Contents
Introduction
Epidemiology
Natural history
Pathogenetic mechanism(s), genetic and epigenetic
factors
Clinical features
Diagnosis, screening and surveillance
Treatment
Introduction
Esophageal cancer (EC) is an important worldwide
health problem because of its poor prognosis and a relatively high
incidence in some parts of the world. Advances in surgical
techniques, chemotherapy and radiotherapy have not substantially
modified its prognosis over the last 25 years. The large majority
of esophageal tumors are accounted for by squamous cell carcinomas
(SCCs: 60–70%) or adenocarcinomas (ACs: 20–30%), whereas melanomas,
leiomyosarcomas, carcinoids and lymphomas are rarely diagnosed
(1).
SCC occurs most often in patients with histories of
tobacco consumption or ethanol intake. Additional risk factors
include prior caustic ingestion, chronic achalasia, and
non-epidermolytic palmo-plantar keratoderma (tylosis), a rare
autosomal dominant disorder associated with genetic abnormalities
at chromosome 17q25, hyperkeratosis of the palms and soles, and
thickening of oral mucosa. Plummer-Vinson syndrome, characterized
by dysphagia, iron-deficiency anemia and esophageal webs, is
another risk condition for SCC. Previous radiotherapy to the
mediastinum, for example in patients with breast cancer and
lymphoma, is also linked to the development of EC, usually 10 or
more years after exposure to radiation (1).
Esophageal AC, on the other hand, can complicate
longstanding acid reflux, and the main condition predisposing to
its onset is Barrett’s esophagus (BE), an acquired disorder whose
prevalence is rapidly increasing worldwide. The aim of this review
is to provide an update of the epidemiology, clinical features,
pathogenetic mechanisms and new diagnostic and therapeutic
approaches to BE and EC.
Epidemiology
EC is the eighth most common malignant tumor
worldwide. In 2010 an estimated 16,640 new cases and 14,500 deaths
due to EC occurred in the United States (2). It is associated with a 5-year
survival rate of 15 to 20%. The lifetime risk of developing this
cancer is 0.8% for men and 0.3% for women. The risk increases with
age and the mean age at diagnosis is 67 years. Although its
incidence is largely variable among different geographical areas,
it is endemic in many parts of the world, particularly in Asia,
Southern and Eastern Africa (3).
The area with the highest reported incidence of EC is the so-called
Asian ‘esophageal cancer belt’, which stretches from eastern Turkey
through north-eastern Iran, northern Afghanistan and southern
Russia to northern China (4).
Approximately 75% of all ACs are localized in the
distal tract of esophagus, whereas SCCs are usually distributed
between the middle and lower third. SCC is more common in the
endemic areas of the world. On the contrary, AC is more frequent in
regions such as North America and Western Europe. Although SCC is
the most common histotype, over the past three decades a slight
decline in its prevalence and, conversely, a dramatic increase in
the prevalence of AC have been recorded, especially in the United
States, United Kingdom and Western Europe (4,5). Any
factor that causes chronic inflammation of the esophageal mucosa,
especially alcohol intake in combination with smoking, may increase
the risk of esophageal SCC (2).
Patients with BE have a 30 to 40 times higher risk
of developing EC. In the United States, approximately 1.5-2 million
people are affected with BE, and its prevalence in people without
symptoms of gastro-esophageal reflux (GER) is about 0.4–6%
(6). Additional information has
been provided by the Kalixanda and SILC studies (7,8) that
involved about 1,000 volunteers, a representative sample of local
residents who underwent endoscopy. The Kalixanda study, performed
in Northern Sweden, showed that 10.3% of them had columnar-lined
esophagus on endoscopic examination, and 1.6% had histologically
confirmed BE. In the SILC study, performed in Shanghai,
endoscopically suspected BE was present in 1.9% of the subjects
examined, 26% of whom having an extent of metaplasia ≥3 cm.
The risk of malignant progression has been examined
in over 8,500 patients with BE using the Northern Ireland BE
Register and followed-up for a mean of 7 years (9). Incidence of EC or gastric cardia
cancer or high-grade dysplasia combined was found to be 0.22% per
year. In patients with specialized intestinal metaplasia (SIM), the
combined incidence was 0.38% per year. A statistically significant
elevation of cancer risk was observed in patients with vs. those
without SIM at index biopsy (0.30% per year vs. 0.07% per year), in
men compared with women (0.28% per year vs. 0.13% per year) and in
patients with low-grade dysplasia compared with those lacking
dysplasia (1.4% per year vs. 0.17% per year). Moreover, in a
nationwide, population-based, cohort study (10) that included all patients with BE
collected in Denmark in the period 1992 through 2009, the relative
risk of AC among patients with BE, compared with the risk in the
general population, was 11.3 and the annual risk of esophageal AC
was 0.12%. Low-grade dysplasia on the index endoscopy was
associated with an incidence rate for AC of 5.1 cases per 1,000
person-years, whereas the incidence rate for patients without
dysplasia was 1.0 case per 1,000 person-years. These two large
population-based studies indicate that BE remains a strong risk
factor for esophageal AC, but the absolute annual risk is
remarkably lower than the previously assumed risk of 0.5% (11) and suggest that current surveillance
guidelines should be revised (9,10,12).
A major risk factor for BE is gastro-esophageal
reflux disease (GERD), whose prevalence, defined as at least weekly
heartburn and/or acid regurgitation, is estimated to be 10–20% in
the Western world (13). However,
only 10–15% of patients with chronic GERD develop BE, indicating
that additional genetic and/or environmental factors are likely to
be involved. Other risk factors associated with the development of
esophageal AC include advanced age, male gender, white race and
elevated body mass index. GERD is associated with obesity, that
increases intra-abdominal pressure and may favor reflux (14). Notably, an inverse association has
been described between the presence of BE and H. pylori
infection, probably linked to the reduction in acid reflux
associated with gastric atrophy (15).
Natural history
The metaplastic transformation of the normal
squamous epithelium lining the esophageal mucosa into a
differentiated epithelium with crypt architecture that resembles
the intestinal columnar epithelium was first described by Norman
Barrett in 1950 (16), correlated
with GERD in 1953 (17) and linked
to esophageal AC in 1975 (18). On
the basis of meta-analysis assessment, the incidence of esophageal
AC among individuals with BE has been reported to be about 0.5% per
patient-year (11) although, as
already mentioned, a much lower absolute annual risk (0.12%) has
been calculated in a recent cohort study (10). Esophageal AC is an uncommon cause
of death in people with BE and, therefore, the majority of patients
affected with this last condition die with their disease, not
because of it.
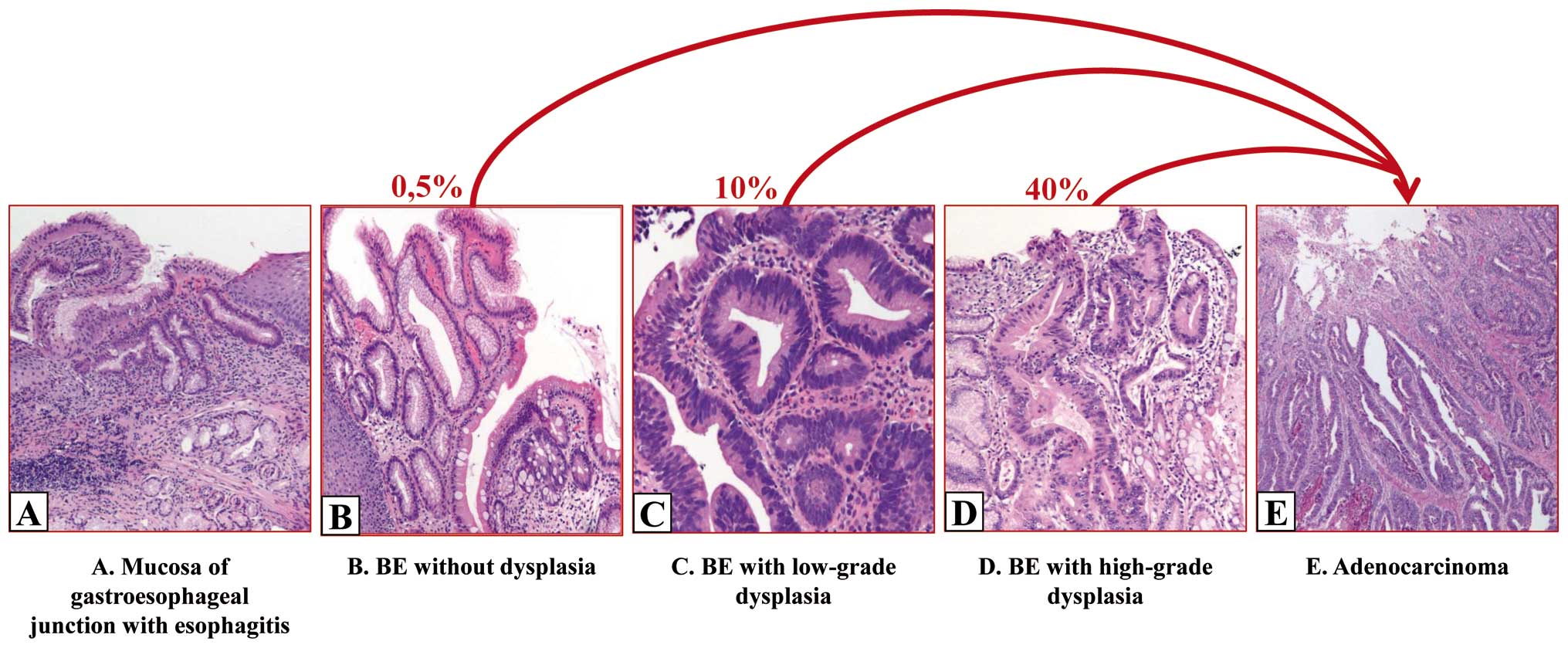
Neoplastic transformation of BE is a stepwise
process that includes non-dysplastic disease, low-grade dysplasia,
high-grade dysplasia and AC, although patients with BE under
endoscopic surveillance often develop cancer without prior biopsy
detection of each of these stages (Fig. 1). In 2000, the World Health
Organization International Agency, according to Vienna
classification system, changed the term ‘dysplasia’ into
‘intraepithelial’ or ‘noninvasive’ neoplasia, a definition that
more appropriately identifies a neoplastic lesion in its early
pre-invasive stage (19,20).
Pathogenetic mechanism(s), genetic and
epigenetic factors
BE is considered an acquired pre-malignant lesion of
the gastrointestinal tract, but there are still major gaps in the
understanding of pathogenetic factors that lead to its development
and trigger progression to dysplasia and esophageal AC (Fig. 2). Since only 10–15% of patients
with GER will eventually develop BE, additional factors capable of
inducing its onset may be inferred. The risk of both BE and
esophageal AC has long been related to body-mass index. However,
the association between body-mass index and BE has been recently
shown to be relatively low, whereas central adiposity does indeed
represent a significant risk factor (21).
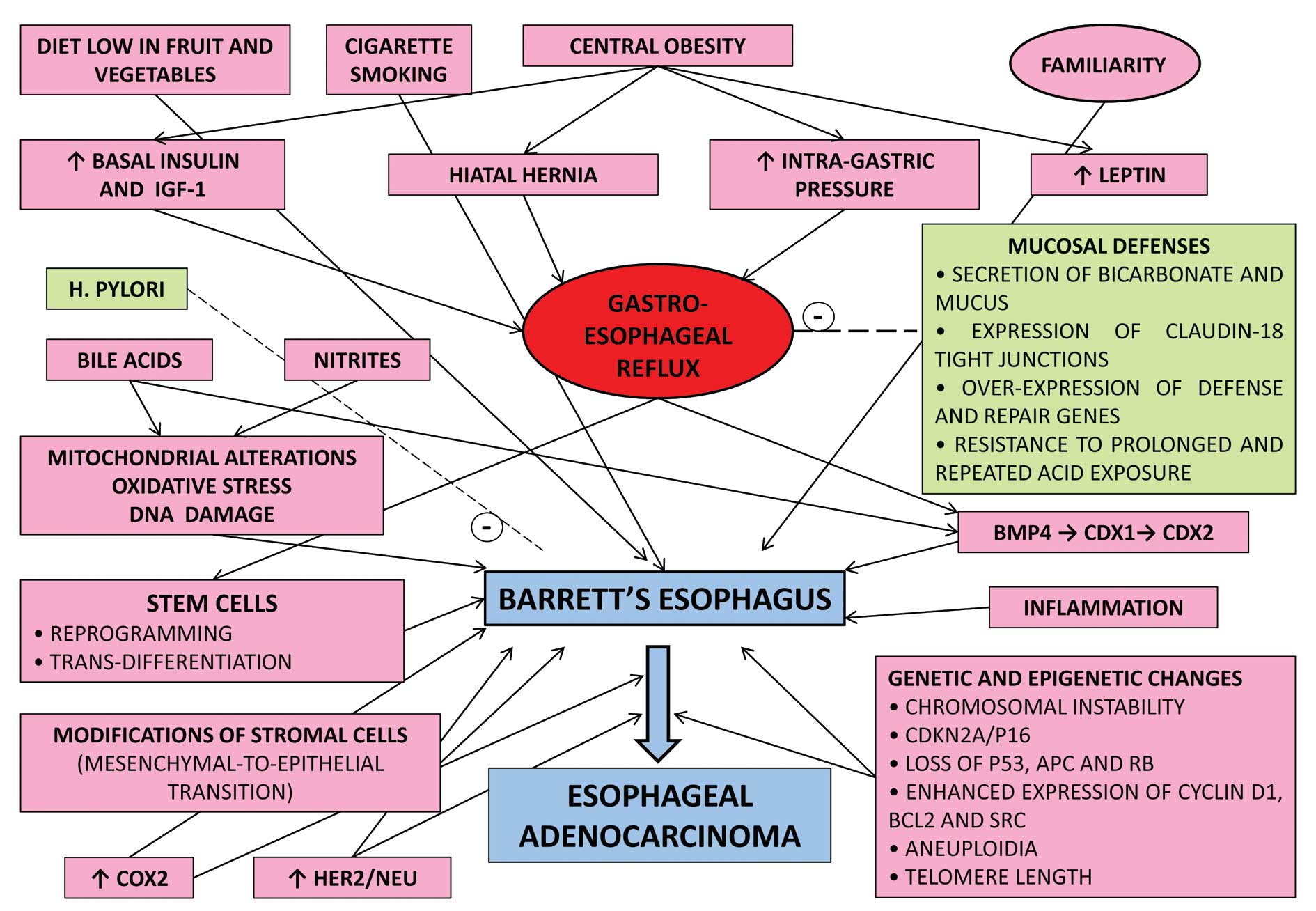 | Figure 2Hypothetical representation of risk
factors and pathways for the development of Barrett’s esophagus and
esophageal adenocarcinoma. Gastro-esophageal reflux plays a central
role in the pathogenesis of BE and several factors are thought to
contribute to its occurrence, including central obesity, which
causes hiatal hernia, increased intragastric pressure, high levels
of insulin and IGF-1 and leptin elevation. Additional risk factors
are diet low in fruit and vegetables, cigarette smoking, elevated
levels of nitrites and bile acids. Familiarity may be an important
predisposing factor to the development of BE and eventually
oesophageal adenocarcinoma. Genetic and epigenetic studies have
shown modifications of intestinal-specific transcription factors
(CDX1 and CDX2) and protein (BMP4), that regulate the development
and differentiation of the intestinal columnar epithelium.
Overexpression of COX2 and HER2/neu have also been claimed to lead
to the development of BE. Reprogramming and/or trans-differentation
of stem cells situated in the basal layer of the normal squamous
epithelium, as well as modifications of stromal cells characterized
by mesenchimal-to-epithelial transition, are presently the object
of intense investigation for their presumed role in the
pathogenesis of BE. In contrast to the mentioned enhancing factors,
H. pylori infection and gastro-oesophageal mucosal defenses
(dotted arrows) exert a protective role against the formation of
BE. |
Patients with central obesity are more predisposed
to hiatal hernia and present an increased intra-gastric pressure
that enhances reflux. In addition, they usually have higher basal
insulin and insulin-like-growth factor-1 (IGF-1) levels, which
promote cell proliferation and determine cell differentiation.
Three IGF-1 gene polymorphisms have been shown to be associated
with esophageal AC or its precursors, and can be considered as risk
markers (22). Moreover, these
patients exhibit higher serum levels of leptin, a hormone secreted
by visceral fat that possibly promotes carcinogenesis by mitogenic
and angiogenic means (23). On the
other hand, in a cross-sectional study increased levels of low
molecular weight adiponectin were associated with a decreased risk
of BE among patients with GERD (24).
Esophageal mucosal injury from acid reflux is
considered a prerequisite for the development of BE although, as
already stated, additional factors besides acid reflux are likely
responsible for the development of intestinal metaplasia. The
distal esophagus is chronically exposed to acid reflux even in
healthy volunteers, with no evidence of esophagitis or metaplasia
(25). Whether the esophageal
mucosa in patients with BE has a higher permeability to physiologic
amounts of acid is still unclear. In an experimental study carried
out in mice (26), acute stress
was found to increase esophageal mucosa permeability by itself.
Thus, there is a connection between stress and exposure of the
esophageal mucosa to acid pepsin, leading to increased permeability
and dilated intercellular spaces. Probably, intestinal metaplasia
develops as a protective mechanism against chronic acid reflux at
the molecular level. Several mucosal defenses have been identified,
including the secretion of bicarbonate and mucus, expression of
claudin-18 tight junctions, over-expression of defense and repair
genes, and resistance to prolonged and repeated acid exposure
(27).
Elevated concentrations of nitric oxide, that are
potentially mutagenic, are detected at the gastro-esophageal
junction in this acidic microenvironment. Nitrites are present in
the saliva and derive from reduction of dietary nitrates effected
by oral bacteria. Nitrites are reduced into nitric oxide by gastric
juice with higher concentrations in correspondence of the
gastro-esophageal junction and gastric cardias, thus inducing a
potentially increased risk of metaplasia and carcinogenesis. Nitric
oxide has also been shown to induce DNA double-strand breaks in
primary BE cells (28). In
addition, different bile acids have been identified in the GER,
including glycocholic acid, taurocholic acid, glycodeoxycholic acid
and glycochenodeoxycholic acid. Bile acids can cause injury to the
esophageal epithelium and lead to the development of metaplasia by
inducing mitochondrial alterations, oxidative stress or DNA damage
(29).
It is not clearly established whether BE is a
hereditary condition. No single causative gene has been identified,
although the condition is more prevalent in first-degree relatives
of patients with BE. Familiarity can be detected in about 7% of
patients in whom BE and AC have been diagnosed, a percentage which
is higher than that reported in general population surveys
(30,31). Since the heritability is weak and
does not correspond to any commonly recognized pattern, the genetic
condition underlying BE is probably a polygenic trait, rather than
a single gene mutation. Studies assessing gene expression in BE
compared with squamous tissue suggest that different cellular
pathways are activated in metaplasia, but the cellular origin of
the columnar cells of BE is not yet clear (11).
Formerly Barrett’s metaplasia was considered the
result of migration of gastric columnar cells to the
gastro-esophageal junction (32).
It is now widely accepted that columnar cells arise within the
esophagus, possibly as the result of a modification in the stem
cells responsible for the constant replenishing of the esophageal
lining epithelium, such that they are reprogrammed to produce
columnar, rather than squamous cells. Stem cells of the esophageal
epithelium possibly home in the intra-papillary regions of the
basal layer (33), or else reside
in the submucosal esophageal glands (34), connected to the surface by a
cuboidal cell-lined duct that penetrates the epithelium and opens
into the esophageal lumen. It seems reasonable to hypothesize that
pluripotent stem cells located distally within the duct lining
become exposed following erosive esophagitis consequent to chronic
reflux and promote the differentiation into intestinal-type
columnar cells that migrate out to repopulate the injured
epithelium. Bone marrow-derived stem cells have also been reported
to contribute to metaplasia in a rat model of BE (35).
Alternatively, rather than an abnormality of stem
cells, the acidic environment determined by chronic GER may induce
cellular trans-differentiation through an epigenetic effect on
post-mitotic cells. During development, the esophagus is initially
lined by a columnar-type epithelium, that is replaced by the mature
squamous epithelium during late embryogenesis through
trans-differentiation (36). This
suggests that columnar cells which characterize Barrett’s
metaplasia may result from a change in the developmental program.
Another possibility is that Barrett’s metaplasia arises indirectly
as a consequence of mutational and/or environmental modifications
in the stromal cells (eg. myofibroblasts, inflammatory cells, etc.)
of the submucosa. Cytokines and other regulatory signals from the
stromal cells could potentially influence the differentiation and
development of cells within the epithelial layer (37). It has also been suggested that the
columnar epithelium of BE arises directly from stromal cells via a
mesenchymal-to-epithelial transition (38).
The mechanisms directly driving
trans-differentiation likely involve important transcriptional
regulators such as the homeobox genes, a family of DNA-binding
proteins. In a rat model of BE (39), acid reflux induces the sequential
expression of CDX1, followed by CDX2, in esophageal epithelial
cells. CDX1 and CDX2 are intestinal-specific transcription factors
that regulate the development and differentiation of the intestinal
columnar epithelium, and are likely to play a role in the
development of BE (40). Neither
factor is expressed in the normal esophagus and stomach, whereas
both are highly expressed in regions of intestinal metaplasia
(41). Strikingly, transgenic
expression of CDX2 in the stomach leads to the development of
intestinal metaplasia in mice (42), while the loss of CDX2 in intestinal
tissue leads to the formation of stratified squamous epithelium
similar to that found in the esophagus (43). Furthermore, chronic exposure to
acid induces expression of CDX2 in normal mouse esophageal cells.
Additional data indicate that epigenetic changes are partly
responsible for abnormal genetic expression that might determine
the development of BE. For example, demethylation of the promoter
regions of CDX2 might allow the expression of this gene in a
previously quiescent cell line, inducing intestinal-like
differentiation of the progeny cells. Such a mechanism might be a
link between genetic factors and environmental exposures. In animal
models, epithelial exposure to the duodenal content affects the
expression of CDX2 (44).
One of the proteins exclusively expressed in BE
epithelium is bone morphogenetic protein 4 (BMP4). It belongs to
the transforming growth factor-β (TGF-β) family, that seems to be
involved in controlling cellular differentiation, migration and
proliferation. BMP4 and its downstream targets are present in
biopsy specimens from BE mucosa. BMP4 is also present in squamous
epithelium in the area of esophagitis, but not in normal squamous
esophageal mucosa (45). A
possible link between CDX2 and BMP4 leading to BE has been
proposed. Pro-inflammatory factors, such as acid and bile, lead to
upregulation of BMP4 expression in esophageal mesenchyme. In turn,
BMP4 activates stem cells in the basal layer of the esophageal
epithelium, initiating gene transcription, which leads to the
development of columnar epithelium. If CDX2 is activated, the
columnar epithelium will be a specialized columnar cell type,
whereas lack of activation of CDX2 will lead to a non-specialized
columnar cell type (46).
Inflammation, in particular upregulation of IL-6, which activates
the signal transducer and activator of transcription 3 (STAT3)
pathway, is also important for progression of BE. Bile acids
activate the CDX2 promoter via nuclear factor-κB (NF-κB), resulting
in the production of intestinal type mucin in esophageal
keratinocytes (47).
Genomic instability seems to be a fundamental
property of neoplastic progression. Acid and bile in the GER,
either directly or indirectly, induce genetic and/or epigenetic
changes that lead to the onset of BE and its progression to
esophageal AC. Multiple genetic changes are indeed present in BE.
Whole genome studies have demonstrated that the majority of BE
samples show some level of chromosomal instability, including copy
number gains, copy number losses and loss of heterozygosity (LOH).
Genetic abnormalities increase during different stages of
carcinogenesis: from less than 2% of the genome in early stages
Barrett’s metaplasia to over 30% in late stages (48). The most frequent genetic alteration
is loss of the short arm of chromosome 9, including 9p21.3
(CDKN2A/p16). In early stage BE, additional abnormalities have been
detected, such as copy loss on 3p and 16q. Among genetic
alterations usually associated with cancer, loss of p53, APC and RB
and overexpression of cyclin D1, Bcl2 and SRC should be mentioned
(49).
Flow cytometric, cytogenetic, comparative genomic
hybridization (CGH) and other studies have shown that aneuploidy,
LOH, and cell cycle alterations are more frequent when grades of
dysplasia are higher. Aneuploid cell populations are found in
approximately two-thirds of patients with high-grade dysplasia and
in about 90% of those with esophageal AC. Tumor suppressor genes,
like other genes, may be inactivated by mutation, LOH, or by
epigenetic suppression of gene expression by DNA hypermethylation,
which involves the abnormal addition of methyl (CH3) groups to
cytosine bases at particular sites (CpG dinucleotides) in gene
promoter regions (39). The
relative risk of developing esophageal AC at 5 years in those with
baseline 9pLOH and 17pLOH and a DNA content abnormality was 79%,
compared to no case of AC in patients with none of these
abnormalities at baseline (50).
In a longitudinal study, baseline analysis of blood
samples has revealed that a shorter telomere length was associated
with increased risk of progression to esophageal AC (51), and in a case-control study, overall
telomere length, as well as 17p and 12q telomere lengths, but not
11q and 2p telomere lengths, were associated with increased
esophageal AC (52). These results
suggest that chronic systemic inflammation is important for the
development of BE and esophageal AC, independently of smoking,
obesity, and the use of non-steroidal anti-inflammatory drugs
(NSAIDs). Therefore, telomere length may be a useful biomarker to
stratify risk in people with BE (53).
H. pylori infection represents an important
environmental factor that possibly interferes with carcinogenesis
in the esophagus. The infection might decrease intra-gastric
acidity by generating large amounts of ammonia, or cause severe
corpus gastritis with destruction of parietal cells, thereby
reducing acid output, or both. Infection with CagA+
strains of H. pylori is associated with high grades of
gastric inflammation and increased propensity to develop gastric
atrophy and intestinal metaplasia. Particularly in East Asia, but
also in the United States and Europe, H. pylori infection
has protective effect from the development of erosive esophagitis
and even BE. The decreased prevalence of H. pylori infection
in developed countries is temporally associated with an increased
incidence of gastro-esophageal complications, including BE
(54).
There is a tissue overexpression of
cyclo-oxygenase-2 (COX-2) in BE and esophageal AC, but it is not
known at what stage they may act in the esophageal
inflammation-metaplasia-AC sequence. Therefore, the use of aspirin
and NSAIDs is probably associated to a reduced risk of BE
development (55). Other factors
with protective effect are dietary factors, such as high intake of
fibers, fruit and vegetables, and meat (56), and nutrients, including high intake
of vitamin C, beta-carotene and vitamin E (57).
HER-2/neu oncogene is also overexpressed/amplified
in about 15–30% of patients with BE or esopha geal AC. HER2/neu has
a possible role in the early transition from dysplasia to AC and
correlates with a poor prognosis. Thus, it could help to identify
patients at high risk of malignant transformation and could be a
target for treatment of esophageal premalignant and malignant
lesions (58).
Clinical features
BE is by itself an asymptomatic disorder, but its
clinical presentation is associated with GERD in 10–15% of cases.
The Montreal consensus conference (59) defined GERD as ‘a condition which
develops when the reflux of gastric contents causes troublesome
symptoms and/or complications’. The severity of reflux esophagitis
is commonly classified into four grades according to the Los
Angeles Classification System (60), depending on the presence, length
and circumferential extent of clearly visible breakages on the
mucosal surface. Grades A and B can be considered mild and
moderate, respectively; grades C and D are severe and associated
with a much higher risk of developing major complications such as
peptic stricture, deep ulceration and BE.
The patient with BE is typically a middle-aged,
overweight, white man; the average age at diagnosis is 55 years.
The most common symptoms, which result from abnormal reflux of the
acidic gastric content, are heartburn, regurgitation, dyspepsia and
epigastric pain. In the Western world heartburn affects up to 40%
of adults, but approximately 20% of them experience it on a weekly
basis. Heartburn occurs in about 70% of GERD patients and is
considered a good marker for the disease, although many patients
with biopsy-proven BE report no symptoms (61).
In some patients, GERD may induce chest pain that
can mimic angina pectoris. A variety of ear, nose and throat
complications can also be associated with GERD, which primarily
result directly or indirectly from refluxed gastric acid. In
addition, hoarseness, chronic cough, globus, pharyngitis,
sinusitis, vocal chord granuloma, subglottic stenosis, and even
laryngeal cancer are reported with variable frequency. Less
commonly associated conditions include asthma, dental erosions due
to acid in the mouth, and acid aspiration, which can cause
pulmonary damage.
The diagnosis of GERD and BE can be problematic for
at least two reasons. First, patients can complain of any or none
of a diverse range of esophageal and extra-esophageal symptoms.
Second, the severity of symptoms is often unrelated to the severity
of the disease, that correlates more often with frequency and
chronicity (total number of years with reflux symptoms) of GERD
clinical features. The different degrees of dysplasia do not modify
clinical presentation of BE patients; therefore, only endoscopy
permits to identify various stages in the evolution of BE up to
transformation of Barrett’s metaplasia into EC. Consequently, the
prognosis of EC is quite poor, because most patients present with
advanced disease, whose symptoms can be attributed to the direct
effects of the local tumor, regional or distant complications
rather than to GERD clinical features (present in only 21% of EC
patients).
Most patients with EC complain of dysphagia (74%),
and odynophagia (17%) at the time of diagnosis. Weight loss is also
common (57%) and is an independent indicator of a poor prognosis if
there is a loss of more than 10% of body mass. Dyspnea, cough,
hoarseness and pain (retrosternal, back or right upper abdominal)
occur less often but may reflect the presence of extensive,
unresectable disease.
Diagnosis, screening and surveillance
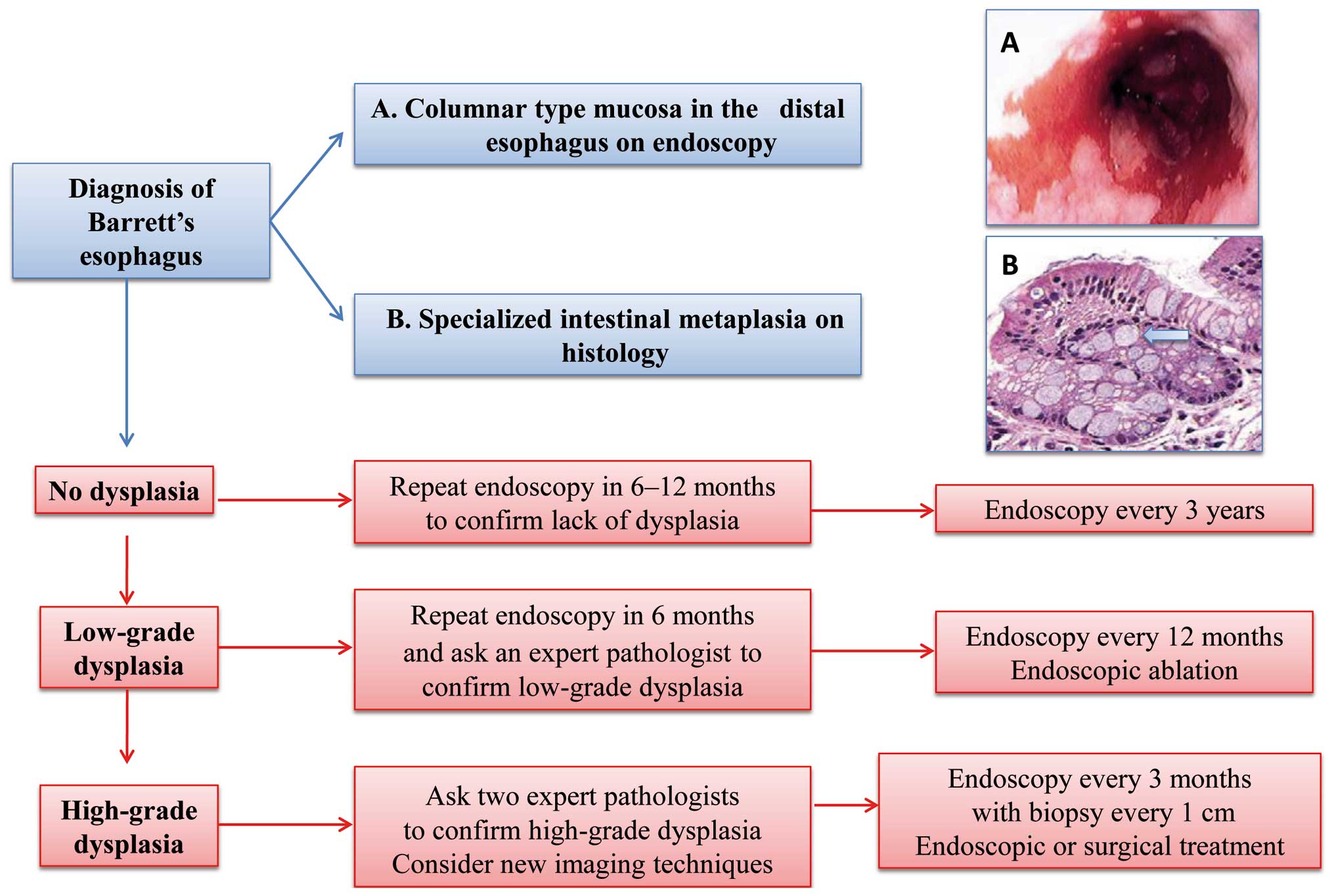
The diagnosis of BE should meet two criteria
(62,63). The first is based on upper
endoscopy, that shows the displacement of the squamo-columnar
junction. While the intersection of the squamous epithelium of the
esophagus and the columnar epithelium of the stomach occurs at the
level of the ‘Z line’, in BE salmon-colored epithelium projects
into the tubular esophagus, such projections appearing as tongues
of tissue or as circumferential involvement of the mucosa, or both.
The second diagnostic criterion is based on the histopathology of
bioptic specimens of the tubular esophagus showing the presence of
SIM, with or without goblet cells. At variance from the position of
the American Society of Gastroenterology (63), but according to the British Society
of Gastroenterology guidelines, the identification of goblet cells
in the metaplastic epithelium is not required for diagnosis
(64). Provided that biopsy
samples derive from endoscopically recognized abnormalities and in
spite of inter-observer variability, histologic assessment of
dysplasia (Fig. 1) remains a
crucial and largely irreplaceable method of diagnosis and
surveillance (62).
If the histological diagnosis is lacking but the
endoscopic picture is suggestive, BE should be more appropriately
defined as ‘esophageal metaplasia’. There are three main
histological subtypes of BE, namely: i) a gastric fundus subtype
with parietal and chief cells; ii) a junctional (cardias) subtype
with mucus-secreting glands; and iii) a distinctive metaplastic
columnar epithelium with intestinal-type goblet cells. These
subtypes occupy different zones of the esophagus, in that the
intestinal-type metaplasia with goblet cells is found most
proximally to the squamous epithelium, followed by the junctional
(cardias) subtype in the middle, and the gastric fundus subtype
most distally. Histological subgroupings of BE are associated with
a different capability to develop malignancy. The fundic subtype
implies a very low risk of developing esophageal AC, whereas the
metaplastic columnar epithelium with intestinal-type goblet cells
and the junctional (cardias) type have a more significant risk of
malignant transformation (62). BE
can also be classified as short-segment disease (<3 cm) or
long-segment disease (≥3 cm) in relation to the length of the
metaplastic epithelium on endoscopic examination. Short segments
are not considered clinically relevant, even if the risk of
developing cancer is increased as compared with the general
population. More recently, BE has been defined as ‘salmon-colored
mucosa of any length in an esophagus harboring goblet cells’
(64,65).
Endoscopy remains a constant requirement for the
routine diagnosis of esophageal columnar metaplasia. The main
objectives of endoscopic assessment are the recognition of BE and
the grading of its extent. Other technical assessments, such as
barium study or CT, are biased by low sensitivity. Despite several
technical improvements, endoscopic evaluation and grading of BE
remain difficult and poorly reliable because of inter-observer
variability. The first systematic study on the endoscopic
recognition of BE was carried out by the International Working
Group for the Classification of Oesophagitis (IWGCO), that led to
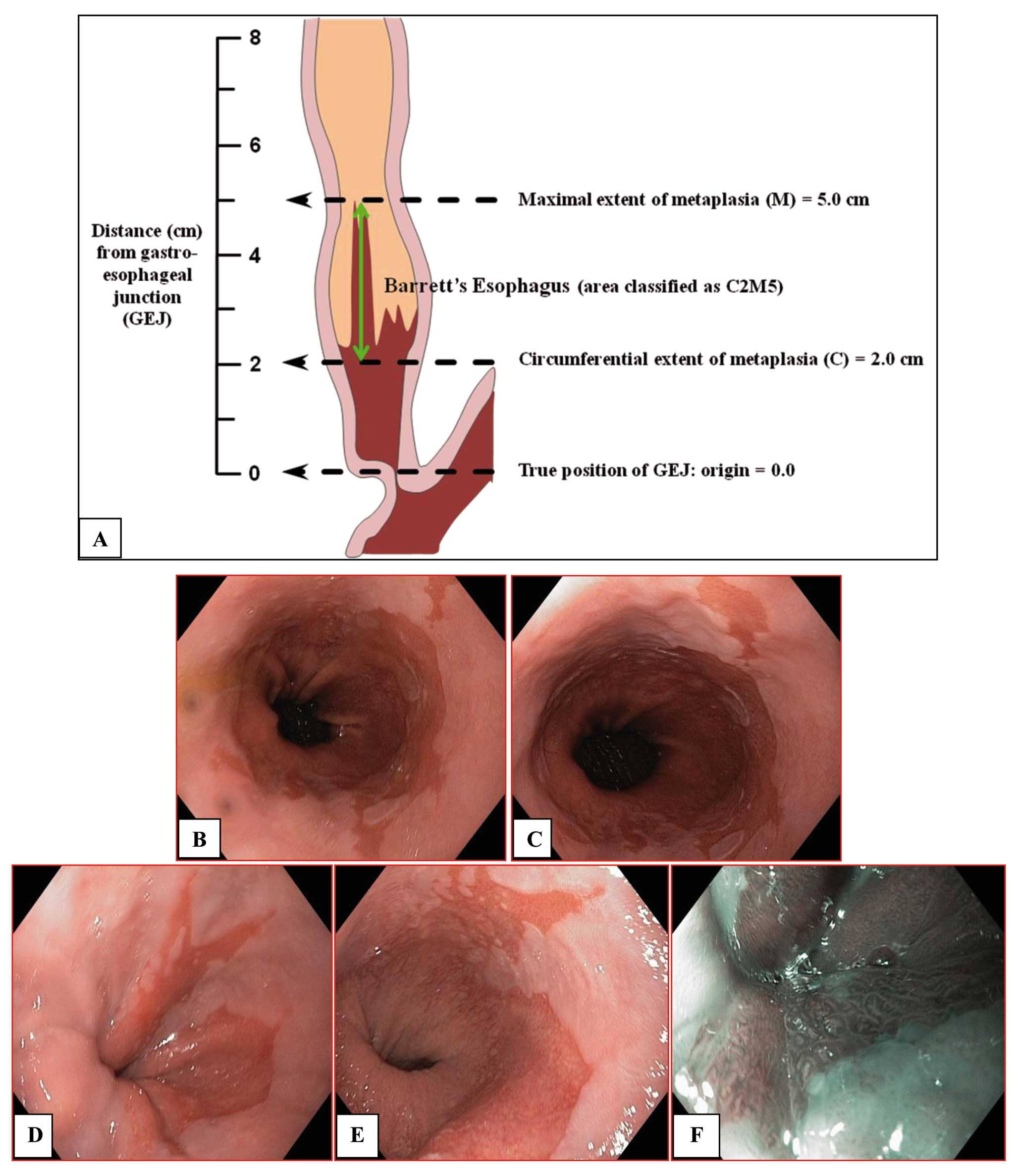
the Prague C & M Criteria (66) illustrated in Fig. 3. In this classification, the first
step for the endoscopic diagnosis of BE is the correct
determination of the position of the anatomical junction between
the stomach and the esophagus. Both the maximal length (M)
(including tongue) of BE, and the length of circumferential Barrett
segment (C) are measured during endoscopy. The diagnosis of BE
should be confirmed by random biopsies throughout the length of the
suspected regions to identify intestinal metaplasia with goblet
cells in samples classically stained with hematoxylin and eosin.
Some studies suggest that eight random biopsies should be performed
to increase the probability of identifying intestinal metaplasia
(11). Histological confirmation
varies with the length of columnar epithelium. This classification
system has a high degree of overall reliability when the
endoscopically visualized segment of BE is longer than 1 cm.
In 2008, the American College of Gastroenterology
Guidelines (Fig. 4) withdrew
recommendations for endoscopic screening of patients with GERD.
There is no direct evidence supporting endoscopic screening for
either BE or AC in individuals with GERD. Recommendations should be
individualized for the single patient, and usually screening is
considered for patients with chronic long-standing GERD (63). Moreover, no randomized controlled
trial has evaluated the efficacy of surveillance, and it is not
clear whether surveillance reduces the mortality from esophageal
AC. Furthermore, several factors limit the expected benefits of
current surveillance strategies, including the low overall
incidence of cancer in patients with BE, the absence of a previous
diagnosis of BE in most patients with esophageal AC, and
difficulties in the diagnosis of dysplasia because of random biopsy
specimens and high variations among pathologists in the
interpretation of biopsy findings.
We have already mentioned that in large
population-based cohort studies cancer incidence has been shown to
be significantly lower (9,10) than 0.5% per year (11) for non-dysplastic BE. The risk of
progression to cancer increases gradually from 1% to 10% per year
in low-grade dysplasia. The majority of patients with low-grade
dysplasia detected on endoscopic surveillance do not have evidence
of dysplasia on the subsequent endoscopic examination (67). The surveillance in patients with BE
without dysplasia is based on two esophageal examinations with
biopsy within 1 year and follow-up with endoscopy every 3 years. In
patients with BE with low-grade dysplasia, surveillance consists of
two esophageal examinations with biopsy within 1 year and follow-up
with endoscopy every year (63).
The risk of developing esophageal AC rises up to 40%
per year among patients with high-grade dysplasia, with an
estimated incidence of 6.6 cases per 100 patients/years (5). In these patients, if mucosal
irregularities are detected, a new esophageal examination with
biopsy within 3 months is mandatory to exclude the possibility of
cancer. Should the pathologist confirm such mucosal irregularity,
then strict follow-up and possible endoscopic mucosal resection
would be requested. In case of flat mucosa, endoscopic surveillance
is intensive (every 3 months) and esophagectomy or ablation should
be considered (63,64).
Enhanced imaging techniques have been found to
improve the efficiency and accuracy of endoscopic surveillance.
Although most of these techniques are not directly comparable with
standard endoscopy, preliminary results with the use of narrow-band
imaging and chromo-endoscopy indicate that they have a high
sensitivity (85 to 92%) in the diagnosis of neoplasia in patients
with BE (68). Narrow-band imaging
improves contrast by narrowing the band of white light, filtering
it into two major colors (blue and green) which are then better
absorbed by blood vessels in the mucosa and submucosa. Combined
with high-resolution endoscopy, narrow-band imaging produces
detailed images of the mucosal and vascular surface patterns within
the BE segment, and identifies characteristic patterns for
nondysplastic intestinal metaplasia, high-grade dysplasia, and
early cancer (11).
Chromo-endoscopy is a simple technique involving the application of
chemical agents to improve the visualization of mucosal surfaces
either by selective uptake (vital staining with methylene blue or
Lugol’s solution) or enhancement of mucosal surface pattern
(contrast staining with indigo carmine and acetic acid). Methylene
blue is the most common of these stains that colors non-dysplastic
SIM in blue, whereas it does not bind to high-grade dysplastic or
neoplastic mucosa (69).
Treatment
As already mentioned, patients with non-dysplastic
disease or low-grade dysplasia evolve to high-grade dysplasia or
cancer in less than 10% of cases (11,18),
and should therefore undergo periodic endoscopic surveillance
without therapy (Fig. 4),
according to internationally accepted recommendations (63,64).
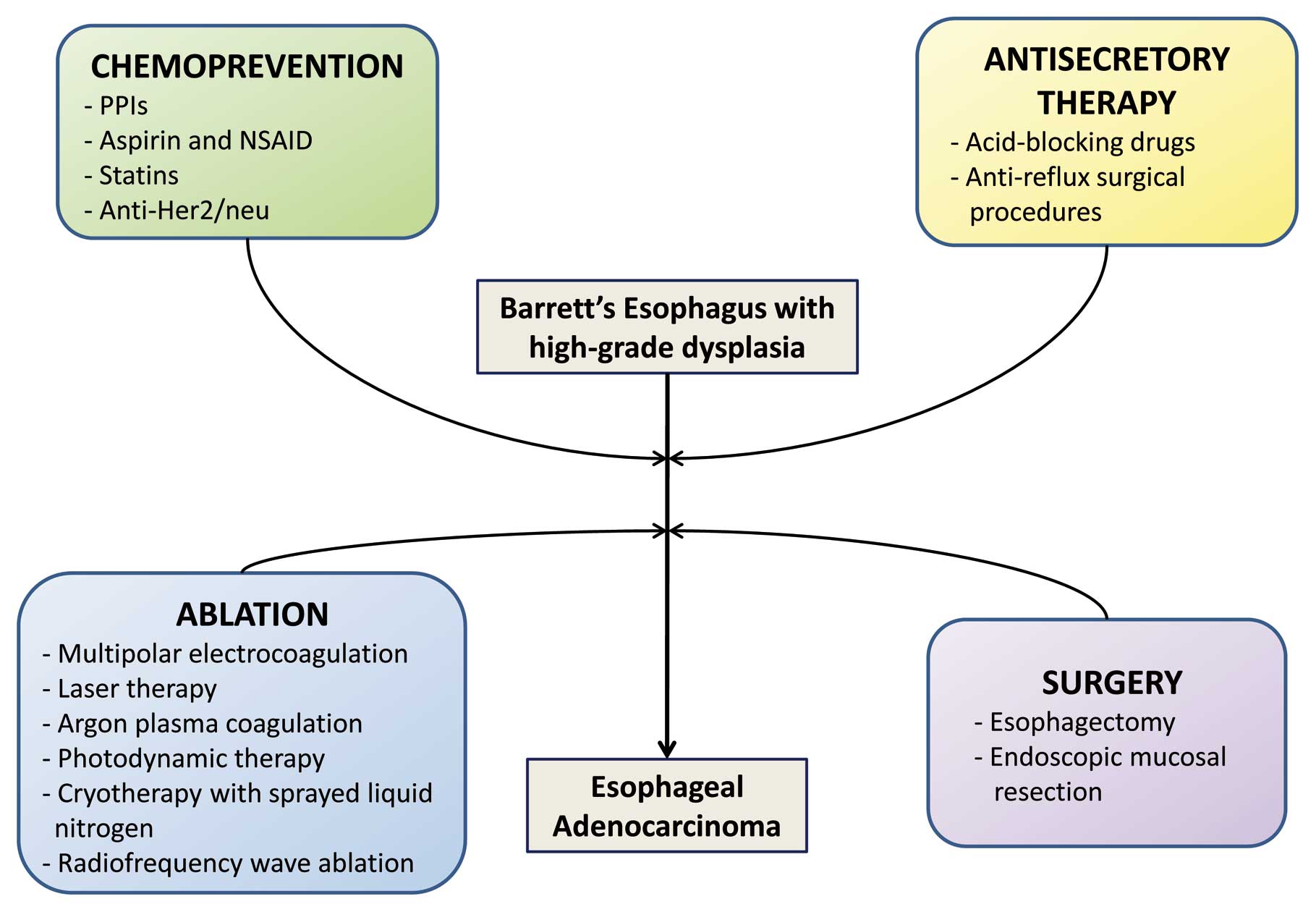
On the contrary, patients with high-grade dysplasia,
and hence at high risk of malignant transformation, obviously
require a more aggressive approach. Esophagectomy should be the
standard of care for them (Fig.
5). This approach is based on the results of several studies in
which neoplasia was demonstrated in approximately 40% of resected
samples (70). The mortality and
morbidity rates of esophagectomy are 3–5% and 20–50%, respectively,
even when performed in largely attended expert centers (71). An alternative surgical approach is
endoscopic mucosal resection, that allows for a definite histologic
diagnosis and is also potentially curative. This procedure implies
the excision of a piece of mucosa of approximately 1.5–2 cm in
diameter (72) and is recommended
for lesions confined to the mucosa and to the upper third of the
submucosa. It can be used for the definitive treatment of some
intra-mucosal cancers arising in BE, or circumferentially by using
multiple excisions to remove the whole metaplastic tract.
Circumferential resection is followed by esophageal stricture
formation in 10–40% of patients. The risk increases in relation to
the number of mucosal resections undertaken. Therefore, endoscopic
mucosal excision of the full segment of BE is usually limited to
shorter lesions (11). Moreover,
it is associated with the risk of neoplastic recurrence in up to
20% of cases during follow-up (73).
The introduction of new endoscopic treatments
characterized by a lower incidence of complications, including the
risk of lymphatic or hematogenous dissemination, has reduced the
indications of surgery to patients with high grade dysplasia or
early AC (74,75). The endoscopic procedures, either by
resection or by ablation of the inner lining of the esophagus,
usually result in regenerated squamous epithelium. Although the
malignant potential of regenerating epithelium has not been fully
assessed, this reversion seems to result in a substantially
decreased risk of esophageal AC. Endoscopic ablation techniques
include photo-dynamic therapy, multipolar electrocoagulation, laser
therapy, argon plasma coagulation, cryotherapy with sprayed liquid
nitrogen and radiofrequency wave ablation (11) (Fig.
5). Because comparative studies among these techniques are not
available so far, their choice is mostly empirical and largely
dependent on the patient’s characteristics and preferences, as well
as on the expertise of the operator.
Photodynamic therapy has been extensively
investigated. It involves the administration of a photosensitizer
substrate which accumulates in the tumor tissue and is subsequently
activated by a laser impulse endoscopically applied to the
malignant site. This results in the formation of free oxygen
radicals in the tumor tissue, leading to ischemic necrosis of the
tumor cells. The strongest evidence for the effectiveness of
photodynamic therapy derives from a prospective, randomized study
in which, after a five years’ follow-up, a significant difference
was shown between a group of patients treated with photodynamic
therapy plus omeprazole vs. a group treated with omeprazole alone.
In this study, the percentage of ablation in high grade dysplasia
was 77% vs. 39% (p=0.004), whereas the recurrence of neoplasia was
15% vs. 29% (p=0.027) (75). The
occurrence of esophageal stenosis is a relatively frequent adverse
event of photodynamic therapy (27–34% of cases), which typically
develops within one month (76).
Radiofrequency ablation is also used to treat BE. It
consists of a high-power radiofrequency energy generator, sizing
balloon and ablation catheters. In patients with dysplastic BE,
radio-frequency ablation is associated with a high rate of complete
eradication of both dysplasia and intestinal metaplasia, and a
reduced risk of disease progression (77). When circumferential radiofrequency
ablation was applied to patients with BE without dysplasia, its
complete elimination occurred in 70% of cases assessed after 1 year
follow-up and there were no subsequent strictures or buried glands
(78,79).
Since acid reflux is thought to play an important
role in the development of esophageal intestinal metaplasia, most
treatment strategies focus on acid-blocking drugs or anti-reflux
surgical procedures, though no prospective trials are available
showing that controlling reflux symptoms and even esophagitis can
indeed result in the prevention of esophageal AC. The association
between the use of proton pump inhibitors (PPIs) and the risk of
esophageal AC has been examined in observational studies, but
controversial results have been achieved (80,81).
For example, a retrospective cohort study evaluated the use of PPIs
in 344 individuals without any dysplasia at baseline endoscopy. No
association with the development of dysplasia was reported; on the
contrary, a statistically significant reduction in the risk of
high-grade dysplasia and/or esophageal AC was observed (82).
Several studies have considered aspirin and NSAID as
chemopreventive drugs. Their use is associated with a 50% reduced
risk of developing esophageal AC (83). A large European study (84) aimed at defining the role of
low-dose aspirin and PPIs, alone or in combination, seems to
delineate a potential role for these drugs and their dosing in the
chemoprevention of esophageal AC. On the basis of emerging evidence
showing increased COX2 levels in BE (55), COX2 inhibitors have also been
reported to decrease the incidence of esophageal AC in BE, although
conflicting data have been published on their role as
chemopreventive drugs and on the optimum dose of these drugs
(85). Statins are not
significantly associated with the risk of neoplasia in patients
with BE (82) and, in addition to
NSAIDs and PPIs, they can even exert (86) a chemopreventive effect in
esophageal AC.
The complexity of the process involved in neoplastic
progression suggests that no single therapeutic measure is likely
to be sufficient in clinical practice. The best strategy of care
for patients with BE needs further elucidation. Currently, a risk
stratification is needed. Given the overall low risk of neoplastic
progression of BE, there is an obvious interest to search for
biomarkers that might identify people at particular cancer risk.
Among biomarkers predictive of neoplastic progression,
abnormalities in the tumor-suppressor genes CDKN2A (which encodes
the cyclin-dependent kinase inhibitor p16INK4a) and TP53 (which
encodes tumor protein p53), and the presence of tetraploidy or
aneuploidy in epithelial cells should be mentioned (87). Tailored risk stratification, based
on interactions among environmental and genetic factors, is
essential to deliver personalized care to patients with high risk
of developing esophageal AC.
Abbreviations:
|
AC
|
adenocarcinoma;
|
|
BE
|
Barrett’s esophagus;
|
|
BMP4
|
bone morphogenetic protein 4;
|
|
CGH
|
comparative genomic hybridization;
|
|
COX-2
|
cyclo-oxygenase-2;
|
|
EC
|
esophageal cancer;
|
|
GER
|
gastro-esophageal reflux;
|
|
GERD
|
gastro-esophageal reflux disease;
|
|
H. pylori
|
Helicobacter pylori;
|
|
IGF-1
|
insulin-like-growth factor-1;
|
|
LOH
|
loss of heterozygosity;
|
|
NBI
|
Narrow Band Imaging;
|
|
NF-κB
|
nuclear factor-κB;
|
|
NSAIDs
|
non-steroidal anti-inflammatory
drugs;
|
|
PPIs
|
proton-pump inhibitors;
|
|
SCC
|
squamous cell carcinoma;
|
|
SIM
|
specialized intestinal metaplasia;
|
|
STAT3
|
signal transducer and activator of
transcription 3;
|
|
TGF-β
|
transforming growth factor β
|
Acknowledgements
This study was supported in part by
grants from the Italian Association for Cancer Research (AIRC,
Milan, Italy), the Italian Foundation ‘Cassa di Risparmio di
Puglia’ (Bari, Italy), and the strategic project ‘BIOTECNOTER’ of
the Apulia Region (Bari, Italy).
References
|
1
|
Enzinger P and Mayer R: Esophageal cancer.
N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ajani JA, Barthel JS, Bentrem DJ, et al:
Esophageal and esophago-gastric junction cancers. J Natl Compr Canc
Netw. 9:830–887. 2011.PubMed/NCBI
|
|
3
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
4
|
Parkin DM: International variation.
Oncogene. 23:6329–6340. 2004. View Article : Google Scholar
|
|
5
|
Pohl H and Welch HG: The role of
overdiagnosis and reclassification in the marked increase of
esophageal adenocarcinoma incidence. J Natl Cancer Inst.
97:142–146. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharma P: Clinical practice. Barrett’s
esophagus. N Engl J Med. 361:2548–2556. 2009.
|
|
7
|
Ronkainen J, Aro P, Storskrubb T, et al:
Prevalence of Barrett’s esophagus in the general population: an
endoscopic study. Gastroenterology. 129:1825–1831. 2005.
|
|
8
|
Zou D, He J, Ma X, et al: Helicobacter
pylori infection and gastritis: the systematic investigation of
gastrointestinal diseases in China (SILC). J Gastroenterol Hepatol.
26:908–915. 2011. View Article : Google Scholar
|
|
9
|
Bhat S, Coleman HG, Yousef F, Johnston BT,
McManus DT, Gavin AT and Murray LJ: Risk of malignant progression
in Barrett’s esophagus patients: results from a large
population-based study. J Natl Cancer Inst. 103:1049–1057.
2011.
|
|
10
|
Hvid-Jensen F, Pedersen L, Drewes AM,
Sørensen HT and Funch-Jensen P: Incidence of adenocarcinoma among
patients with Barrett’s esophagus. N Engl J Med. 365:1375–1383.
2011.
|
|
11
|
Shaheen NJ and Richter JE: Barrett’s
esophagus. Lancet. 373:850–861. 2009.
|
|
12
|
Kahrilas PJ: The problems with
surveillance of Barrett’s esophagus. N Engl J Med. 365:1437–1438.
2011.
|
|
13
|
Garud SS, Keilin S, Cai Q and Willingham
FF: Diagnosis and management of Barrett’s esophagus for the
endoscopist. Ther Adv Gastroenterol. 3:227–238. 2010.
|
|
14
|
Dent J, El-Serag HB, Wallander MA and
Johansson S: Epidemiology of gastro-oesophageal reflux disease: a
systematic review. Gut. 54:710–717. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Whiteman DC, Parmar P, Fahey P, et al:
Association of Helicobacter pylori infection with reduced
risk for esophageal cancer is independent of environmental and
genetic modifiers. Gastroenterol. 139:73–83. 2010.
|
|
16
|
Barrett NR: Chronic peptic ulcer of the
oesophagus and ‘oesophagitis’. Br J Surg. 38:175–182. 1950.
|
|
17
|
Allison PR and Johnstone AS: The
oesophagus lined with gastric mucous membrane. Thorax. 8:87–101.
1953. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naef AP, Savary M and Ozzello L:
Columnar-lined lower esophagus: an acquired lesion with malignant
predisposition. Report on 140 cases of Barrett’s esophagus with 12
adenocarcinomas. J Thorac Cardiovasc Surg. 70:826–835.
1975.PubMed/NCBI
|
|
19
|
Hamilton SR and Aaltonen LA; World Health
Organization of Tumours Classification: Pathology and Genetics of
Tumours of the Digestive System. IARC Press; Lyon: 2000
|
|
20
|
Schlemper RJ, Riddell RH, Kato Y, et al:
The Vienna classification of gastrointestinal epithelial neoplasia.
Gut. 47:251–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lagergren J: Influence of obesity on the
risk of esophageal disorders. Nat Rev Gastroenterol Hepatol.
8:340–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McElholm AR, McKnight AJ, Patterson CC,
Johnston BT, Hardie LJ and Murray LJ: A population-based study of
IGF axis polymorphisms and the esophageal inflammation, metaplasia,
adenocarcinoma sequence. Gastroenterology. 139:204–212. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kendall BJ, Macdonald GA, Hayward NK, et
al: Leptin and the risk of Barrett’s oesophagus. Gut. 57:448–454.
2008.
|
|
24
|
Rubenstein JH, Kao JY, Madanick RD, et al:
Association of adiponectin multimers with Barrett’s oesophagus.
Gut. 58:1583–1589. 2009.PubMed/NCBI
|
|
25
|
Fletcher J, Wirz A, Henry E and McKoll KE:
Studies of acid exposure immediately above the gastro-oesophageal
squamocolumnar junction: evidence of short segment reflux. Gut.
53:168–173. 2004. View Article : Google Scholar
|
|
26
|
Farrè R, De Vos R, Geboes K, et al:
Critical role of stress in increased oesophageal mucosa
permeability and dilated inter-cellular spaces. Gut. 56:1191–1197.
2007.PubMed/NCBI
|
|
27
|
Jovov B, Van Itallie CM, Shaheen NJ,
Carson JL, Gambling TM, Anderson JM and Orlando RC: Claudin-18: a
dominant tight junction protein in Barrett’s esophagus and likely
contributor to its acid resistance. Am J Physiol Gastrointest Liver
Physiol. 293:1106–1113. 2007.PubMed/NCBI
|
|
28
|
Clemons NJ, McColl KE and Fitzgerald RC:
Nitric oxide and acid induce double-strand DNA breaks in Barrett’s
esophagus carcinogenesis via distinct mechanisms. Gastroenterology.
133:1198–1209. 2007.
|
|
29
|
Dvorak K, Payne CM, Chavarria M, et al:
Bile acids in combination with low pH induce oxidative stress and
oxidative DNA damage: relevance to the pathogenesis of Barrett’s
oesophagus. Gut. 56:763–771. 2007.PubMed/NCBI
|
|
30
|
Sun X, Elston R, Barnholtz-Sloan J, et al:
A segregation analysis of Barrett’s esophagus and associated
adenocarcinomas. Cancer Epidemiol Biomarkers Prev. 19:666–674.
2010.
|
|
31
|
Chak A, Ochs-Balcom H, Falk G, et al:
Familiality in Barrett’s esophagus, adenocarcinoma of the
esophagus, and adenocarcinoma of the gastroesophageal junction.
Cancer Epidemiol Biomarkers Prev. 15:1668–1673. 2006.
|
|
32
|
Croagh D, Phillips WA, Redvers R, Thomas
RJ and Kaur P: Identification of candidate murine esophageal stem
cells using a combination of cell kinetic studies and cell surface
markers. Stem Cells. 25:313–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Croagh D, Thomas RJ, Phillips WA and Kaur
P: Esophageal stem cells - a review of their identification and
characterization. Stem Cell Rev. 4:261–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Leedham SJ, Preston SL, McDonald SA, et
al: Individual crypt genetic heterogeneity and the origin of
metaplastic glandular epithelium in human Barrett’s oesophagus.
Gut. 57:1041–1408. 2008.PubMed/NCBI
|
|
35
|
Sarosi G, Brown G, Jaiswal K, et al: Bone
marrow progenitor cells contribute to esophageal regeneration and
metaplasia in a rat model of Barrett’s esophagus. Dis Esophagus.
21:43–50. 2008.PubMed/NCBI
|
|
36
|
Yu WY, Slack JM and Tosh D: Conversion of
columnar to stratified squamous epithelium in the developing mouse
oesophagus. Dev Biol. 284:157–170. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kong J, Crissey MA, Funakoshi S, Kreindler
JL and Lynch JP: Ectopic Cdx2 expression in murine esophagus models
an intermediate stage in the emergence of Barrett’s esophagus. PloS
One. 6:e182802011.PubMed/NCBI
|
|
38
|
Kosinski C, Stange DE, Xu C, et al: Indian
hedgehog regulates intestinal stem cell fate through
epithelial-mesenchymal interactions during development.
Gastroenterology. 139:893–903. 2010. View Article : Google Scholar
|
|
39
|
Kazumori H, Ishihara S and Kinoshita Y:
Roles of caudal-related homeobox gene Cdx1 in oesophageal
epithelial cells in Barrett’s epithelium development. Gut.
58:620–628. 2009.
|
|
40
|
Phillips WA, Lord RV, Nancarrow DJ, Watson
DJ and Whiteman DC: Barrett’s esophagus. J Gastroenterol Hepatol.
26:639–648. 2011.
|
|
41
|
Colleypriest BJ, Farrant JM, Slack JM and
Tosh D: The role of Cdx2 in Barrett’s metaplasia. Biochem Soc
Trans. 38:364–369. 2010.
|
|
42
|
Mutoh H, Sakurai S, Satoh K, Osaka H,
Hakamata Y, Takeuchi T and Sugano K: Cdx1 induced intestinal
metaplasia in the transgenic mouse stomach: comparative study with
Cdx2 transgenic mice. Gut. 53:1416–1423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Barros R, da Costa LT, Pinto-de-Sousa J,
Duluc I, Freund JN, David L and Almeida R: CDX2 autoregulation in
human intestinal metaplasia of the stomach: impact on the stability
of the phenotype. Gut. 60:290–298. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huo X, Zhang HY, Zhang XI, et al: Acid and
bile salt-induced CDX2 expression differs in esophageal squamous
cells from patients with and without Barrett’s esophagus.
Gastroenterology. 139:194–203. 2010.PubMed/NCBI
|
|
45
|
Chen X, Qin R, Liu B, Ma Y, et al:
Multilayered epithelium in a rat model and human Barrett’s
esophagus: similar expression patterns of transcription factors and
differentiation markers. BMC Gastroenterol. 8:1–9. 2008.PubMed/NCBI
|
|
46
|
Badreddine RJ and Wang KK: Barrett
esophagus: an update. Nat Rev Gastroenterol Hepatol. 7:369–378.
2010. View Article : Google Scholar
|
|
47
|
Wang DH, Clemons NJ, Miyashita T, et al:
Aberrant epithelialmesenchymal Hedgehog signaling characterizes
Barrett’s metaplasia. Gastroenterology. 138:1810–1822.
2010.PubMed/NCBI
|
|
48
|
Liu W, Hahn H, Odze RD and Goyal RK:
Metaplastic esophageal columnar epithelium without goblet cells
shows DNA content abnormalities similar to goblet cell-containing
epithelium. Am J Gastroenterol. 104:816–824. 2009. View Article : Google Scholar
|
|
49
|
Chaves P, Crespo M, Ribeiro C, et al:
Chromosomal analysis of Barrett’s cells: demonstration of
instability and detection of the metaplastic lineage involved. Mod
Pathol. 20:788–796. 2007.
|
|
50
|
Li X, Galipeau PC, Sanchez CA, et al:
Single nucleotide polymorphism-based genome-wide chromosome copy
change, loss of heterozygosity, and aneuploidy in Barrett’s
esophagus neoplastic progression. Cancer Prev Res. 1:413–423.
2008.PubMed/NCBI
|
|
51
|
Wu IC, Zhao Y, Zhai R, et al: Association
between polymorphisms in cancer-related genes and early onset of
esophageal adenocarcinoma. Neoplasia. 13:386–392. 2011.PubMed/NCBI
|
|
52
|
Galipeau PC, Li X, Blount PL, et al:
NSAIDs modulate CDKN2A, TP53, and DNA content risk for progression
to esophageal adenocarcinoma. PLoS Med. 4:e672007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xing J, Ajani JA, Chen M, et al:
Constitutive short telomere length of chromosome 17p and 12q but
not 11q and 2p is associated with an increased risk for esophageal
cancer. Cancer Prev Res. 2:459–465. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang C, Yuan Y and Hunt RH:
Helicobacter pylori infection and Barrett’s esophagus: a
systematic review and meta-analysis. Am J Gastroenterol.
104:492–500. 2009. View Article : Google Scholar
|
|
55
|
Anderson LA, Johnston BT, Watson RG, et
al: Nonsteroidal anti-inflammatory drugs and the esophageal
inflammation-metaplasia-adenocarcinoma sequence. Cancer Res.
66:4975–4982. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kubo A, Block G, Quesenberry CP, Buffler P
and Corley DA: Effects of dietary fiber, fats, and meat intakes on
the risk of Barrett’s esophagus. Nutr Cancer. 61:607–616.
2009.PubMed/NCBI
|
|
57
|
Kubo A, Levin TR, Block G, et al: Dietary
antioxidants, fruits, and vegetables and the risk of Barrett’s
esophagus. Am J Gastroenterol. 103:1614–1623. 2008.
|
|
58
|
Rossi E, Grisanti S, Villanacci V, et al:
HER-2 overexpression/amplification in Barrett’s oesophagus predicts
early transition from dysplasia to adenocarcinoma: a
clinico-pathologic study. J Cell Mol Med. 13:3826–3833. 2009.
|
|
59
|
Vakil N, van Zanten SV, Kahrilas P, Dent J
and Jones R: The Montreal definition and classification of
gastroesophageal reflux disease: a global evidence-based consensus.
Am J Gastroenterol. 101:1900–1920. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lundell LR, Dent J, Bennett JR, et al:
Endoscopic assessment of oesophagitis: clinical and functional
correlates and further validation of the Los Angeles
classification. Gut. 45:172–180. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
American Gastroenterological Association;
Spechler SJ, Sharma P, Souza RF, Inadomi JM and Shaheen NJ:
American Gastroenterological Association medical position statement
on the man agement of Barrett’s esophagus. Gastroenterology.
140:1084–1091. 2011.
|
|
62
|
Voltaggio L, Montgomery EA and Lam-Himlin
D: A clinical and histopathologic focus on Barrett esophagus and
Barrett-related dysplasia. Arch Pathol Lab Med. 135:1249–1260.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang KK and Sampliner RE; Practice
Parameters Committee of the American College of Gastroenterology:
Updated guidelines 2008 for the diagnosis, surveillance and therapy
of Barrett’s esophagus. Am J Gastroenterol. 103:788–797.
2008.PubMed/NCBI
|
|
64
|
Playford RJ: New British Society of
Gastroenterology (BSG) guidelines for the diagnosis and management
of Barrett’s oesophagus. Gut. 55:4422006.PubMed/NCBI
|
|
65
|
Sikkema M, Looman CWN, Steyerberg EW, et
al: Predictors for neoplastic progression in patients with
Barrett’s esophagus: a prospective cohort study. Am J
Gastroenterol. 106:1231–1238. 2011.
|
|
66
|
Sharma P, Dent J, Armstrong D, et al: The
development and validation of an endoscopic grading system for
Barrett’s esophagus: the Prague C & M Criteria.
Gastroenterology. 131:1392–1399. 2006.PubMed/NCBI
|
|
67
|
Wani S, Mathur S and Sharma P: How to
manage a Barrett’s esophagus patient with low-grade dysplasia. Clin
Gastroenterol Hepatol. 7:27–32. 2009.
|
|
68
|
Mannath J, Subramanian V, Hawkey CJ and
Raqunath K: Narrow band imaging for characterization of high grade
dysplasia and specialized intestinal metaplasia in Barrett’s
esophagus: a meta-analysis. Endoscopy. 42:351–359. 2010.
|
|
69
|
Wasielica-Berger J, Baniukiewicz A,
Wroblewski E, Chwiesko A and Dabrowski A: Magnification endoscopy
and chromoendoscopy in evaluation of specialized intestinal
metaplasia in Barrett’s esophagus. Dig Dis Sci. 56:1987–1995.
2011.
|
|
70
|
Prasad GA, Wu TT, Wigle DA, et al:
Endoscopic and surgical treatment of mucosal (T1a) esophageal
adenocarcinoma in Barrett’s esophagus. Gastroenterology.
137:815–823. 2009.
|
|
71
|
Yachimski P, Nishioka NS, Richards E and
Hur C: Treatment of Barrett’s esophagus with high-grade dysplasia
or cancer: predictor of surgical versus endoscopic therapy. Clin
Gastroenterol Hepatol. 6:1206–1211. 2008.
|
|
72
|
Fleischer DE, Overholt BF, Sharma P, et
al: Endoscopic ablation of Barrett’s esophagus: a multicenter study
with 2.5-year follow-up. Gastrointest Endosc. 68:867–876. 2008.
|
|
73
|
Pech O, Behrens A, May A, et al: Long-term
results and risk factor analysis for recurrence after curative
endoscopic therapy in 349 patients with high-grade intraepithelial
neoplasia and mucosal adenocarcinoma in Barrett’s oesophagus. Gut.
57:1200–1206. 2008.PubMed/NCBI
|
|
74
|
Gaddam S and Sharma P: Advances in
endoscopic diagnosis and treatment of Barrett’s esophagus. J Dig
Dis. 11:323–333. 2010.
|
|
75
|
Nealis TB, Washington K and Keswani RN:
Endoscopic therapy of esophageal premalignancy and early
malignancy. J Natl Compr Cancer Netw. 9:890–899. 2011.PubMed/NCBI
|
|
76
|
Yachimski P, Puricelli WP and Nishioka NS:
Patient predictors of esophageal stricture development after
photodynamic therapy. Clin Gastroenterol Hepatol. 6:302–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ulsiewicz WJ and Shaheen NJ: The role of
radiofrequency ablation in the management of Barrett’s esophagus.
Gastrointest Endosc Clin N Am. 21:95–109. 2011.
|
|
78
|
Shaheen NJ, Overholt BF, Sampliner RE, et
al: Durability of radiofrequency ablation in Barrett’s esophagus
with dysplasia. Gastroenterology. 141:460–468. 2011.
|
|
79
|
Shaheen NJ, Sharma P, Overholt BF, et al:
Radiofrequency ablation in Barrett’s esophagus with dysplasia. N
Engl J Med. 360:2277–2288. 2009.
|
|
80
|
Dent J: Barrett’s esophagus: A historical
perspective, an update on core practicalities and predictions on
future evolutions of management. J Gastroenterol Hepatol. 26:11–30.
2011.
|
|
81
|
El-Serag HB, Aguirre TV, Davis S, Kuebeler
M, Bhattacharyya A and Sampliner RE: Proton pump inhibitors are
associated with reduced incidence of dysplasia in Barrett’s
esophagus. Am J Gastroenterol. 99:1877–1883. 2004.
|
|
82
|
Nguyen DM, El-Serag HB, Henderson L, Stein
D, Bhattacharyya A and Sampliner RE: Medication usage and the risk
of neoplasia in patients with Barrett’s esophagus. Clin
Gastroenterol Hepatol. 7:1299–1304. 2009.PubMed/NCBI
|
|
83
|
Jankowski JA and Hooper PA:
Chemoprevention in Barrett’s esophagus: A pill a day? Gastrointest
Endosc Clin N Am. 21:155–170. 2011.
|
|
84
|
Neumann H, Mönkemüller K, Vieth M and
Malfertheiner P: Chemoprevention of adenocarcinoma associated with
Barrett’s esophagus: potential options. Dig Dis. 27:18–23.
2009.
|
|
85
|
Heath EI, Canto MI, Piantadosi S, et al:
Secondary chemo-prevention of Barrett’s esophagus with celecoxib:
results of a randomized trial. J Natl Cancer Inst. 99:545–557.
2007.
|
|
86
|
Das D, Chilton AP and Jankowski JA:
Chemoprevention of oesophageal cancer and the AspECT trial. Recent
Results Cancer Res. 181:161–169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ong CA, Lao-Sirieix P and Fitzgerald RC:
Biomarkers in Barrett’s esophagus and esophageal adenocarcinoma:
Predictors of progression and prognosis. World J Gastroenterol.
16:5669–5681. 2010.
|