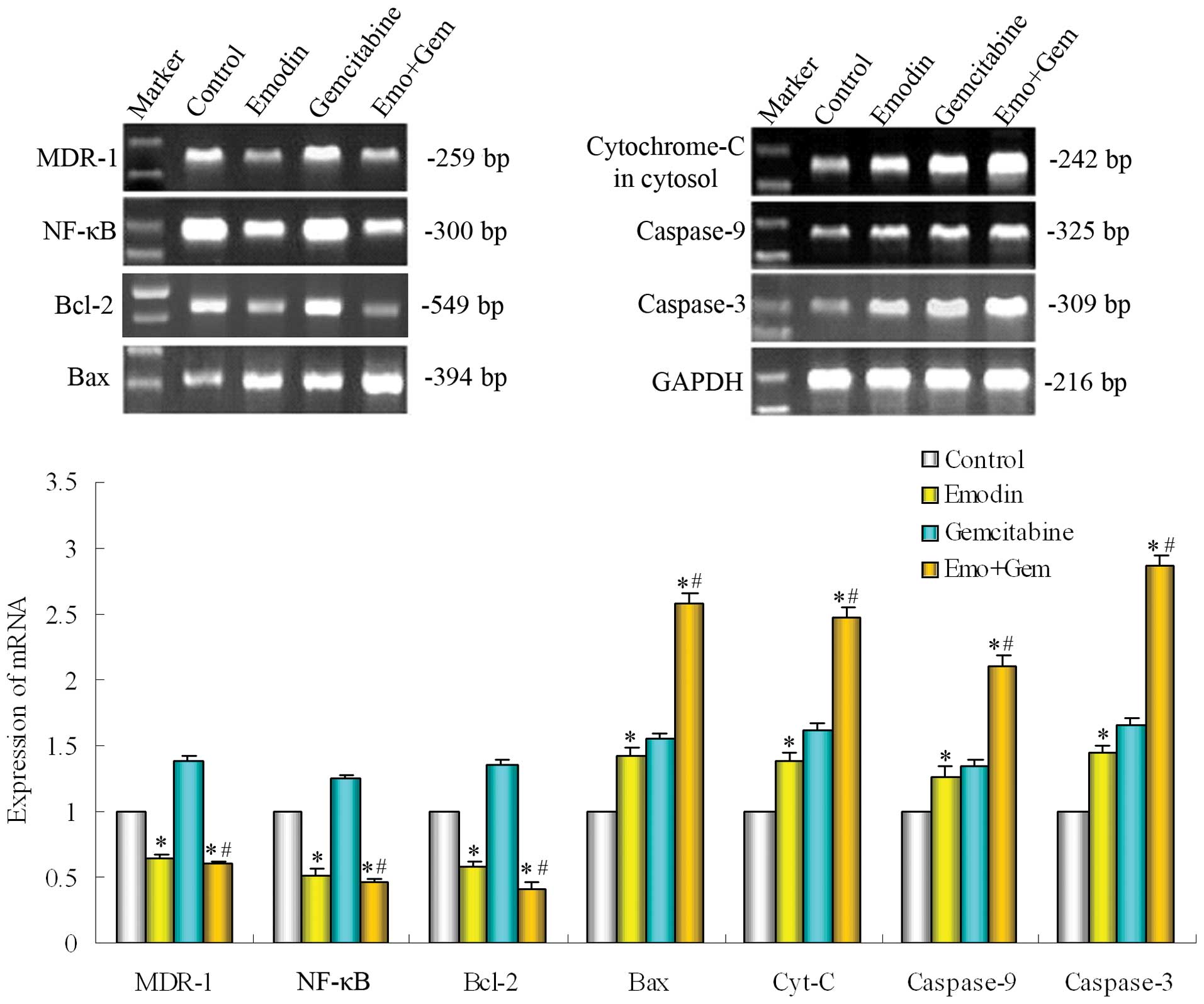
Effect of emodin on the mRNA of NF-κB and
its regulated genes in SW1990/Gem cells
The SW1990/Gem cells were treated with emodin (10
μM) alone or combined with gemcitabine (20 μM) for 48 h. The
expression of gene was measured by PT-PCR. As shown in Fig. 6, emodin alone or combined with
gemcitabine both down-regulated the expression of NF-κB, then
down-regulated the gene expression of MDR-1 and Bcl-2, but
up-regulated the expression of Bax, cytochrome-C, caspase-9 and -3,
these were in line with the proteins change and validated the
effect of emodin further.
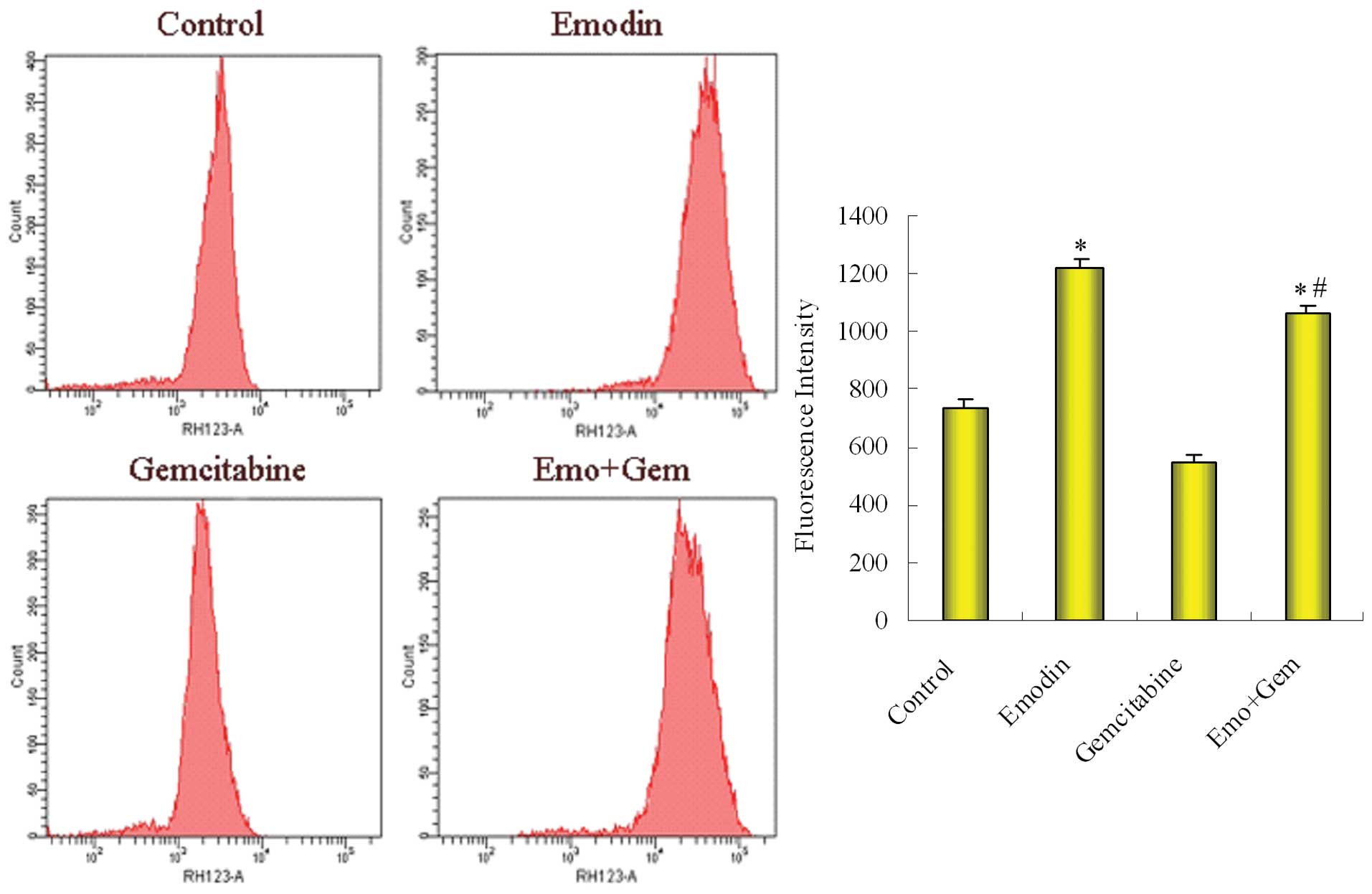
Effect of emodin on the the function of
P-gp in SW1990/Gem cells
The level of rhodamine efflux function was
determined by FCM after SW1990/Gem cells were treated with emodin
alone and then combined with gemcitabine. As shown in Fig. 7, the fluorescence intensity was
734.62±25.74 in control group cells, 1225.28±28.55 in
emodin-treated group, 545.23±28.27 in gemcitabine-treated group
cells and 1068.44±22.85 in the combined group. Compared with
control group, emodin decreased the function of P-gp; compared with
gemcitabine-treated group, the combined group also decreased
function of P-gp.
Discussion
Pancreatic cancer is a common gastrointestinal
tumor, because most patients are diagnosed at advanced stage, the
surgical resection rate is low and chemo-therapy is the most
critical treatment. Gemcitabine is the standard chemotherapeutic
agent for advanced pancreatic cancer, but in clinical work the
treatment effect is not ideal due to the increase of
gemcitabine-induced drug resistance. The rate of 5-year overall
survival is less than 5% and drug resistance is the main reason for
the failure of the chemo-treatment in pancreatic cancer (6). According to reports, the mechanism of
resistance to chemotherapy may be associated with increased drug
cellular efflux by overexpressed P-gp encoded by multidrug
resistance gene (MDR-1) and deregulated expression of
anti-apoptotic or pro-apoptotic molecules (20,21),
may also be associated with multidrug resistance-associated protein
(MRP) (22).
As a traditional Chinese medicine, emodin not only
has anti-tumor effect (15), but
also can enhance the anti-tumor effect of the chemo-therapy drugs
(16,23,24).
It has been reported that emodin sensitizes the ovarian cancer
cells and the gallbladder cancer cells to chemotherapeutic agents
(25,26). However, reports on whether emodin
can reverse the chemotherapeutic drug resistance in pancreatic
cancer are rare. Our group first reported that emodin sensitized
resistant cell to gemcitabine through inhibiting the expression of
NF-κB in SW1990/Gem cells (18).
In this study, we established the gemcitabine-resistant cell line
SW1990/Gem with intermittently increasing the concentration of
gemcitabine in the culture medium for 10 months, then calculated
the resistance index and observed cell morphology. In the
follow-up, SW1990/Gem cells were pretreated with emodin (10 μM) for
different periods and then treated with gemcitabine (20 μM) for 48
h, cell viability was detected by MTT. The results showed that
inhibition effect of gemcitabine on proliferation of drug-resistant
cell line SW1990/Gem was significantly enhanced after cells were
pretreated with emodin. FCM results showed that emodin could
promote cell apoptosis in drug-resistant cell line SW1990/Gem.
Western blotting detected the basal expression of P-gp, NF-κB,
Bcl-2, Bax, cytochrome-C (cytosol), caspase-9 and -3 in SW1990/Gem
cells and SW1990 cells, it was found that compared with parental
cell line SW1990, the expression of P-gp, NF-κB and Bcl-2 was
increased in the SW1990/Gem cells, while the expression of Bax,
cytochrome-C, caspase-9 and -3 was decreased. Based on this, we
furher investigated the potential molecule mechanism of reversing
the resistance effect of gemcitabine-resistant pancreantic cancer
cell line SW1990/Gem by emodin, possibly it is via decreasing the
function of P-gp and the mitochondrial apoptosis pathway.
P-gp encoded by multidrug resistance gene-1 (MDR-1)
is a kind of transmembrane glycoprotein, belongs to transporter
protein superfamily ABC and has the ATP-dependent drug efflux
function (22). P-gp can induce
drug resistance due to decrease in the cellular chemotherapeutic
drug under effective concentration through pumping the
chemotherapeutic agents out of the cells against the concentration
(27). Therefore, MDR-1/P-pg plays
an important role in tumor chemotherapeutic resistance (28,29).
According to reports, nuclear transcription factor NF-κB induces
resistance of tumor cells through down-regulating the expression of
MDR-1 mRNA (30). Other reports
stated that the decrease of P-gp in expression and function
reversed the chemotherapeutic resistance in breast cell line MCF-7,
through the inhibition of MDR-1 expression induced by the decrease
of expression and activity of NF-κB (31). In this study, the levels of gene
and protein of the NF-κB and MDR-1 (P-gp) were decreased both in
emodin group and in gemcitabine group. Rhodamine efflux experiments
indicated that the function of P-gp was decreased both in emodin
group and combination group. The decrease of expression and
function of P-gp directly increased the intracellular drug
concentration, and this may be partly the response to the reversion
of the gemcitabine-resistance in pancreatic cancer.
Apoptosis defection is another important reason for
drug resistance. Chemotherapeutic agents, as one of the major
treatments of cancer, kill tumor cells mainly through inducing
apoptosis, and the defection of apoptosis is one of the important
reasons for drug-resistance due to insensitive of the tumor to
chemotherapy (32).
Bcl-2 protein family is very important in apoptosis,
the anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bax are
the major members in this family. Bcl-2 and Bax also play very
important roles in mitochondrial pathway (33), the down-regulation of Bcl-2 and the
up-regulation of Bax can induce the release of cytochrome-C
(cytosol) from mitochondria, trigger the activity of caspase-3 and
-9 and finally cause cell apoptosis (34). This study verified that the
expression of Bcl-2 in SW1990/Gem was significantly higher than
SW1990, but the expression of Bax and cytochrome-C were
significantly lower suggesting that the mitochondrial receptor
pathway may be involved in the formation of gemcitabine-resistance.
From the level of gene and protein expression, we further found
that emodin decreased the expression of Bcl-2, increased the
expression of Bax and cytochrome-C, this was most obvious in
combination group suggesting that low concentration of emodin could
reverse the increase of Bcl-2 and the decrease of Bax induced by
resistance, but had no significant pro-apoptotic effect, and
therefore enhanced the sensitivity of gemcitabine-resistant
pancreatic cells to gemcitabine.
NF-κB is a family of ubiquitous transcription
factors involving immunity, inflammation, regulation of cell
growth, differentiation, apoptosis, and tumor metastasis. In recent
reports, NF-κB is shown closely related to tumor resistance to
chemotherapy. As previous studies show, down-regulation of
anti-apoptotic protein is one of the mechanisms that NF-κB takes
part in apoptosis and induces apoptosis (35). Banerjee et al have reported
that NF-κB caused the resistance of pancreatic cancer through
up-regulating the expression of anti-apoptotic proteins (XIAP,
Bcl-xL, Survivin) (21). Another
report shows that NF-κB induced the resistance of breast cancer by
increasing the expression of anti-apoptotic protein Bcl-2 and
decreasing the expression of pro-apoptotic protein Bax (36). Also, there are reports that NF-κB
can overcome the chemotherapeutic resistance through
down-regulating of expression of anti-apoptotic protein Bcl-2
family (37). Our study suggested
that NF-κB participated in the formation of tumor resistance via
multidrug resistance encoding protein P-gp and Bcl-2, with Bax that
existed in mitochondrial apoptosis pathway. In this study we found
that emodin reversed the gemcitabine resistance effect in
pancreatic cancer, the action might be associating with
down-regulation of NF-κB expression, and lowering the expression of
P-gp and Bcl-2, increasing Bax expression.
In conclusion, emodin can effectively reverse the
resistance effect of pancreatic cancer to gemcitabine. The
potential mechanisms are 1) the decrease in the expression and
function of P-gp, thus causing decrease of the efflux of drug and
then increasing the intracellular drug concentration, thus the
treatment effect was enhanced, 2) the down-regulation of Bcl-2
expression in mitochondrial apoptosis pathway and the up-regulation
of Bax in mitochondrial pathway, followed by the occurrence of
apoptosis.
Acknowledgements
We are grateful for funding support from the
Administration of Traditional Chinese Medicine of Zhengjing
Province, China (Grant No. 2011ZZ010) and The National Natural
Science Foundation of China (Grant No. 81173606). We thank the
entire staff of the Animal Experimental Center in Wenzhou Medical
College and of Scientific Research platform of the Second
Affiliated Hospital of Wenzhou Medical College for helpful
assistance.
References
|
1
|
Rivera F, Lopez-Tarruella S, Vega-Villegas
MA and Salcedo M: Treatment of advanced pancreatic cancer: from
gemcitabine single agent to combinations and targeted therapy.
Cancer Treat Rev. 35:335–339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Murray T, Xu J
and Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 57:43–66.
2007. View Article : Google Scholar
|
|
3
|
Jemal A, Siegel R, Xu J and Ward E: Cancer
statistics, 2010. CA Cancer J Clin. 60:277–300. 2010. View Article : Google Scholar
|
|
4
|
Burris HA, Moore MJ, Andersen J, Green MR,
et al: Improvements in survival and clinical benefit with
gemcitabine as first-line therapy for patients with advanced
pancreas cancer: a randomized trial. J Clin Oncol. 15:2403–2413.
1997.PubMed/NCBI
|
|
5
|
Yu C, Zhang X, Sun G, Guo X, et al: RNA
interference-mediated silencing of the polo-like kinase 1 gene
enhances chemosensitivity to gemcitabine in pancreatic
adenocarcinoma cells. J Cell Mol Med. 12:2234–2249. 2008.PubMed/NCBI
|
|
6
|
El Maalouf G, Le Tourneau C, Batty GN,
Faivre S and Raymond E: Markers involved in resistance to
cytotoxics and targeted therapeutics in pancreatic cancer. Cancer
Treat Rev. 35:167–174. 2009.PubMed/NCBI
|
|
7
|
Tada M, Arizumi M, Nakai Y, Sasaki T, et
al: Efficacy of gemcitabine for locally advanced pancreatic cancer:
comparsion with 5-fuorouracil-based chemoradiotherapy.
Chemotherapy. 54:302–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Basu S, Ghosh A and Hazra B: Evaluation of
the antibacterial activity of Ventilago madraspatana Gaertn., Rubia
cordifolia Linn and Lantana camara Linn: isolation of emodin and
physcion as active antibacterial agents. Phytother Res. 19:888–894.
2005. View
Article : Google Scholar
|
|
9
|
Manojlovic NT, Solujic S, Sukdolak S, et
al: Antifungal activity of Rubia tinctorum, Rhamnus frangula and
Caloplaca cerina. Fitoterapia. 76:244–246. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alves DS, Perez-Fons L, Estepa A, et al:
Membrane-related effects underlying the biological activity of the
anthraquinones emodin and barbaloin. Biochem Pharmacol. 68:549–561.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mbwambo ZH, Apers S, Moshi MJ, et al:
Anthranoid compounds with antiprotozoal activity from Vismia
orientalis. Planta Med. 70:706–710. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li HL, Chen HL, Li H, et al: Regulatory
effects of emodin on NF-kappaB activation and inflammatory cytokine
expression in RAW 264.7 macrophages. Int J Mol Med. 16:41–47.
2005.PubMed/NCBI
|
|
13
|
Kitano A, Saika S, Yamanaka O, Ikeda K,
Okada Y, et al: Emodin suppression of ocular surface inflammatory
reaction. Invest Ophthalmol Vis Sci. 48:5013–5022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuo YC, Tsai WJ, Meng HC, Chen WP, et al:
Immune reponses in human mesangial cells regulated by emodin from
Polygonum hypoleucum Ohwi. Life Sci. 68:1271–1286. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cai J, Razzak A, Hering J, Saed A, et al:
Feasibility evaluation of emodin (rhubarb extract) as an inhibitor
of pancreatic cancer cell proliferation in vitro. J Parenter
Enteral Nutr. 32:190–196. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen H, Wei WT, Guo YF, Liu A, et al:
Enhanced effect of gemcitabine by emodin against pancreatic cancer
in vivo via cytochrome C-regulated apoptosis. Oncol Rep.
25:1253–1261. 2011.PubMed/NCBI
|
|
17
|
Jung HA, Chung HY, Yokozawa T, et al:
Alaternin and emodin with hydroxyl radical inhibitory and/or
scavenging activities and hepatoprotective activity on
tacrine-induced cytotoxicity in HepG2 cells. Arch Pharm Res.
27:947–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu A, Chen H, Tong H, et al: Emodin
potentiates the antitumor effects of gemcitabine in pancreatic
cancer cells via inhibition of nuclear factor-kappaB. Mol Med Rep.
4:221–227. 2011.PubMed/NCBI
|
|
19
|
Hansen MB, Nielsen SE and Berg K:
Re-examination and further development of a precise and rapid dye
method for measuring cell growth/cell kill. J Immunol Methods.
119:203–210. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hao ZM, Li XH, Qiao TD, Du R, Hong L and
Fan DM: CIAPIN1 confers multidrug resistance by upregulating the
expression of MDR-1 and MRP-1 in gastric cancer cells. Cancer Biol
Ther. 5:261–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Banerjee S, Wang Z, Kong D and Sarkar FH:
3,3′-Diindolyl-methane enhances chemosensitivity of multiple
chemotherapeutic agents in pancreatic cancer. Cancer Res.
69:5592–5600. 2009.
|
|
22
|
Gao S, Liu Q, Wang X, Lin B and Zhang S:
Effects of Lewis Y antigen on the gene expression of multiple drug
resistance-associated proteins in human ovarian cancer RMG-I-H
cells. Med Oncol. 27:960–967. 2010. View Article : Google Scholar
|
|
23
|
Wang ZH, Chen H, Guo HC, Tong HF, et al:
Emodin antitumor efficacy by the combination of emodin and
gencitabine against human pancreatic cancer cells via
downregulation of the expression of XIAP in vitro and in vivo. Int
J Oncol. 39:1123–1131. 2011.PubMed/NCBI
|
|
24
|
Wei WT, Chen H, Ni ZL, Liu HB, et al:
Antitumor and apoptosis-promoting properties of emodin, an
anthraquinone derivative from Rheum officinale Baill, against
pancreatic cancer in mice via inhibition of Akt activation. Int J
Oncol. 39:1381–1390. 2011.PubMed/NCBI
|
|
25
|
Wang W, Sun YP, Huang XZ, He M, Chen YY,
et al: Emodin enhances sensitivity of gallbladder cancer cells to
platinum drugs via glutathion depletion and MRP1 downregulation.
Biochem Pharmacol. 79:1134–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Liu PS, Mao HL, et al: Emodin
sensitizes paclitaxel-resistant human ovarian cancer cells to
paclitaxel-induced apoptosis in vitro. Oncol Rep.
21:1605–1610. 2009.PubMed/NCBI
|
|
27
|
Leonessa F and Clarke R: ATP binding
cassette transporters and drug resistance in breast cancer. Endocr
Relat Cancer. 10:43–73. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takakuwa O, Oguri T, Ozasa H, Uemura T,
Kasai D, et al: Over-expression of MDR1 in amrubicinol-resistant
lung cancer cells. Cancer Chemother Pharmacol. 68:669–676. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong Y, Shao S, Hu J and Yang P: Reversal
effect of Raf-1/Mdr-1 siRNAs co-transfection on multidrug
resistance in KBv200 cell line. Oral Oncol. 45:991–997. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bentires Alj M, Barbu V, Fillet M, et al:
NF-kappaB transcription factor induces drug resistance through MDR1
expression in cancer cells. Oncogene. 22:90–97. 2003.PubMed/NCBI
|
|
31
|
Chen C, Shen HL, Yang J, Chen QY and Xu
WL: Preventing chemoresistance of human breast cancer cell line,
MCF-7 with celecoxib. J Cancer Res Clin Oncol. 137:9–17. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hannun YA: Apoptosis and the dilemma of
cancer chemotherapy. Blood. 89:1845–1853. 1997.PubMed/NCBI
|
|
33
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qi FH, Inagaki Y, Gao B, Cui X and Xu HL:
Bufalin and cinobufagin induce apoptosis of human hepatocellular
carcinoma cells via Fas- and mitochondria-mediated pathways. Cancer
Sci. 102:951–958. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Leger DY, Liagre B and Beneytout JL: Role
of MAPKs and NF-kappaB in diosgenin-induced megakaryocytic
differentiation and subsequent apoptosis in HEL cells. Int J Oncol.
28:201–207. 2006.PubMed/NCBI
|
|
36
|
Bachmeier B, Fichtner I, Killian PH,
Kronski E, et al: Development of resistance towards artesunate in
MDA-MB-231 human breast cancer cells. PLoS One. 6:e205502011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumar MV, Shirley R, Ma Y and Lewis RW:
Role of genomics-based strategies in overcoming chemotherapeutic
resistance. Curr Pharm Biotechnol. 5:471–480. 2004. View Article : Google Scholar : PubMed/NCBI
|