Introduction
Hepatocellular carcinoma (HCC) represents the fifth
most common malignancy and the third most frequent cause of cancer
death around the world (1).
Hepatitis B and C virus infections, exposure to aflatoxin, and
excessive intake of alcohol have been identified as major risk
factors of HCC. Surgery is the most effective option, but
unfortunately the majority of patients with HCC are not amenable to
surgery at the time of diagnosis. Presently, one of the main
approaches in treating inoperable HCC is to use cytotoxic
chemotherapy, but sometimes HCC is less sensitive to or becomes
resistant to anticancer drugs after consecutive treatments; most
tests failed to find a therapy which can produce a response rate
>25% among hepatoma patients (2,3).
Despite recent progress in diagnosis and treatment, HCC is still
diagnosed at an advanced stage where prognosis is poor. Important
efforts should therefore be directed toward developing effective
and less toxic chemotherapeutic strategies.
Aspirin (acetylsalicylic acid) is the best-known
salicylate and belongs to the pharmacologic category of the
nonsteroidal anti-inflammatory drugs (NSAIDs). Despite wide use
being made for more than 100 years, clinical uses of aspirin have
been changed over time and knowledge on its mechanisms of action
and therapeutic entities continually evolve. During the first fifty
years since it was developed, aspirin was primarily used as an
analgesic, anti-pyretic, and anti-inflammatory agent based on its
main mechanism of action of inhibiting prostaglandin synthesis.
However, currently aspirin is more commonly used as an
anti-thrombotic agent, in primary and secondary prevention of
thromboembolic events. The suggestion that aspirin could be of
benefit against cancer initially arose from the observation that
tumor metastases were reduced in rats with thrombocytopenia
(4–6). Subsequently, prostaglandin
concentration proved to be raised in rat colorectal tumor tissues
(7,8), which strengthened the expectation
that anticancer benefit might be gained through the inhibition of
cyclooxygenase (COX).
One obvious molecular target for aspirin is COX-2
which is the enzyme highly and rapidly induced in response to
mediators of inflammation, growth factors, cytokines, or
endotoxins, and is involved in cell proliferation and tumor
promotion (9). This is supported
by the fact that aspirin can decrease the production of potentially
neoplastic prostaglandins produced from COX-2-mediated catalysis of
arachidonic acid (10). The
carcinogenic contribution of prostaglandins has generated much
interest; their deleterious effects include promotion of cell
survival, stimulation of cell proliferation, and promotion of
angiogenesis (11,12). These effects can also enhance
cancer spread and thus underscore the cancer fighting potential of
aspirin.
However, the anticancer effect of aspirin and NSAIDs
cannot be solely explained by the inhibition of prostaglandin
synthesis, since several NSAIDs have antiproliferative effects in
cells that have no COX activity. High doses of aspirin have been
reported to induce apoptosis through COX-independent mechanisms, by
regulating several different targets (13), such as 15-LOX-1 (14), a proapoptopic gene Par-4
(15), and an antiapoptopic gene
Bcl-XL (16). Additionally, NSAIDs including
aspirin also induce apoptosis by means of the activation of
caspases (17,18), the activation of p38 MAP kinase
(19), release of mitochondrial
cytochrome c (18–20), and activation of the ceramide
pathway (21). These effects might
not be universal to all cell types, however, and the dose range of
aspirin needed in such COX-independent pathways could be higher
than that for the inhibition of COX-2 (22). Other mechanisms contributing to the
potential anticancer effects of aspirin could be attributed to the
upregulation of tumor suppressor gene, such as Bax, and the
downregulation of antiapoptotic gene, such as Bcl-2
(23). Apoptosis (programmed cell
death) has been recognized as an important physiological event in
the development and pharmacology of anticancer agents and cancer
therapies (24,25). In recent years, the regulation of
apoptosis has become an area of extensive study in cancer research
as the life span of both normal and cancer cells within a living
system is regarded to be significantly affected by the rate of
apoptosis (26,27).
Raza et al (18) recently demonstrated that aspirin
treatment (5–10 μmol/ml) induced oxidative stress, cell cycle
arrest in the G0/G1 phase, apoptosis, and mitochondrial dysfunction
in HepG2 cells in vitro. In this study, we evaluated the
effects of aspirin on apoptosis induction in human hepatocellular
carcinoma cell line in vitro and antitumor activity in HepG2
cell xenograft of nude mouse model.
Materials and methods
Chemicals
Aspirin was purchased from Sigma-Aldrich Co. (St.
Louis, MO). Aspirin was freshly prepared before each experiment and
solubilized as described elsewhere (28). All other chemicals and reagents
were from standard commercial sources and of the highest
purity.
Cell culture and cell viability
assay
The human hepatocellular carcinoma cell line HepG2
(wild-type p53; Rb-positive; Ras-mutated; and HBV-negative) cells
were purchased from the American Type Culture Collection (Manassas,
VA), and maintained at 37°C in a humidified condition of 95% air
and 5% CO2 in DMEM (Gibco-BRL, Gaithersburg, MD)
supplemented with 10% heat inactivated fetal bovine serum (FBS), 2
mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cell
proliferation was assessed using MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide,
Sigma-Aldrich Co.] assay, as described before (29), which is based on the conversion of
MTT to MTT-formazan by mitochondrial enzymes.
DNA fragmentation assay
DNA fragmentation was performed as described
previously (30). Briefly, after
treatment with aspirin, cells were rinsed twice in cold PBS and
resuspended in lysis buffer [5 mM Tris-HCl (pH 7.5), 5 mM ethylene
diamine tetraacetic acid and 0.5% Triton X-100] at 4°C for 30 min.
After centrifugation at 27,000 × g for 15 min, the supernatant was
treated with RNase, followed by proteinase K digestion,
phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v) extraction and
isopropanol precipitation. DNA was separated through a 1.5% agarose
gel, and stained with 0.1 μg/ml ethidium bromide (EtBr,
Sigma-Aldrich Co.), and was visualized by ultraviolet light
source.
Caspase activity assay
Activities of caspase-3, -8 and -9 were determined
using the corresponding caspase activity detection kits (R&D
Systems, Minneapolis, MN) as described by the manufacturers.
Briefly, cells were harvested and cold lysis buffer was added, and
then incubated on ice for 10 min and centrifuge for 1 min in a
microcentrifuge (10,000 × g). The supernatant was transferred to a
fresh tube and protein concentration was determined using a
standard colorimetric assay (Bio-Rad Laboratories, Hercules, CA).
The protein concentration of each sample was adjusted to 200 μg per
50 μl of cell lysate using chilled cell lysis buffer. Then 50 μl of
2X reaction buffer was added and 5 μl of substrates of
DEVD-pNA (for caspase-3), IETD-pNA (for caspase-8),
and LEHD-pNA (for caspase-9), respectively. Samples were
incubated at 37°C for 2 h and the enzyme-catalyzed release of
pNA was quantified at 405 nm using a microtiter plate
reader. The values of aspirin treated samples were normalized to
the untreated controls, allowing determination of the fold increase
in caspase activity.
Gel electrophoresis and western blot
analysis
The cells were harvested, lysed, and protein
concentrations were quantified using the Bio-Rad protein assay
(Bio-Rad Laboratories), following the procedure described by the
manufacturer. Western blot analysis was performed as described
previously (31). Briefly, an
equal amount of protein was subjected to electrophoresis on
SDS-polyacrylamide gels and transferred to nitrocellulose membranes
(Schleicher & Schuell, Keene, NH) by immunoblotting. Blots were
probed with the desired antibodies for overnight, incubated with
diluted enzyme-linked secondary antibodies and then visualized by
the enhanced chemiluminescence (ECL) according to the recommended
procedure (Amersham Corp., Arlington Heights, IL). The primary
antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa
Cruz, CA). Peroxidase-labeled goat anti-rabbit and goat anti-mouse
immunoglobulin were purchased from Santa Cruz Biotechnology Inc.
and Amersham.
In vivo xenograft model
Six-week-old male BALB/c nude mice (obtained from
Japan SLC, Inc., Japan) were used for in vivo animal
experiments. The animals were housed in constant laboratory
conditions of a 12-h light/dark cycle and specific pathogen-free
conditions and fed with water and food ad libitum. The
animal protocol used in this study has been reviewed by the Pusan
National University-Institutional Animal Care and Use Committee
(PNU-IACUC, Busan, Korea) on their ethical procedures and
scientific care, and it has been approved. For xenograft study,
mice were inoculated subcutaneously into the right-back with
7.5×106 HepG2 cells in 200 μl PBS and Matrigel (1:1).
The mice were randomly assigned into two groups of 6 each; one
group received oral aspirin, suspended in 0.5% sodium carboxymethyl
cellulose (Na-CMC), at 100 mg/kg/day; the other group was used as
the control that received same amount of Na-CMC in water orally.
The body weight and tumor volume [(major axis) × (minor
axis)2 × 1/2] of every mouse were monitored biweekly
after 4 weeks up to the end of the experiment (7 weeks).
Histopathological findings
Xenograft tumor and stomach tissue samples were
fixed in 10% neutral buffered formalin, dehydrated, and embedded in
paraffin. Samples were subsequently sectioned at 5 μm thickness,
and stained with hematoxylin and eosin (H&E) for
histopathology.
Statistical analysis
The data are presented as means ± SEM of three
independent determinations. ANOVA was conducted to analyze
significant differences among all groups. Other statistical
analyses were carried out by Student's t-test. p<0.05 and
p<0.01 were considered to be significant.
Results
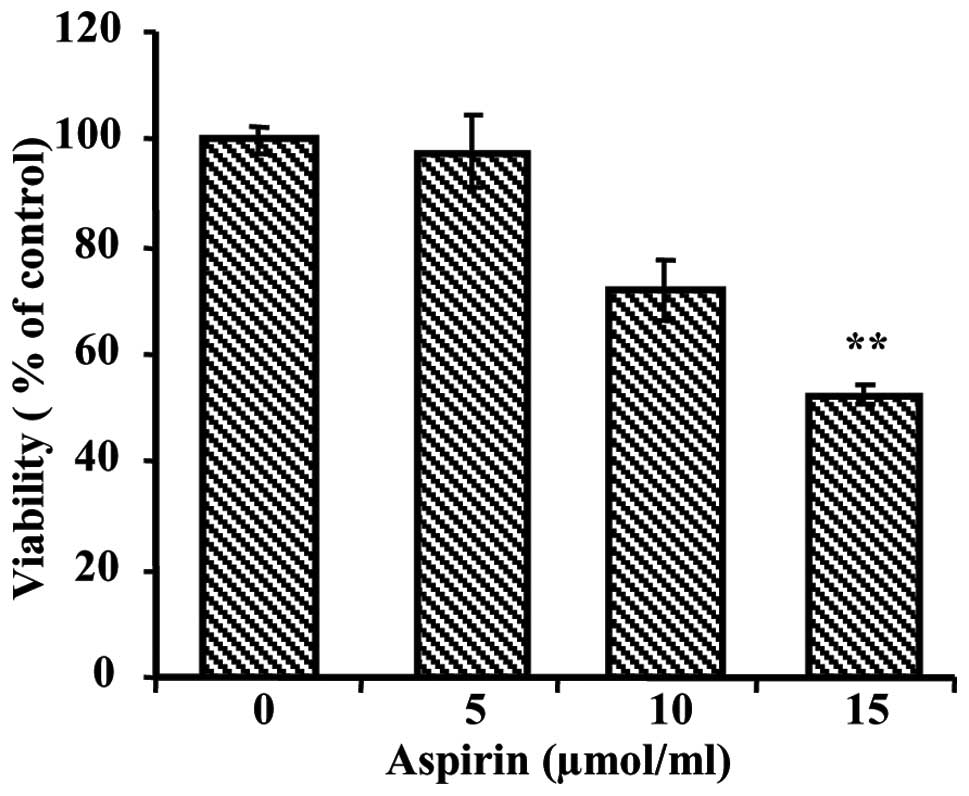
Aspirin reduced the viability of
cells
To evaluate the growth inhibiting effects of aspirin
on human hepatocellular carcinoma cell line, we initially performed
an MTT assay. The MTT assay showed increasing cytotoxicity of
aspirin in HepG2 cells after 24 h of treatment. As shown in
Fig. 1, cell viability was
significantly decreased by treatment of aspirin in a dose-dependent
manner. The dose required for half-maximal inhibition
(IC50) of HepG2 cell growth was ~15 μmol/ml.
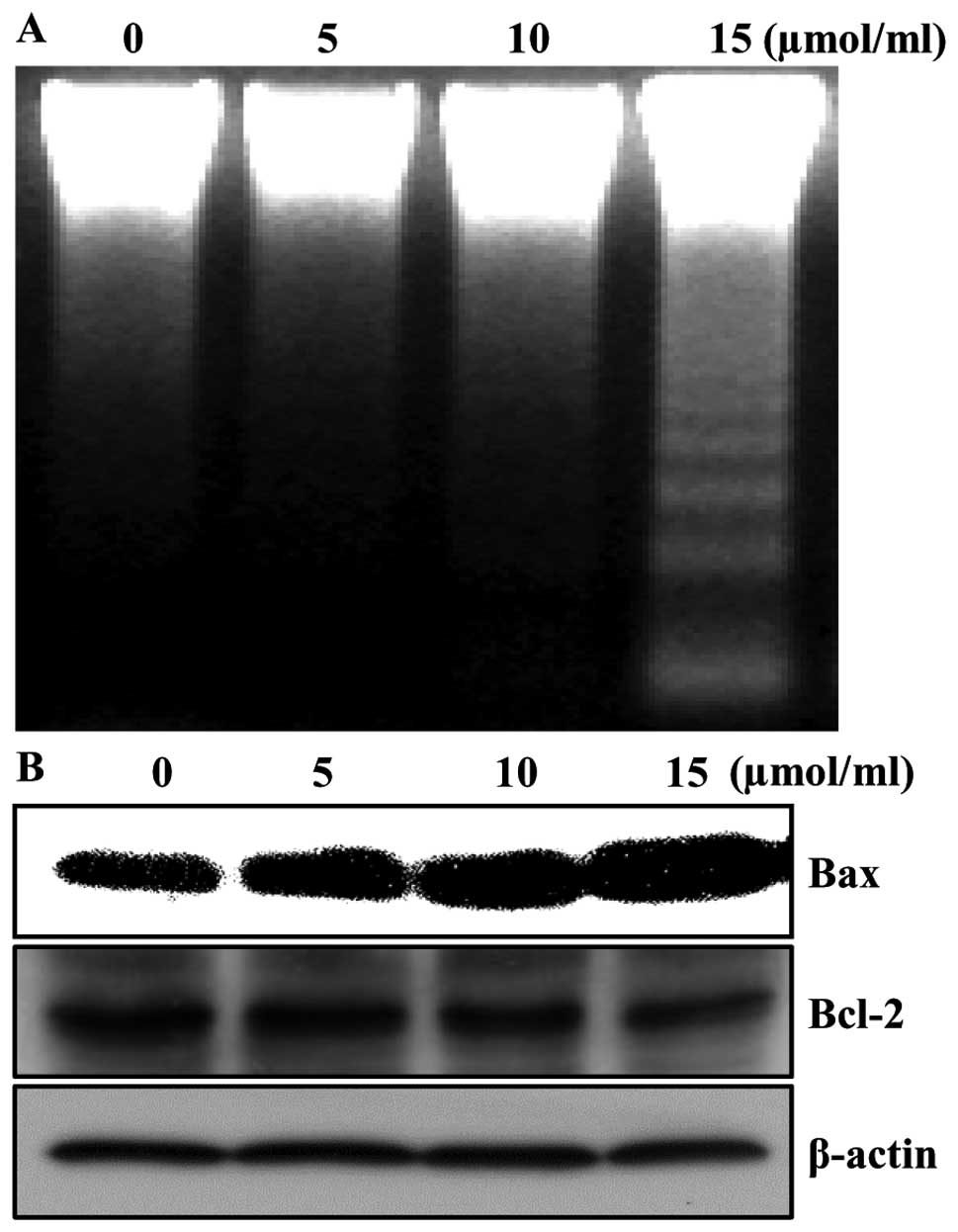
Aspirin induces apoptosis
In order to determine whether the growth inhibition
by aspirin was associated with the induction of apoptosis in HepG2
cells, we analyzed DNA fragmentation which is a hallmark of
apoptosis. Following agarose gel electrophoresis of the cells
treated with 5, 10 and 15 μmol/ml of aspirin for 24 h, a typical
ladder pattern of internucleosomal fragmentation was observed
(Fig. 2A). Since apoptosis might
be regulated by the alteration in the ratio of Bax/Bcl-2 family
protein expression, we tested whether aspirin-induced apoptosis was
accompanied by the change of the expression levels of Bax and
Bcl-2. The results from western blotting showed that treatment with
aspirin resulted in the up-regulation of Bax and slightly decreased
in Bcl-2 expression in cells, suggesting aspirin treatment alters
the Bax/Bcl-2 ratio in HepG2 cells (Fig. 2B). Taken together, these results
implied that the cytotoxic effects observed in response to aspirin
were most likely to be associated with the induction of apoptotic
cell death.
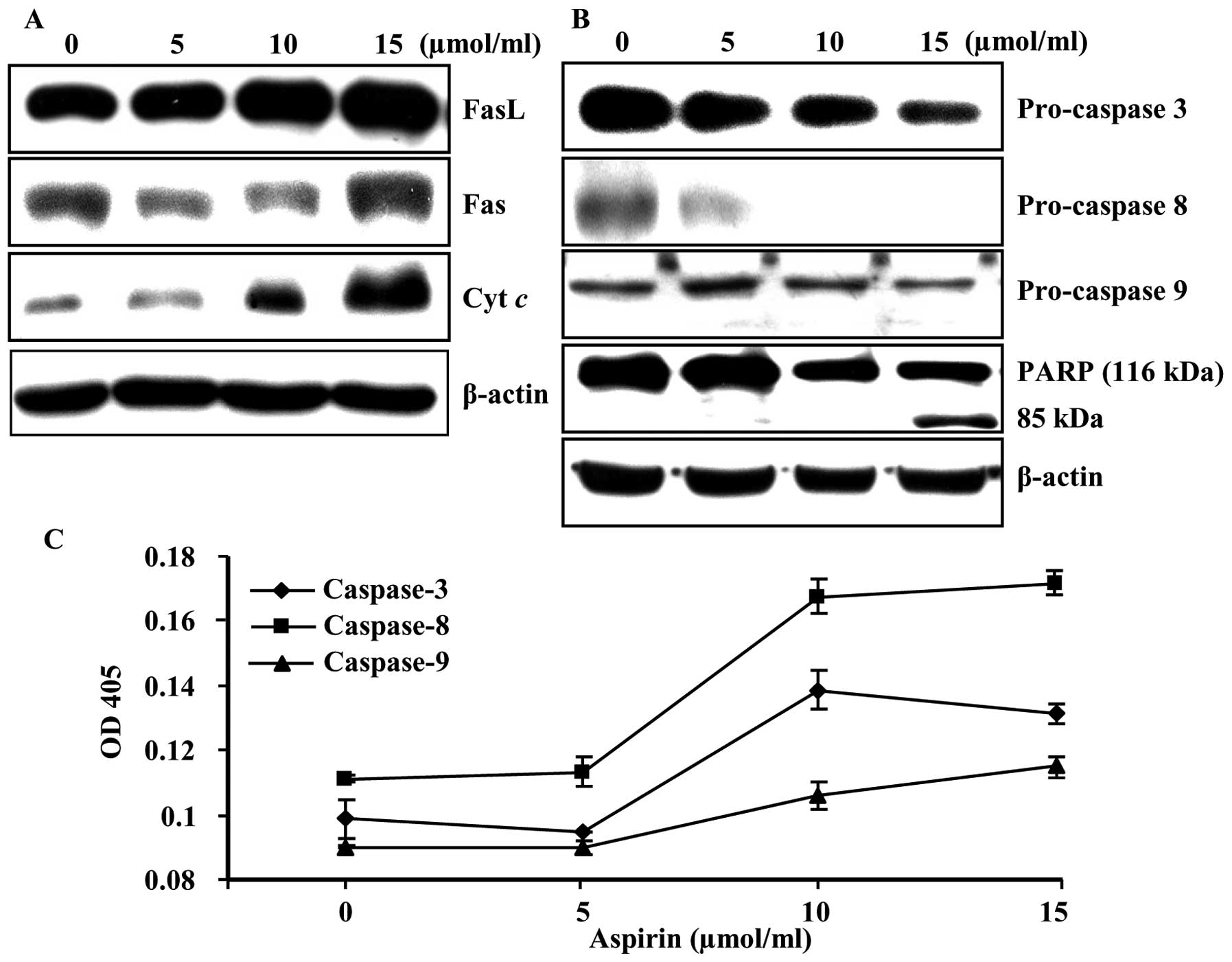
Aspirin induces apoptosis through
extrinsic and intrinsic pathways and caspase cascade
To investigate the mechanisms whereby apoptosis is
induced, we examined the death receptor and death receptor mediated
proteins as well as lethal stimuli of mitochondria inside the cells
using immunoblot analysis. A significant increase in the expression
levels of FasL, Fas, and cytochrome c were observed
(Fig. 3A). In addition, induction
of procaspase-3, -8, and -9 and the subsequent proteolytic cleavage
of poly(ADP-ribose) polymerase (PARP) were also observed in a
dose-dependent manner (Fig. 3B).
Moreover, aspirin treatment significantly increased caspase-3, -8,
and -9 activities in a dose-dependent manner (Fig. 3C). These results indicate that
aspirin treatment induces apoptotic death in HepG2 cells, at least
in part through a caspase-dependent pathway.
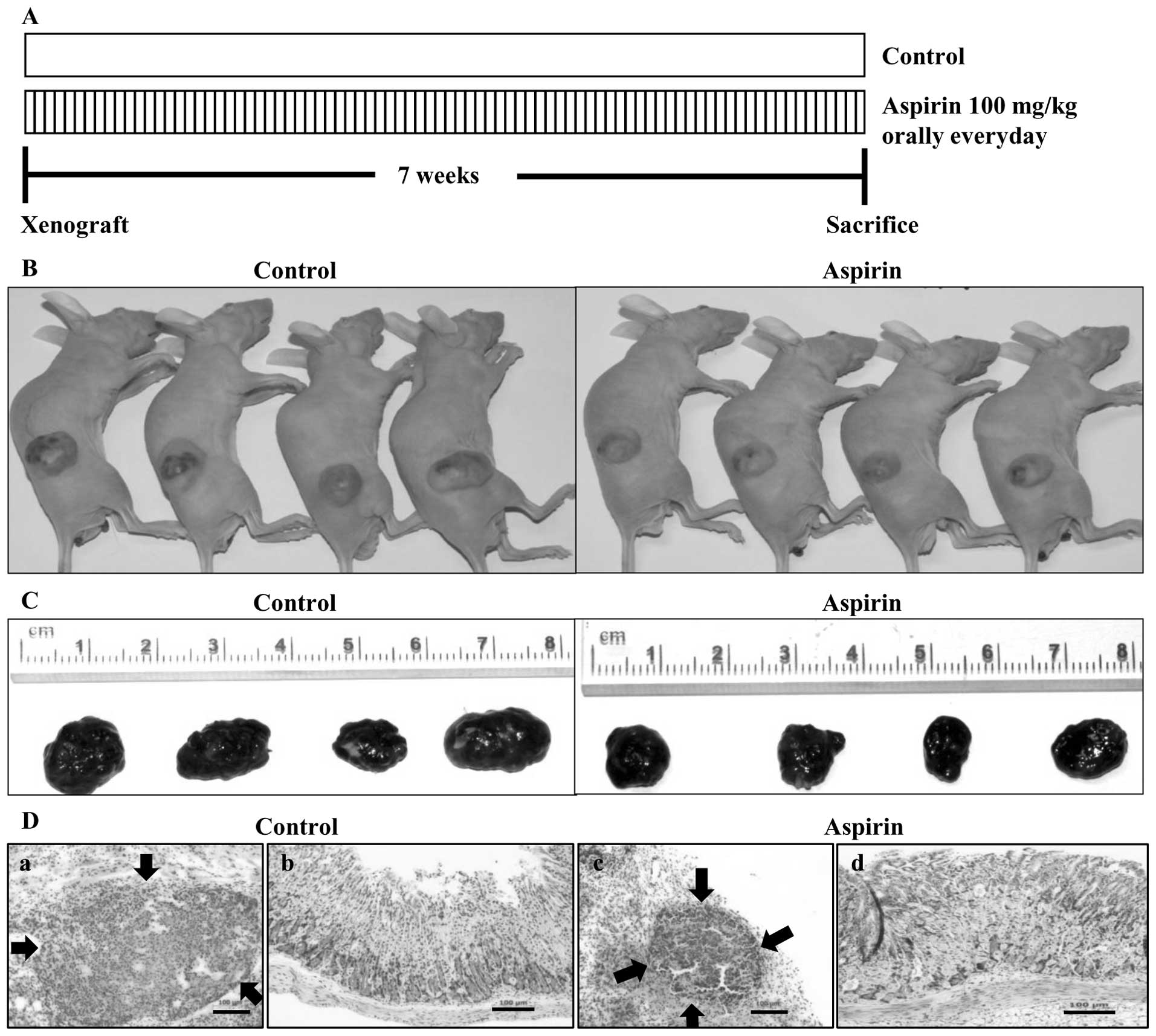
Aspirin administration reduces the growth
of HepG2 xenografts
Our in vitro observation suggested a
potential role of aspirin in the treatment of liver cancer.
Therefore, we examined the ability of aspirin to inhibit tumor
growth of HepG2 cells in a nude mouse xenograft model. Aspirin was
administered orally everyday at the dose of 100 mg/kg. After 7
weeks of treatment, mice were sacrificed and the tumors were
collected (Fig. 4A). Image of
tumors before (Fig. 4B) and after
(C) necropsy showed that aspirin treatment resulted in shrinkage of
tumor size. Histopathological sections of xenograft tumors in nude
mice were invariably encapsulated by connective tissue and there
was no histopathological difference except the size of tumors
between control (Fig. 4D-a) and
aspirin-treated mice (D-c). Histopathological examination of
stomachs of control mice (Fig.
4D-b) and aspirin-treated mice (D-d) revealed that the aspirin
dose (100 mg/kg) used in this study showed no degenerative changes
in the stomach tissues compared to the control group.
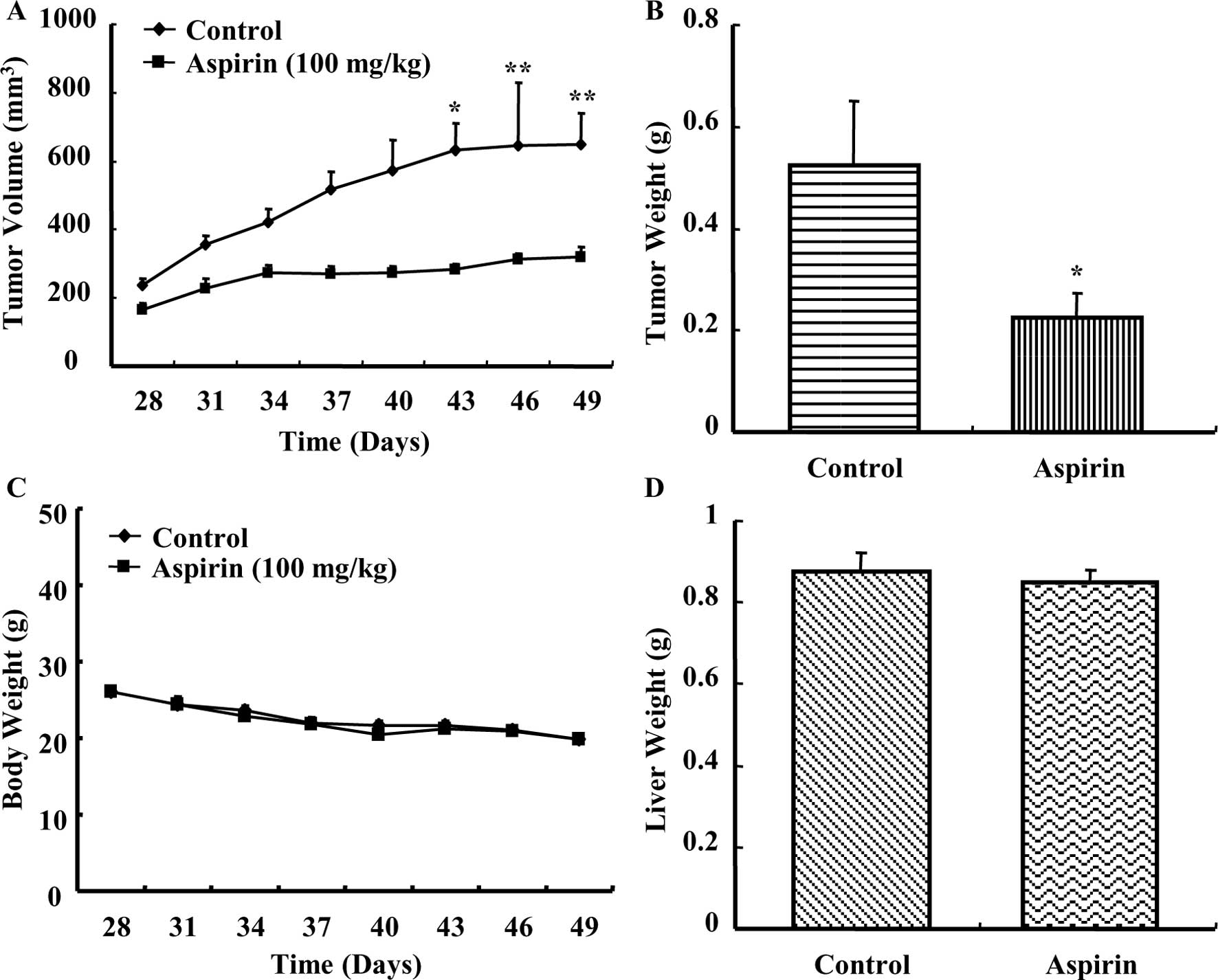
Aspirin treatment caused a time-dependent regression
of HepG2 xenograft tumors in nude mice as compared to the control
group (Fig. 5A). A considerable
reduction in tumor weight (Fig.
5B) was observed in mice treated with aspirin. The average body
weight of control or aspirin-treated mice did not vary
significantly throughout the experiment (Fig. 5C). Additionally, the average liver
weight of both control and aspirin-treated mice remained almost the
same after the experiment (Fig.
5D). There were no observable signs of distress in
aspirin-treated animals compared with control. These results
indicated that aspirin reduced the growth of HepG2 xenografts in
nude mice without causing any observable side effects.
Discussion
The ability of aspirin to trigger apoptosis in
cancer cells is well known and is consistent with the clinical and
epidemiological evidence on its anticancer effects. The aim of the
study was to investigate the anticancer activity of aspirin in
HepG2 human hepatocellular carcinoma cells. This study revealed
that aspirin had significant apoptotic activity against human
hepatocellular carcinoma cell line in culture and suppressed growth
of HepG2 cell xenograft tumors in nude mice.
Understanding of cell death signaling pathways is
relevant to understanding cancer and to developing more effective
anticancer agents. The induction of apoptosis by aspirin was
confirmed by DNA fragmentation. The regulation of apoptosis is a
complex process and involves a number of gene products including
Bcl-2 family protein. It has been shown that the Bcl-2 family plays
important regulatory roles in apoptosis, either as inhibitor or as
activator. Thus, it has been suggested that the ratio between the
level of pro-apoptotic Bax protein and that of the anti-apoptotic
factor Bcl-2 protein determines how a cell responds to an apoptotic
signal (16,32). In our study, there was a
concentration-dependent increase of Bax protein levels in HepG2
cells treated with aspirin and the levels of Bcl-2 slightly
decreased, and this consequently led to an increase in the ratio of
Bax/Bcl-2 as we previously described (31).
The apoptotic signaling pathway that leads to
caspase activation is subdivided into two major categories,
extrinsic or intrinsic pathways. The anti-apoptotic Bcl-2 protein
has been found to be associated with the mitochondrial membrane and
to prevent both the loss of the mitochondrial membrane potential
and the efflux of cytochrome c (33). Substantial evidence has been
accumulated suggesting that release of cytochrome c from
mitochondria is an important step in apoptosis. Once released in
the cytosol, cytochrome c binds to Apaf-1 in a
dATP/ATP-dependent manner, an event that triggers oligomerization
of Apaf-1/cytochrome c in complexes that activate
procaspase-9 (34). The ensuing
recruitment and activation of caspase-9 result in activation of
caspase-3, -6, and -7, which function as downstream effectors of
the cell death program. Caspase-3 is an executioner caspase that
also associated with death receptor pathway involving caspase-8
(33).
The extrinsic pathway is initiated by binding of the
transmembrane death receptors with their specific ligands
(Fas/FasL). Once activated, the intracellular domains of these
receptors (DD) bind to the adaptor protein Fas-associated death
domain (FADD) or TRADD (TNFR1-associated death domain protein) to
form the death inducing complex (DISC) with recruitment of
procaspase-8. Procaspase-8 is in turn proteolytically activated and
serves as the ‘initiator’ caspase, further activating downstream
effector proteins such as caspase-3, -6 and -7 to initiate cell
degradation, and thereby causing inevitable apoptosis (35). Activation of caspases results in
cleavage and inactivation of key cellular proteins, including the
DNA repair enzyme PARP (36).
Therefore, we evaluated the involvement of various caspases, death
receptor with their ligand, cytochrome c and PARP during
aspirin-mediated apoptosis in HepG2 cells. Our results implied that
the involvement of caspase-3, -8, -9, FasL, Fas, cytochrome
c, and PARP in execution of aspirin-induced apoptosis. Of
note, we reported that oral administration of aspirin caused a
considerable reduction in the growth of HepG2 cell xenograft tumors
in nude mice. The dose used in this study (100 mg/kg/day) can be
translated to a clinical dose of 520 mg for average body surface
area or approximately one aspirin tablet taken for analgesic
purposes in humans (37). There
was no morbidity due to treatment, nor was there drastic variation
in activity level or significant weight loss or gain between
control and aspirin-treated animals indicating low toxicity of
aspirin in vivo.
Experimental data presented herein showed that
aspirin induced cell death and reduced growth of human
hepatocellular carcinoma cells in vitro as well as in
vivo. Overall, aspirin shows great promise as a potent
anticancer agent.
Acknowledgements
This work was supported by the Bio-Scientific
Research Grant funded by the Pusan National University (PNU,
Bio-Scientific Research Grant) (PNU-2008-101-20080598000). We thank
Aging Tissue Bank for providing research information.
References
|
1
|
GLOBOCAN. 2008, Cancer fact sheet.
http://globocan.iarc.fr/factsheets/cancers.
|
|
2
|
Simonetti RG, Liberati A, Angiolini C and
Pagliaro L: Treatment of hepatocellular carcinoma: a systematic
review of randomized controlled trails. Ann Oncol. 8:117–136. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weiman A, Oldhafer KJ and Pichlmayr R:
Primary liver cancer. Curr Opin Oncol. 7:387–396. 1995. View Article : Google Scholar
|
|
4
|
Gasic GJ, Gasic TB and Stewart CC:
Anti-metastatic effects associated with platelet reduction. Proc
Natl Acad Sci USA. 61:46–52. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gasic GJ, Gasic TB and Murphy S:
Anti-metastatic effect of aspirin. Lancet. 2:932–933. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kolenich JJ, Mansour EG and Flynn A:
Haematological effects of aspirin. Lancet. 2:7141972. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jaffe BM: Prostaglandin and cancer: an
update. Prostaglandins. 6:453–461. 1974. View Article : Google Scholar
|
|
8
|
Bennett A and Del TM: Proceedings:
prostaglandins in human colonic carcinoma. Gut.
16:4091975.PubMed/NCBI
|
|
9
|
Herschman HR: Prostaglandin synthase 2.
Biochim Biophys Acta. 1299:125–140. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vane JR: Inhibition of prostaglandin
synthesis as a mechanism of action for aspirin-like drugs. Nat New
Biol. 231:232–235. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chell S, Kadi A, Williams AC and Paraskeva
C: Mediators of PGE2 synthesis and signalling downstream of COX-2
represent potential targets for the prevention/treatment of cancer.
Biochim Biophys Acta. 1766:104–119. 2006.PubMed/NCBI
|
|
12
|
Wang D and DuBois RN: Prostaglandins and
cancer. Gut. 55:115–122. 2006. View Article : Google Scholar
|
|
13
|
Kashfi K and Rigas B: Non-COX-2 targets
and cancer: expanding the molecular target repertoire of
chemoprevention. Biochem Pharmacol. 70:969–986. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shureiqi I, Xu X and Chen D: Nonsteroidal
anti-inflammatory drugs induce apoptosis in esophageal cancer cells
by restoring 15-lipoxygenase-1 expression. Cancer Res.
61:4879–4884. 2001.PubMed/NCBI
|
|
15
|
Zhang Z and DuBois RN: Par-4, a
proapoptotic gene, is regulated by NSAIDs in human colon carcinoma
cells. Gastroenterol. 118:1012–1017. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L, Yu J, Park BH, Kinzler KW and
Vogelstein B: Role of BAX in the apoptotic response to anticancer
agents. Science. 290:989–992. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bellosillo B, Pique M and Barragan M:
Aspirin and salicylate induce apoptosis and activation of caspases
in B-cell chronic lymphocytic leukaemia cells. Blood. 92:1406–1414.
1998.PubMed/NCBI
|
|
18
|
Raza H, John A and Benedict S:
Acetylsalicylic acid-induced oxidative stress, cell cycle arrest,
apoptosis and mitochondrial dysfunction in human hepatoma HepG2
cells. Eur J Pharmacol. 668:15–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schwenger P, Bellosta P, Vietor I,
Basilico C, Skolnik Y and Vilcek J: Sodium salicylate induces
apoptosis via p38 mitogen activated protein kinase but inhibits
tumour necrosis factor-induced c-Jun N-terminal
kinase/stress-activated protein kinase activation. Proc Natl Acad
Sci USA. 94:2869–2873. 1997. View Article : Google Scholar
|
|
20
|
Zimmermann KC, Waterhouse NJ, Goldstein
JC, Schuler M and Green DR: Aspirin induces apoptosis through
release of cytochrome c from mitochondria. Neoplasia.
2:505–513. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chan TA, Morin PJ, Vogelstein B and
Kinzler KW: Mechanisms underlying non-steroidal anti-inflammatory
drug-mediated apoptosis. Proc Natl Acad Sci USA. 95:681–686. 1998.
View Article : Google Scholar
|
|
22
|
Zhou XM, Wong BC and Fan XM: Non-steroidal
anti-inflammatory drugs induce apoptosis in gastric cancer cells
through up-regulation of bax and bak. Carcinogenesis. 22:1393–1397.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahdi JG, Alkarrawi MA, Mahdi AJ, Bowen ID
and Humam D: Calcium salicylate-mediated apoptosis in human HT01080
fibrosarcoma cells. Cell Prolif. 39:249–260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chinnaiyan AM and Dixit VM: The cell-death
machine. Curr Biol. 6:555–562. 1996. View Article : Google Scholar
|
|
25
|
Kasibhatla S and Tseng B: Why target
apoptosis in cancer treatment? Mol Cancer Ther. 2:573–580.
2003.PubMed/NCBI
|
|
26
|
Makin G: Targeting apoptosis in cancer
chemotherapy. Expert Opin Ther Targets. 6:73–84. 2002. View Article : Google Scholar
|
|
27
|
Johnstone RW, Ruefli AA and Lowe SW:
Apoptosis: a link between cancer genetics and chemotherapy. Cell.
108:153–164. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiang S, Sun Z, He Q, Yan F, Wang Y and
Zhang J: Aspirin inhibits ErbB2 to induce apoptosis in cervical
cancer cells. Med Oncol. 27:379–387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee JH, Park SE, Hossain MA, Kim MY, Kim
MN, Chung HY, Choi JS, Yoo YH and Kim ND:
2,3,6-Tribromo-4,5-dihydroxybenzyl methyl ether induces growth
inhibition and apoptosis in MCF-7 human breast cancer cells. Arch
Pharm Res. 30:1132–1137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SE, Park C, Skim SH, Hossain MA, Kim
MY, Chung HY, Son WS, Kim GY, Choi YH and Kim ND: Korean red
ginseng extract induces apoptosis and decreases telomerase activity
in human leukemia cells. J Ethnopharmacol. 121:304–312. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Park SE, Lee SW, Hossain MA, Kim MY, Kim
MN, Ahn EY, Park YC, Suh HS, Kim GY, Choi YH and Kim ND: A
chenodeoxycholic derivative, HS-1200, induces apoptosis and cell
cycle modulation via Egr-1 gene expression control on human
hepatoma cells. Cancer Lett. 270:77–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oltvai ZN, Milliman CL and Korsmeyer SJ:
Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that
accelerates programmed cell death. Cell. 74:609–619. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rosse T, Olivier R, Monney L, Rager M,
Conus S and Fellay I: Bcl-2 prolongs cell survival after
Bax-induced release of cytochrome c. Nature. 391:496–499.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zou H, Li X and Wang X: An Apaf-1,
cytochrome c multimeric complex is a functional apoptosome
that activates procaspase-9. J Biol Chem. 274:11549–11556.
1999.
|
|
35
|
Burz C, Berindan-neagoe I, Balacescu O and
Irimie A: Apoptosis in cancer: key molecular signaling pathways and
therapy targets. Acta Oncol. 48:811–821. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wolf BB and Green DR: Suicidal tendencies:
apoptotic cell death by caspase family proteinases. J Biol Chem.
274:20049–20052. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stark LA, Reid K, Sansom OJ, Din FV,
Guichard S, Mayer I, Jodrell DI, Clarke AR and Dunlop MG: Aspirin
activates the NF-κB signaling pathway and induces apoptosis in
intestinal neoplasia in two in vivo models of human colorectal
cancer. Carcinogenesis. 28:968–976. 2007.
|