Contents
Introduction
Cannabinoids
Cannabinoid-induced apoptotic pathways
Cannabinoid-induced autophagic pathways
Synergistic effects of cannabinoids in combination
with other drugs
Conclusion
Introduction
The health and survival of every living organism is
based on the crucial balance between cellular life and death
pathways. To maintain a normal size, each tissue must balance the
plethora of opposite signals which arrive on its membrane surface
and finely control each to prevent pathological conditions such as
cancer.
For many decades, the molecular bases of cancer
transformation were related to the gain of function of oncogenes or
the inactivation of tumor suppressor genes, two classes of genes
involved in regulating cell proliferation (1,2).
More recently, this classification has been extended when it became
clear that neoplastic transformation is related not only to the
uncontrolled proliferative thrust but also to the loss of death
ability (3,4). Stimulation and/or restoration of cell
death can lead to the suppression of transformation and
tumorigenesis (4). From then on,
the list of tumor suppressors with death-inducing action towards
tumor cells has lengthened progressively. Moreover, mutations which
affect specific genes controlling death signalling can compromise
the effectiveness of chemotherapy (5).
Recent studies have shown that tumor cells can
activate different pathways of death, such as apoptosis, necrosis,
mitotic catastrophe and autophagy which very often are activated by
the same oncogenic factors related to proliferative signals
(6). Signalling molecules and
pathways involved in these alternative forms of cell death have not
yet been defined and the activation of a specific cell death
mechanism may depend on the metabolic context, the genotype and/or
phenotype of the cell, or the kind of damage. Thus, the relative
contribution of these different modes of cell death remains to be
defined.
Apoptosis, the first identified process of
genetically programmed cell death, has been extensively studied and
its contribution to the pathogenesis of cancer is well documented.
Classical studies on apoptotic pathways evidenced the activation of
caspase family proteases as a key step in apoptotic cell death
(7). Caspases can be initiated
through the extrinsic (receptor) pathway or at the mitochondria by
stimulating the intrinsic pathway (8,9) and
following the activation they are responsible for the cleavage of a
number of different substrates leading to many of the morphologic
features of apoptotic cell death.
Another interesting event in cancer is the balance
between the expression of survival factors and the induction of
apoptotic ones. It is well known that cancer cells show
constitutive activation of survival factors which sustain
deregulated cell proliferation and counteract death stimuli
(10). The survival factors which
are very often related to tumorigenicity are survivin, a member of
inhibitor of apoptosis protein (IAP) family and AKT. Survivin
sustains cell survival maintaining inactivated the executioner
caspases (11). The other survival
factor, the serine/threonine protein kinase AKT, is widely
recognised as a key mediator of growth factor-promoted cell
survival. It sustains nutrient uptake, macromolecular synthesis and
ATP production. Also, this factor which is constitutively active in
many types of human cancer (12),
plays an anti-apoptotic role in the cell. In fact, it
phosphorylates and inactivates some members of Bcl-2 and caspase
families.
Surprisingly, a new mechanism of cell death in
cancer cells has been demonstrated, which has been related to the
activation of autophagic pathway (13,14).
Originally known as a housekeeping process responsible for the
protein and organelle turnover, autophagy has also been indicated
to play a significant role in cell death.
Autophagic process is achieved through two pathways:
microautophagy and macroautophagy, which can be further classified
into canonical and non-canonical forms (14). They differ in the transport of the
material to be degraded within the lysosomal lumen, the type of
transported material and its regulation. Microautophagy has been
traditionally regarded as a form of active autophagy to ensure the
turnover of long-lived proteins in basal conditions (15). During microautophagy, lysosomes
incorporate and digest regions of cytosol, including proteins and
cytoplasmic organelles, by invagination or protrusion without the
intermediate formation of autophagic vacuoles.
Macroautophagy (or autophagy) is responsible for the
degradation of soluble proteins and organelles under stress
conditions (16). It involves the
formation of double-membrane vacuoles (autophagosomes) that
sequester portions of the cytoplasm and translocate them to the
lysosomes (17).
To date, >30 autophagy-related genes (ATG)
required for autophagy and their related pathways have been
identified and analysed as markers of the different steps of
autophagic process. Some, such as Atg1, are involved in upstream
steps of autophagosome formation; others, such as Beclin-1 (Atg6),
perform their role as part of a core complex that contains vacuolar
sorting protein 34 (VPS34); others, such as LC-3, a protein which
is lipidated and inserted into the outer and inner surface of
autophagosomes, are essential for the biogenesis of the large
double-membrane vesicles (18).
Once digested by autophagosome, the substances are reabsorbed in
the vacuole and made available to the cells.
At the molecular level, autophagy is regulated by
many protein factors which are involved in key steps of the
mechanism. The PI3K/AKT/mTOR axis and AMP kinase are the two major
pathways involved in autophagy induction. mTOR is a repressor of
autophagy because it is responsible for Atg1 hyper-phosphorylation,
an event that inhibits the formation of induction complex. A
correlation between AKT and mTOR pathway exists; in fact, AKT is
able to positively regulate mTOR since it phosphorylates and
inhibits TSC1/TSC2 complex which is, in turn, responsible for mTOR
inactivation. This inhibition allows mTOR to act freely thus
inhibiting autophagy (19).
The other crucial pathway involved in autophagy is
sustained by AMPK. This protein is activated by an increase in the
intracellular AMP/ATP ratio as a consequence of energy deficit.
This event is responsible for mTOR inhibition and p53
phosphorylation, inducing increase in autophagy and arrest of cell
cycle progression, respectively (20).
In an attempt to explain the exact role of autophagy
in cancer process, Eisenberg-Lerner and Kimchi (21) recently presented a model in which
the role of autophagy is dependent on the stage of cell life, i.e.
precancerous stage, transformed cell or solid tumor. In the first
two stages autophagy could accelerate tumor development whereas in
solid tumors, when apoptosis is absent, autophagy program could be
antitumorigenic.
It has been demonstrated that autophagy can
undertake a complex interplay with apoptosis (22). In relation to cell type, it could
serve as a cell survival pathway suppressing apoptosis, or in
others, it could trigger a death pathway either in collaboration
with apoptosis or substituting to that when the former is
defective. It is very difficult to distinguish the two pathways
because some death stimuli can activate both of them and which
pathway will be undertaken depends on the molecular expression
profile of the cell type (23). An
interesting observation concerns the role explained by ER-stress
response in determining cell fate. Different injuries, such as
calcium imbalance, oxidative stress or misfolded proteins, trigger
a condition indicated as stress of endoplasmic reticulum
(ER-stress) which can lead to different cell responses. ER-stress
can drive mitochondria-dependent apoptosis when there is the
activation of the transcription factor CHOP; or it can determine
cell survival when it is accompanied by the increase in the level
of GRP78, the main intraluminar HSP70 that protects cells from the
injury. Differently, if the level of the CHOP target TRB-3
significantly increases, ER-stress triggers autophagy.
The cross-talk between apoptosis and autophagy is
therefore quite complex, and sometimes contradictory, but surely
critical to the overall cell fate.
Cannabinoids
Cannabis derivatives (cannabinoids) include the
bioactive constituents of cannabis
(Δ9-tetrahydrocannabinol, cannabidiol), endogenous
lipids with cannabinoid-like activity (anandamide,
2-arachydonoyl-glycerol) and the synthetic analogues (WIN55,212-2,
methanandamide, the stable analogue of anandamide, JWH-015, HU-210,
HU-331) (24).
In the last few years, investigations have suggested
the potential application of cannabinoids as antitumor drugs
because in many in vivo and in vitro tumor models the
activation of the cannabinoid system induces cell cycle arrest,
inhibition of cell survival and activation of programmed cell death
signalling (25). Apart from
regulating tumor cell growth and death, other antitumorigenic
mechanisms of cannabinoids are currently emerging as a focus of
research work, such as their effects on tumor neovascularization,
cell migration, adhesion, invasion and metastasis (26). The present review focuses on the
impact of cannabinoids on apoptosis and autophagy, the two main
death pathways responsible for cancer cell death.
Cannabinoid-induced apoptotic pathways
Although the antitumor properties of cannabinoids
were first observed more than 30 years ago, when Munson et
al (27) demonstrated that
Δ9-tetrahydrocannabinoid (THC) inhibits lung
adenocarcinoma cell growth in vivo, the elucidation of
mechanisms employed by cannabinoids for influencing cancer cell
proliferation and death was developed in the last two decades.
However, there are still many obscure sides on death pathways
activated by these compounds and, in particular, on the different
contribution of apoptosis and autophagy in cell death.
The anticancer potential of this class of compounds
can be very different in the various tumor systems. This depends on
the mechanism used by cannabinoids to interact with the cells, i.e.
the class of receptors to which they bind or on the specific
intracellular activated pathways.
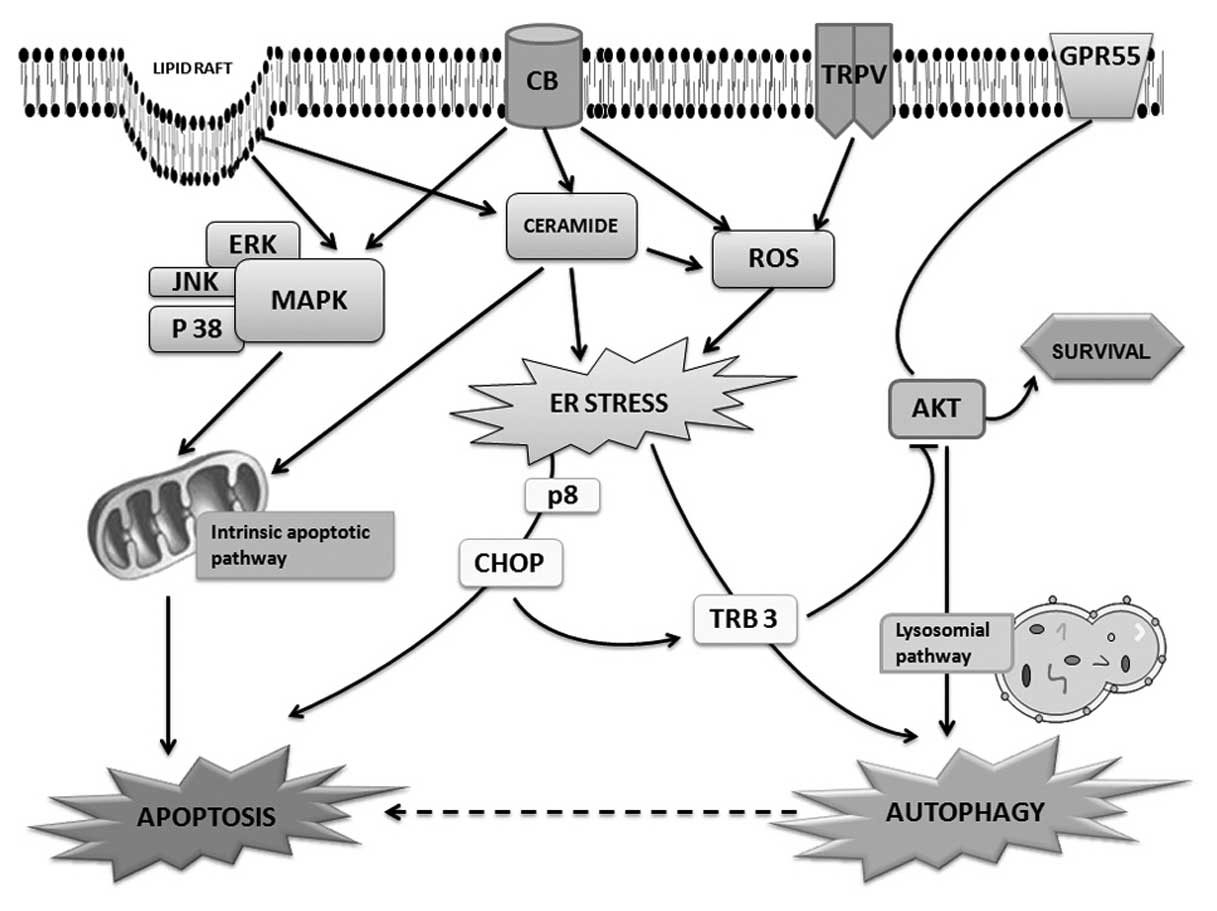
Regarding the interaction with the target cell, it
has been demonstrated that cannabinoids can interact with the
specific type 1 or 2 cannabinoid receptors (CB1 and CB2), which are
differently expressed in neural and peripheral tissues,
respectively, with transient receptor potential channels of the
vanilloid type-1 (TRPV1), or directly with membrane microdomains
rich in cholesterol named lipid rafts (28). The interaction of cannabinoids with
the different receptor types often leads to the same cell fate even
if different intracellular signalling cascades can be activated.
For example, the activation of CB receptors can be responsible for
the increase in the level of ceramide or the decrease of cAMP level
via inhibition of adenilate cyclase. These events result in
canonical apoptosis mediated by the activation of caspase
activities (29,30). It has also been demonstrated that
the interaction of cannabinoids with TRPV receptors causes the
activation of overlapping mechanisms, such as the mitochondrial
apoptotic pathway, although in this case the event is accompanied
by the increase in the level of ROS with the consequent oxidative
stress (31). However, sometimes
the effects of cannabinoids can not be counteracted by the addition
of selective cannabinoid receptor antagonists, hypothesizing that
another type of interaction between cannabinoids and cell membrane
can be present. In hepatic stellate cells, an interaction between
anandamide and lipid rafts, the membrane micro-domains rich in
cholesterol, was found. This relationship induces downregulation of
PI3K/AKT signalling pathway with a possible role in inflammatory
process (32). Differently, in
hepatocellular carcinoma cells, as well as in melanoma cells, the
interaction of the synthetic cannabinoid WIN55,212-2 to lipid rafts
is able to promote a canonical caspase-dependent apoptotic pathway
(33,34). Moreover, it is not possible to
exclude that the activation of CB1 receptors and the consequent
induction of cannabinoid signalling could be mediated by the
association of receptor with lipid rafts, as recently demonstrated
by Oddi et al (35) in
elegant experiments of site-specific mutations of palmitoylated
cysteine residue of CB1 receptors.
An exception to that previously reported is
represented by GPR55, a novel potential cannabinoid receptor, whose
activation is strictly related to cell proliferation (36).
The analysis of intracellular mediators of apoptotic
cell death induced by cannabinoids leads to the identification of
some molecules, such as ceramide, reactive oxygen species (ROS),
mitogenic kinases (MAPK) and some survival factors, that, more than
others, seem to be representative of cannabinoid actions. Ceramide
is one of the first molecules identified as a mediator of
cannabinoid action. The induction of ceramide accumulation mediated
by CB receptors leads to apoptosis in mantle cell lymphoma, glioma,
colon and pancreatic cancer (37–40).
The increase in ceramide level can be dependent on the de
novo synthesis or the release from membrane sphyngolipids
induced by the activation of sphyngomyelinase. The different
origins of ceramide can be disclosed by the employment of specific
inhibitors of the two enzymes, fumonisin B1, the inhibitor of
ceramide synthase, or desipramine, an inhibitor of sphyngomyelinase
(41,42).
In many cannabinoid signalling pathways, ROS can
exert a crucial role in activating both initiator and executioner
caspase activities suggesting that continuous oxidative stress can
occur following cannabinoid exposure. The involvement of ROS in
cannabinoid-induced apoptosis has been widely evidenced in glioma
and leukemia cells (43,44). Moreover, the demonstration of ROS
involvement in cannabinoid-induced apoptosis is also confirmed by
the employement of N-acetyl-cysteine, a thiol antioxidant that
scavenges ROS, or the NAD(P)H oxidase inhibitors, which are able to
attenuate cannabinoid effects. A strong interplay between ceramide
and generation of oxidative stress has been demonstrated. Ceramide
stimulates the formation of ROS and apoptotic mechanisms,
establishing a link between sphingolipid metabolism and oxidative
stress. Moreover, inhibition of ROS generating enzymes or treatment
with antioxidants impairs sphingomyelinase activation and ceramide
production. Therefore, it is plausible to hypothesize that the
contemporaneous activation of CB and TRP vanilloid receptors,
responsible for the triggering of the two intracellular mediators
(ceramide and ROS), can augment the effects induced by cannabinoids
carrying out an amplification of caspase cascades.
The study on downstream events following ROS
generation or ceramide induction has evidenced the involvement of
ER-stress. In many experimental tumor models, high ROS levels
induce ER-stress as demonstrated by the increase in the level of
specific ER-stress mediators (p8, CHOP, TRB-3 and GRP-78) which, in
turn, are responsible for the activation of mitochondrial intrinsic
apoptotic pathway (45). On the
other hand, a relationship between the increase in ceramide level
and ER-stress is also well documented in cannabinoid-induced
apoptosis in tumor cells (46).
The activation of p8/CHOP axis in cannabinoid-induced apoptosis has
also been documented (40,47).
Another pathway, which has been demonstrated to be
modulated following cannabinoid exposure, is represented by
mitogen-activated protein kinase (ERK, JNK and p38/MAPK) cascades.
The main players of these pathways are a group of serine/threonine
protein kinases that convert extracellular stress stimuli into
different, sometimes opposite, cellular responses, including cell
cycle arrest, apoptotic cell death and cytokine production, through
the phosphorylation of specific targets. A plethora of data reports
the activation of stress-activated protein kinases or
extracellular-related signal kinases in cannabinoid-dependent
control of cancer cell growth and survival (43,48).
Interestingly, it has been demonstrated that the duration of the
stimulus can be fundamental for the type of cellular response. A
transient activation of ERK cascade leads to cell survival and
proliferation, while long-term ERK activation results to apoptotic
response as proposed by Galve-Roperh et al (48) in malignant glioma and confirmed in
Kaposi sarcoma cells (49). In
this cell model, WIN55,212-2 induced phosphorylation of ERK1/2 at
the beginning of its action. Then, the noxious stimulus may trigger
activation of stress kinases JNK and p38 which, in turn, activate
downstream mechanisms leading Kaposi’s sarcoma cells to apoptotic
death (49). The activation of ERK
1/2 has been recently demonstrated also in gastric cancer; in this
case, it is related to cell cycle arrest in G0/G1 phase of
proliferative cycle, an event which precedes apoptotic response
(50). Differently, studies on
several prostate and ovarian cancer cell lines evidenced that the
activation of ERK signalling by the putative cannabinoid receptor
GPR55 activates an autocrine loop that sustains cell proliferation
(51).
It is well known that the activation of death
pathways needs to be accompanied by downregulation of survival
factors. A central intracellular pro-survival signalling is
represented by the PI3K/AKT pathway, whose importance in different
cancers is also corroborated by clinical studies. This kinase is
the central node of PI3K/AKT/mTOR signalling pathway that activates
crucial processes such as cell survival, growth, proliferation,
angiogenesis, and cell migration and invasion (52). On the contrary, inhibition of pAKT
leads to cell cycle arrest which precedes apoptotic response very
often mediated by the involvement of intrinsic
mitochondria-dependent pathway. Downregulation of AKT is involved
in cannabinoid antitumoral action. In human gastric cancer cells,
it has been reported that cannabinoid receptor agonists induce cell
cycle arrest which is a consequence of AKT inhibition related to
the activation of MAPK pathway (50). Interesting results were also
obtained in astrocytoma (53). In
this model, cannabinoids induce apoptosis only in cells expressing
low levels of CB receptors, while in cells with high CB receptor
levels, cannabinoids are ineffective because of the contemporaneous
high amount of the phosphorylated pro-survival AKT. These results
suggest that the coupling of CB receptors to AKT pathway (when
these receptors are expressed at mid and high levels) eliminates
the ability of cannabinoids to induce apoptosis in astrocytoma
cells. Therefore, AKT pathway is an attractive target for
anticancer agents and clinical trials with PI3K or AKT inhibitors
could reserve encouraging results.
Cannabinoid-induced autophagic pathways
As previously indicated, autophagy can assume
different and opposite roles in cell fate. In fact, this process
may trigger survival pathways, collaborate with apoptosis to induce
cell death or substitute itself to apoptosis to start an autonomous
death pathway.
Numerous recent studies have indicated the
cannabinoid-mediated induction of autophagy in different
experimental cancer models. Noteworthy is the observation that the
activation of autophagic pathway is often mediated by the same
molecules that are involved in apoptosis.
In human glioma cells, the main cancer model for
studying cannabinoid action, it has been observed that
Δ9-tetrahydrocannabinol (THC) induces cell death through
stimulation of autophagy (54).
Data indicate that THC induces ceramide accumulation,
phosphorylation of elF2α and ER stress which activates autophagy
via TRB3-dependent inhibition of the AKT/mTOR axis. In these cells
autophagy seems to be upstream of apoptosis in cannabinoid-induced
cancer cell death. The stimulation of autophagy-mediated apoptosis
induced by cannabinoids has also been demonstrated in animal models
of cancer, including glioma (54,55).
Similar studies performed to investigate the effects of
cannabinoids on the growth of hepatocellular carcinoma cells have
demonstrated that THC markedly reduces the viability of the human
hepatocellular carcinoma cell lines through induction of autophagy
mediated, also in this case, by upregulation of TRB3 and subsequent
inhibition of the serine-threonine kinase AKT and AMPK stimulation.
As in glioma animal model, in vivo studies on hepatocellular
carcinoma subcutaneous xenografts have revealed that THC-dependent
growth inhibition is reduced when autophagy is genetically or
pharmacologically blocked, thus confirming that the induction of
autophagy can be a key step in cannabinoid-induced cell death
(56).
Further biochemical pathways have been suggested in
the attempt to clarify the molecular mechanisms of cell death
induced by cannabinoids. In breast cancer cells, cannabidiol
induces ER-stress and subsequently inhibition of AKT and mTOR
signalling, indicating autophagy activation. In addition, data have
also shown the activation of intrinsic apoptotic pathway. The
coexistence of autophagy and apoptosis has been confirmed by
different observations: the inhibition of cannabidiol-dependent ROS
production blocked the induction of both processes and the increase
in the level of beclin-1, a marker of autophagy, plays a central
role in the induction of cannabidiol-mediated apoptosis (57).
A new recent study on the interplay between
autophagy and apoptosis induced by cannabinoids showed a surprise
in mantle cell lymphomas (58).
Interestingly, this study shows that the response to cannabinoid
treatment decreases cell viability which does not involve the
caspase-3 cascade. Moreover, mantle cell lymphoma primary cells
respond to cannabinoid treatment through the formation of
cycloheximide-sensitive cytoplasmic vacuoles. However, the lack of
enhanced autophagosome formation and lysosomal contribution also
excludes the involvement of a canonical autophagic process. The
authors hypothesize that the observed features resemble
paraptosis-like cell death, a third type of programmed cell death,
not previously described in response to cannabinoids. Activation of
more types of cell death by cannabinoids widens their potential
therapeutic usefulness in cells overexpressing cannabinoid
receptors.
Synergistic effects of cannabinoids in
combination with other drugs
Recent studies have demonstrated the ability of
cannabinoids to synergize with other molecules to trigger death
pathways in cancer cells. It is well known that in cancer therapy
the employment of combinations of drugs rather than a single drug
represents a therapeutic strategy with distinct advantages. On the
one hand, the contemporaneous activation of different biochemical
pathways can achieve synergistic effects; on the other hand, the
combination can result in a reduction of the dose of each single
drug thereby reducing side effects. Thus, the ability of
cannabinoids in synergizing with other drugs to improve their
anticancer activity has been investigated. In particular, we have
recently demonstrated that the synthetic cannabinoid WIN55,212-2
sensitizes hepatocellular carcinoma cells to apoptosis, mediated by
tumor necrosis-related apoptosis inducing ligand (TRAIL). The
molecular mechanism induced by WIN/TRAIL combined treatment,
involves activation of the extrinsic apoptotic pathway through
upregulation of TRAIL death receptor DR5 (47). This event seems to be related to
the increase in the level of p8 and CHOP, two factors implicated in
ER stress response and apoptosis. Moreover, WIN55,212-2 treatment
also induces a marked downregulation of some survival factors.
Therefore, both the induction of DR5 and the decrease of survival
factors explain synergistic effects of the drugs in hepatocellular
carcinoma cells. Our unpublished data obtained in osteosarcoma
cells seem to indicate that WIN also triggers an autophagic pathway
with the increase in the level of beclin-1 and LC-3 II, but this
pathway is not carried out because of the lysosomal membrane
permeabilization. Also, in pancreatic cancer cells the combination
of cannabinoids with gemcitabine, a pyrimidine analog largely
employed in anticancer therapy, induces synergistic effects via
activation of autophagy (59). In
this case, gemcitabine induces upregulation of both CB receptors
thus sensitizing cells to cannabinoid effects. A central role in
this pathway seems to be related to the increase in ROS production,
induction of ER-stress which carried out to specific cell death
pathway of type II (autophagy). A curious observation about the
therapeutic potential of cannabinoids is that in the first example
cannabinoids sensitize the cells to TRAIL-induced cell death while
in the latter the cannabinoid death action is potentiated by
gemcitabine addition in pancreatic cancer cells. Another example of
synergistic effects of cannabinoids with other drugs has also been
reported by Gustafsson et al (60) who demonstrated that the synthetic
cannabinoid HU210, anandamide and its other derivatives induce
synergistic and cytotoxic, rather than antiproliferative, effects
when employed in combination with the classic pyrimidine antagonist
5-fluorouracil (5-FU) in the colorectal carcinoma cells. The
authors report that the effect does not seem to involve cannabinoid
receptors and suggest the involvement of ER-stress because the
employment of common antioxidants attenuates cannabinoid
cytotoxicity.
Autophagy is also involved in the strong antitumoral
effects induced in glioma xenografts by combined administration of
THC and temozolomide, the benchmark agent for the management of
glioblastoma multiforme, an effect that is also observed in tumors
which are resistant to temozolomide treatment. THC/temozolomide
combined treatment enhances autophagy, whereas pharmacologic or
genetic inhibition of this process prevents the effects of combined
treatment, supporting that the activation of autophagy plays a
crucial role in the mechanism of action of this drug combination
(55).
Conclusion
Data reported in this review seem to confirm the
ability of cannabinoids to induce cell death in different tumor
models. Moreover, it can be seen from the brief literature overview
presented here that these compounds are effective in inducing the
main cell death modes, i.e. apoptosis and autophagy, and that cell
fate will depend on the role and the interplay among these
different death signals. Unfortunately, to simplify the mechanisms
induced by cannabinoids to carry out cells to death and
discriminate the role played by each intracellular mediator appears
to be very difficult for different reasons: i) the two pathways
often employ the same molecules (Fig.
1) and ii) the identification of autophagy as cell death
program of type II is very recent as well as the discovery that
many apoptotic pathways are preceded by the activation of
autophagy. Therefore, it is not possible to exclude that a
cannabinoid-triggered death pathway which was initially identified
as a canonical apoptotic cell death hides a ‘secret step of
autophagic activation’. Furthermore, the observation that
cannabinoids can synergize with other molecules thus accelerating
death pathway, make these compounds, employed alone or in
combination, promising for clinical outcome.
References
|
1
|
Weinberg RA: The molecular basis of
oncogenes and tumor suppressor genes. Ann NY Acad Sci. 758:331–338.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yuspa SH, Długosz AA, Cheng CK, Denning
MF, Tennenbaum T, Glick AB and Weinberg WC: Role of oncogenes and
tumor suppressor genes in multistage carcinogenesis. J Invest
Dermatol. 103:S90–S95. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lowe SW and Lin AW: Apoptosis in cancer.
Carcinogenesis. 21:485–495. 2000. View Article : Google Scholar
|
|
4
|
Brown JM and Attardi LD: The role of
apoptosis in cancer development and treatment response. Nat Rev
Cancer. 5:231–237. 2005.PubMed/NCBI
|
|
5
|
Brown JM and Wilson G: Apoptosis genes and
resistance to cancer therapy: what does the experimental and
clinical data tell us? Cancer Biol Ther. 2:477–490. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsatsanis C and Spandidos DA: Oncogenic
kinases signaling in human neoplasms. Ann NY Acad Sci.
1028:168–175. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Degterev A, Boyce M and Yuan J: A decade
of caspases. Oncogene. 22:8543–8567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peter ME and Krammer PH: The
CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10:26–35. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Green DR and Kroemer G: The
pathophysiology of mitochondrial cell death. Science. 305:626–629.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fassetta M, D’Alessandro L, Coltella N, Di
Renzo MF and Rasola A: Hepatocyte growth factor installs a survival
platform for colorectal cancer cell invasive growth and overcomes
p38 MAPK-mediated apoptosis. Cell Signal. 18:1967–1976. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Altieri DC: Survivin and IAP proteins in
cell-death mechanisms. Biochem J. 430:199–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tokunaga E, Oki E, Egashira A, Sadanaga N,
Morita M, Kakeji Y and Maehara Y: Deregulation of the AKT pathway
in human cancer. Curr Cancer Drug Targets. 8:27–36. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mathew R: Role of autophagy in cancer. Nat
Rev Cancer. 7:961–967. 2007. View
Article : Google Scholar
|
|
14
|
Kimmelman AC: The dynamic nature of
autophagy in cancer. Genes Dev. 25:1999–2010. 2011. View Article : Google Scholar
|
|
15
|
Krick R, Muehe Y, Prick T, Bremer S,
Schlotterhose P, Eskelinen EL, Millen J, Goldfarb DS and Thumm M:
Microautophagy of the nucleus requires the core macroautophagy
genes. Mol Biol Cell. 19:4492–4505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Farré JC and Subramani S: Peroxisome
turnover by micropexophagy: an autophagy-related process. Cell
Biol. 14:515–523. 2004.PubMed/NCBI
|
|
17
|
Periyasamy-Thandavan S, Jiang M,
Schoenlein P and Dong Z: Autophagy: molecular machinery,
regulation, and implications for renal pathophysiology. Am J
Physiol Renal Physiol. 297:F244–F256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N: The role of the Atg1/ULK1
complex in autophagy regulation. Curr Op Cell Biol. 22:132–139.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of AKT/PKB by the
rictormTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hardie D: AMP-activated/SNF1 protein
kinases: conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–781. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eisenberg-Lerner A and Kimchi A: The
paradox of autophagy and its implication in cancer etiology and
therapy. Apoptosis. 14:376–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Djavaheri-Mergny M, Maiuri MC and Kroemer
G: Cross talk between apoptosis and autophagy by caspase-mediated
cleavage of Beclin 1. Oncogene. 29:1717–1719. 2010. View Article : Google Scholar
|
|
23
|
Wong CH, Iskandar KB, Yadav SK, Hirpara
JL, Loh T and Pervaiz S: Simultaneous induction of non-canonical
autophagy and apoptosis in cancer cells by ROS-dependent ERK and
JNK activation. PLoS One. 5:e99962010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pertwee RG: Cannabinoid receptor ligands:
clinical and neuropharmacological considerations, relevant to
future drug discovery and development. Expt Op Invest Drugs.
9:1553–1571. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guzmàn M: Cannabinoids: potential
anticancer agents. Nat Rev Cancer. 3:745–755. 2003.
|
|
26
|
Freimuth N, Ramer R and Hinz B:
Antitumorigenic effects of cannabinoids beyond apoptosis. J
Pharmacol Exp Therapeutics. 332:336–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Munson AE, Harris LS, Friedman MA, Dewey
WL and Carchman RA: Antineoplastic activity of cannabinoids. J Natl
Cancer Inst. 55:597–602. 1975.PubMed/NCBI
|
|
28
|
Demuth DG and Molleman A: Cannabinoid
signalling. Life Sci. 78:549–563. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Herrera B, Carracedo A, Diez-Zaera M,
Gómez del Pulgar T, Guzmán M and Velasco G: The CB2 cannabinoid
receptor signals apoptosis via ceramide-dependent activation of the
mitochondrial intrinsic pathway. Exp Cell Res. 312:2121–2131. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bifulco M, Malfitano AM, Pisanti S and
Laezza C: Endocannabinoids in endocrine and related tumours. Endocr
Relat Cancer. 15:391–408. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maccarrone M, Lorenzon T, Bari M, Melino G
and Finazzi-Agro A: Anandamide induces apoptosis in human cells via
vanilloid receptors. Evidence for a protective role of cannabinoid
receptors. J Biol Chem. 275:31938–31945. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang Q, Liu HY, Zhang YW, Wu WJ and Tang
WX: Anandamide induces cell death through lipid rafts in hepatic
stellate cells. J Gastroenterol Hepatol. 25:991–1001. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Giuliano M, Pellerito O, Portanova P,
Calvaruso G, Santulli A, De Blasio A, Vento R and Tesoriere G:
Apoptosis induced in HepG2 cells by the synthetic cannabinoid WIN:
involvement of the transcription factor PPARgamma. Biochimie.
91:457–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Scuderi MR, Cantarella G, Scollo M,
Lempereur L, Palumbo M, Saccani-Jotti G and Bernardini R: The
antimitogenic effect of the cannabinoid receptor agonist WIN55212-2
on human melanoma cells is mediated by the membrane lipid raft.
Cancer Lett. 310:240–249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oddi S, Dainese E, Sandiford S, Fezza F,
Lanuti M, Chiurchiù V, Totaro A, Catanzaro G, Barcaroli D, De
Laurenzi V, et al: Palmitoylation of cysteine 415 of helix 8:
effect on membrane localisation and signalling of the CB(1)
cannabinoid receptor. Br J Pharmacol. 165:2635–2651. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu G, Ren G and Shi Y: The putative
cannabinoid receptor GPR55 promotes cancer cell proliferation.
Oncogene. 30:139–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gustafsson K, Christensson B, Sander B and
Flygare J: Cannabinoid receptor-mediated apoptosis induced by
R(+)-methanandamide and Win55,212-2 is associated with ceramide
accumulation and p38 activation in mantle cell lymphoma. Mol
Pharmacol. 70:1612–1620. 2006.
|
|
38
|
Blazquez C, Carracedo A, Salazar M,
Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G and Guzman
M: Down-regulation of tissue inhibitor of metalloproteinases-1 in
gliomas: a new marker of cannabinoid antitumoral activity?
Neuropharmacology. 54:235–243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cianchi F, Papucci L, Schiavone N, Lulli
M, Magnelli L, Vinci MC, Messerini L, Manera C, Ronconi E,
Romagnani P, et al: Cannabinoid receptor activation induces
apoptosis through tumor necrosis factor alpha-mediated ceramide de
novo synthesis in colon cancer cells. Clin Cancer Res.
14:7691–7700. 2008. View Article : Google Scholar
|
|
40
|
Carracedo A, Gironella M, Lorente M,
Garcia S, Guzman M, Velasco G and Iovanna JL: Cannabinoids induce
apoptosis of pancreatic tumor cells via endoplasmic reticulum
stress-related genes. Cancer Res. 66:6748–6755. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Olea-Herrero N, Vara D, Malagarie-Cazenave
S and Díaz-Laviada I: The cannabinoid R+ methanandamide induces
IL-6 secretion by prostate cancer PC3 cells. J Immunotoxicol.
6:249–256. 2009.
|
|
42
|
Giuliano M, Calvaruso G, Pellerito O,
Portanova P, Carlisi D, Vento R and Tesoriere G: Anandamide-induced
apoptosis in Chang liver cells involves ceramide and JNK/AP-1
pathway. Int J Mol Med. 17:811–819. 2006.PubMed/NCBI
|
|
43
|
Massi P, Vaccani A, Bianchessi S, Costa B,
Macchi P and Parolaro D: The non-psychoactive cannabidiol triggers
caspase activation and oxidative stress in human glioma cells. Cell
Mol Life Sci. 63:2057–2066. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
McKallip RJ, Jia W, Schlomer J, Warren JW,
Nagarkatti PS and Nagarkatti M: Cannabidiol-induced apoptosis in
human leukemia cells: a novel role of cannabidiol in the regulation
of p22phox and Nox4 expression. Mol Pharmacol. 70:897–908. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: a vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Salazar M, Carracedo A, Salanueva IJ,
Hernández-Tiedra S, Egia A, Lorente M, Vázquez P, Torres S, Iovanna
JL, Guzmán M, et al: TRB3 links ER stress to autophagy in
cannabinoid anti-tumoral action. Autophagy. 5:1048–1049. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pellerito O, Calvaruso G, Portanova P, De
Blasio A, Santulli A, Vento R, Tesoriere G and Giuliano M: The
synthetic cannabinoid WIN 55,212-2 sensitizes hepatocellular
carcinoma cells to tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL)-induced apoptosis by activating p8/CCAAT/enhancer
binding protein homologous protein (CHOP)/death receptor 5 (DR5)
axis. Mol Pharmacol. 77:854–863. 2010.
|
|
48
|
Galve-Roperh I, Sanchez C, Cortes ML, del
Pulgar TG, Izquierdo M and Guzman M: Anti-tumoral action of
cannabinoids: involvement of sustained ceramide accumulation and
extracellular signal-regulated kinase activation. Nat Med.
6:313–319. 2000. View
Article : Google Scholar
|
|
49
|
Luca T, Di Benedetto G, Scuderi MR,
Palumbo M, Clementi S, Bernardini R and Cantarella G: The CB1/CB2
receptor agonist WIN-55,212-2 reduces viability of human Kaposi’s
sarcoma cells in vitro. Eur J Pharmacol. 616:16–21. 2009.PubMed/NCBI
|
|
50
|
Park JM, Xian XS, Choi MG, Park H, Cho YK,
Lee IS, Kim SW and Chung IS: Antiproliferative mechanism of a
cannabinoid agonist by cell cycle arrest in human gastric cancer
cells. J Cell Biochem. 112:1192–1205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Piñeiro R, Maffucci T and Falasca M: The
putative cannabinoid receptor GPR55 defines a novel autocrine loop
in cancer cell proliferation. Oncogene. 30:142–152. 2011.PubMed/NCBI
|
|
52
|
Hers I, Vincent EE and Tavaré JM: Akt
signalling in health and disease. Cell Signal. 23:1515–1527. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cudaback E, Marrs W, Moeller T and Stella
N: The expression level of CB1 and CB2 receptors determines their
efficacy at inducing apoptosis in astrocytomas. PLoS One.
5:e87022010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Salazar M, Carracedo A, Salanueva IJ,
Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C,
Torres S, García S, et al: Cannabinoid action induces
autophagy-mediated cell death through stimulation of ER stress in
human glioma cells. J Clin Invest. 119:1359–1372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Torres S, Lorente M, Rodríguez-Fornés F,
Hernández-Tiedra S, Salazar M, García-Taboada E, Barcia J, Guzmán M
and Velasco G: A combined preclinical therapy of cannabinoids and
temozolomide against glioma. Mol Cancer Ther. 10:90–103. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Vara D, Salazar M, Olea-Herrero N, Guzmán
M, Velasco G and Díaz-Laviada I: Anti-tumoral action of
cannabinoids on hepatocellular carcinoma: role of AMPK-dependent
activation of autophagy. Cell Death Differ. 18:1099–1111. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shrivastava A, Kuzontkoski PM, Groopman JE
and Prasad A: Cannabidiol induces programmed cell death in breast
cancer cells by coordinating the cross-talk between apoptosis and
autophagy. Mol Cancer Ther. 10:1161–1172. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wasik AM, Almestrand S, Wang X, Hultenby
K, Dackland ÅL, Andersson P, Kimby E, Christensson B and Sander B:
WIN55,212-2 induces cytoplasmic vacuolation in apoptosis-resistant
MCL cells. Cell Death Dis. 2:e2252011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Donadelli M, Dando I, Zaniboni T, Costanzo
C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M,
Abbruzzese A, et al: Gemcitabine/cannabinoid combination triggers
autophagy in pancreatic cancer cells through a ROS-mediated
mechanism. Cell Death Dis. 2:e1522011. View Article : Google Scholar
|
|
60
|
Gustafsson SB, Lindgren T, Jonsson M and
Jacobsson SO: Cannabinoid receptor-independent cytotoxic effects of
cannabinoids in human colorectal carcinoma cells: synergism with
5-fluorouracil. Cancer Chemother Pharmacol. 63:691–701. 2009.
View Article : Google Scholar
|