Introduction
Rhabdomyosarcoma (RMS) is the most common
soft-tissue sarcoma of adolescence and childhood, accounting for 5%
of all malignant tumors in patients under 15 years of age. Most RMS
tumors originate in the head and neck region, urogenital tract, and
extremities (1–6). Based on histology, there are two
major subtypes of RMS: alveolar (A)RMS and embryonal (E) RMS
(7). Clinical evidence indicates
that ARMS is more aggressive and has a significantly worse outcome
than ERMS. Genetic characterization of RMS has identified markers
that show excellent correlation with histologic subtype.
Specifically, ARMS is characterized by the translocation
t(2;13)(q35;q14) in 70% of cases and the variant translocation
t(1;13)(p36;q14) in a smaller percentage of cases. These
translocations generate PAX3-FOXO1 and PAX7-FOXO1
fusion genes that encode the fusion proteins PAX3-FOXO1 and
PAX7-FOXO1, which are believed to act in cell survival and
deregulation of the cell cycle in ARMS cells. Evidence accumulates
that ARMS and ERMS are two different disorders. While ARMS may
originate from primitive uncommitted mesodermal cells, ERMS
originates probably from more differentiated myoblasts (8). This interesting concept however,
needs more evidence.
As with other malignancies, the major clinical
problem with RMS is its tendency to metastasize and infiltrate
various organs. This process is directed by several chemokines,
such as stromal-derived factor-1 (SDF-1), interferon-inducible
T-cell alpha chemoattractant (I-TAC), and hepatocyte growth
factor/scatter factor (HGF/SF). In addition, the family of insulin
factors, including insulin (Ins), insulin-like growth factor-1
(Igf-1), and insulin-like growth factor-2 (Igf-2), plays an
important role both in stimulating proliferation and migration of
RMS cells (9–12). In addition to PAX-FOXO1
fusion genes, aberrant expression of p53,
p16INK4A/p14ARF, and activation of the H-Ras
pathway have been postulated to function in RMS pathogenesis
(13).
The Ras superfamily of guanosine triphosphatases
(GTPases), which includes H-, K-, and N-Ras and other closely
related isoforms, are regulated switches that control many
intra-cellular pathways associated with the control of cell
proliferation and migration (14–16).
The Ras GTPases act by cycling between guanosine triphosphate
(GTP)-bound states that can couple to downstream events and
guanosine diphosphate (GDP)-bound states that do not activate those
events (16). The conversion
between these states is governed by several groups of enzymes,
including GTP-exchange factors (GEFs), which catalyze the release
of GDP and subsequent binding of GTP to activate these proteins,
and GTPase-activating proteins (GAPs), which greatly stimulate the
endogenous GTPase activity of Ras proteins and thereby stimulate
their inactivation.
The potential role of Ras pathway activation is
demonstrated very well for ERMS but not for ARMS cases. To support
this role, it has been demonstrated in a zebrafish model that
expression of mutant H-Ras induced ERMS tumors by day 10 of life
(17). Furthermore, ERMS has been
reported in Neurofibromatosis type 1 (18,19),
Noonan syndrome (20,21) and Costello syndrome patients
(22–24) with increased Ras signaling cascade
caused by mutation in one of several genes encoding proteins in
this pathway - a phenomenon known in the literature as
‘RASopathies’ (25). In sporadic
RMS tumors, Ras family mutations have been found in about 20% of
ERMS but not in any ARMS cases. Since the combination of Ras
activation along with expression of dominant-negative p53 or SV40
early region proteins and PAX-FOXO1 in murine mesenchymal stem
cells (MSCs) leads to formation of ARMS-like tumor cells, we became
interested in a potential role of Ras signaling in the pathogenesis
of ARMS. Because no Ras mutations have been reported in ARMS
patients, we hypothesized that RasGRF1 (or CDC25Mm)
which is a GTP exchange factor for Ras GTPases, plays a role in the
pathogenesis of ARMS.
In addition, it was another reason why we become
interested in a potential role of RasGRF1 in pathogenesis of ARMS.
Namely, as it has been postulated this type of RMS develops in some
primitive uncommitted mesodermal cell (8,26).
On other hand RasGRF1 plays an important role in the development of
primitive very small embryonic-like stem cells (VSELs) residing in
adult tissues (27) as
demonstrated in a recent elegant study are precursors for the
mesodermal and mesenchymal stem cells (19). Therefore, based on this and other
studies (28,29) RMS could develop in stem cells
related to mesenchymal lineage. To support further this hypothesis,
the analysis of epigenetic changes in VSELs identified unique
methylation patterns of differentially methylated regions (DMRs) in
several imprinted genes including RasGRF1, Igf2-H19 and KCNQ1 that
explain dormant state of VSELs in adult tissues (27). At the same time, the same genes
(Igf2 and KCNQ1) due to epigenetic changes at their DMR loci are
overexpressed in rapidly proliferating RMS cells. This involvement
of imprinted genes in pathogenesis of RMS explains why we become
interested to see if RasGRF1 similarly as Igf2 and KCNQ1 could be
also involved in pathogenesis of ARMS.
In this study, our findings indicate that RasGRF1
mediates the chemotactic responsiveness of RMS cells to SDF-1,
HGF/SF, Igf-1, and Ins. Furthermore, knockdown of RasGRF1 in RMS
cells inhibited ARMS cell growth in vitro and tumor
formation in vivo in immunodeficient mice. We therefore
postulate that RasGRF1 plays an important role in ARMS pathogenesis
and is a new potential target to inhibit ARMS growth.
Materials and methods
Animals
This study was performed in accordance with the
guidelines of the Animal Care and Use Committee of the University
of Louisville School of Medicine and with the Guide for the Care
and Use of Laboratory Animals (Department of Health and Human
Services, publication no. NIH 86-23).
Cell lines
We used six human ARMS cell lines (gift of Dr Peter
Houghton, Nationwide Children’s Research Hospital, Columbus, OH)
(RH2, RH4, RH18, RH28, RH30 and RH41). RMS cells used for
experiments were cultured in Roswell Park Memorial Institute medium
(RPMI)-1640 (Sigma, St. Louis, MO), supplemented with 100 IU/ml
penicillin, 10 μg/ml streptomycin, and 50 μg/ml
neomycin (Life Technologies, Inc., Grand Island, NY) in the
presence of 10% heat-inactivated fetal bovine serum (FBS, Life
Technologies). The cells were cultured in a humidified atmosphere
at 37°C in 5% CO2 at an initial cell density of
2.5×104 cells/flask (Corning, Cambridge, MA) and the
media were changed every 48 h.
Real-time quantitative reverse
transcription PCR (RQ-PCR)
Total RNA was isolated from cells treated with
hypoxia and from controls with the RNeasy Kit (Qiagen, Valencia,
CA). The RNA was reverse-transcribed with MultiScribe reverse
transcriptase and oligo-dT primers (Applied Biosystems, Foster
City, CA). Quantitative assessment of mRNA levels was performed by
real-time RT-PCR on an ABI 7500 instrument with Power SYBR Green
PCR Master Mix reagent. Real-time conditions were as follows: 95°C
(15 sec), 40 cycles at 95°C (15 sec), and 60°C (1 min). According
to melting point analysis, only one PCR product was amplified under
these conditions. The relative quantity of a target, normalized to
the endogenous control β-2 microglobulin gene and relative to a
calibrator, is expressed as 2-ΔΔCt (-fold difference), where Ct is
the threshold cycle, ΔCt = (Ct of target genes) - (Ct of endogenous
control gene, β-2 microglobulin), and ΔΔCt = (ΔCt of samples for
target gene) - (ΔCt of calibrator for the target gene). The
following primer pairs were used: RasGRF1 F:
5′-GCCACCAATCGTGTCTTGAA-3′; RasGRF1 R:
5′-CAAAGTCCTGAGAGTGCTTGGA-3′.
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections (4
μm) were stained for RasGRF1. The de-waxed, rehydrated
sections were heated in 0.01 M citrate buffer at pH 6.0 in an
autoclave. Afterwards the endogenous peroxidase activity was
blocked in 3% hydrogen peroxide in PBS for 10 min. After washing
the blocking was performed using the avidin/biotin blocking kit
(Vector Laboratories, Cambridge, UK). The sections were incubated
with the primary antibody targeting RasGRF1 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) in 1:200 dilution at 4°C
overnight. The immunohistochemistry detection was done with the
IDetect Super Stain System HRP (ID Laboratories, London, ON,
Canada). The signal was visualized with 3-amino-9-ethylcarbazole
(ID Laboratories). Afterwards the sections were counterstained with
hematoxylin.
The immunohistochemical staining on 4 samples was
analyzed with the software HistoQuest™ from TissueGnostics GmbH
(Vienna, Austria, www.tissuegnostics.com). The HistoQuest™ software
permits quantification of expression intensities on immunostained
slides. The results are visualized in scattergrams and/or
histograms. Images were taken with a Zeiss AxioImager Z.1
microscope. Statistical analysis of the data was performed using
the t-test (Graph Pad Prism Software, San Diego, CA, USA).
Knockdown of RASGRF1 with short hairpin
RNA
In RNAi experiments, the short hairpin RNA
(shRNA)-generating plasmid pSuper/Puro (Oligoengine, Seattle, WA)
was used. The oligonucleotide-targeting base sequence for human
RasGRF1 was: 5′-GTACCGGAGGATGTCCTTA-3′. RMS cells were plated at
80% confluency and transfected with shRNA vector using
Lipofectamine (Invitrogen) according to the manufacturer’s
protocol. A commercially available scrambled shRNA negative control
plasmid was used (Dharmacon). For stable transfection of
shRNA-producing vectors, single-cell dilutions of lipofected cells
were prepared and further expansion was performed in the presence
of puromycin (1 μg/ml, Invitrogen).
Cell proliferation
Cells were plated in 24-well culture plates at an
initial density of 3×103 cells/well in the presence or
absence of IGF-II (100 ng/ml) or insulin (10 ng/ml). In some
experiments farnesyl transferase inhibitor FTI277 (Sigma) was used
at a concentration of 10 μM. The cell number was calculated
at 24, 48, and 72 h after culture initiation. At the indicated time
points, cells were harvested from the culture flasks by
trypsinization and the number of cells determined using an LSR-II
cell cytometer (BD Biosciences).
Phosphorylation of intracellular pathway
proteins and western blotting
Western blots were performed on extracts prepared
from RMS cell lines (2×106 cells) that were kept in RPMI
medium containing low levels of bovine serum albumin (BSA, 0.5%) to
render the cells quiescent. The cells were divided and stimulated
with optimal doses of SDF-1 (300 ng/ml), I-TAC (100 ng/ml), HGF
(100 ng/ml), IGF-II (100 ng/ml), and insulin (10 ng/ml) for 5 min
at 37°C and then lysed (for 10 min) on ice in M-Per lysing buffer
(Pierce, Rockford, IL), containing protease and phosphatase
inhibitors (Sigma). Subsequently, the extracted proteins were
separated by either 12% or 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and the
fractionated proteins were transferred to a nitrocellulose membrane
(Schleicher & Schuell, Keene, NH) as previously described.
RasGRF1 phosphorylated at Ser929 and total RasGRF1 were detected
using rabbit polyclonal antibodies (Santa Cruz Biotechnology).
Phosphorylation of the intracellular kinases, p42/44
mitogen-activated protein kinase (MAPK) (Thr202/Tyr204) and AKT,
was detected using commercial mouse phospho-specific mAb (p42/44)
or rabbit phospho-specific polyclonal antibodies (all from New
England Biolabs, Beverly, MA) with horseradish peroxidase
(HRP)-conjugated goat anti-mouse IgG or goat anti-rabbit IgG as a
secondary antibody (Santa Cruz Biotechnology). Equal loading in the
lanes was evaluated by stripping the blots and reprobing with
appropriate mAbs: p42/44 anti-MAPK clone no. 9102 and anti-AKT
clone no. 9272 (Santa Cruz Biotechnology). The membranes were
developed with an enhanced chemiluminescence (ECL) reagent
(Amersham Life Sciences, Little Chalfont, UK), dried, and exposed
to film (HyperFilm, Amersham Life Sciences).
Ras activity assay
The Ras activation assay kit (Upstate Inc.) was used
for these studies according to the manufacturer’s protocol. Cells
were serum starved overnight in 0.5% BSA containing RPMI (control)
and stimulated with SDF-1 (300 ng/ml) or IGF-II (100 ng/ml) for 5
min and then lysed using an Mg2+ lysis buffer (125 mM
HEPES, pH 7.5, 750 mM NaCl, 5% NP-40, 50 mM MgCl2, 5 mM
ethylenediaminetetraacetic acid, and 10% glycerol). Raf-1
Ras-binding domain agarose beads were added to lysates followed by
incubation for 1 h at 4°C. Beads were washed twice, and bound
Ras-GTP (active form) was detected by immunoblot with a
pan-anti-Ras antibody (clone RAS 10). Quantitative analysis of Ras
activation bands obtained via western blot analysis was performed
using ImageJ software (http://rsb.info.nih.gov/ij/). Relative Ras activity
was calculated as percentage of control (−) and corrected for
signal intensity with loading controls.
Chemotaxis assay
The 8-μm Transwell polycarbonate membranes
were covered with 50 μl of 0.5% gelatin. Cells were detached
with 0.5 mmol/l ethylenediaminetetraacetic acid (EDTA), washed in
RPMI-1640, resuspended in RPMI-1640 with 0.5% BSA, and seeded at a
density of 3×104 cells in 120 μl into the upper
chambers of Transwell inserts (Costar Transwell; Corning Costar,
Corning, NY). The lower chambers were filled with SDF-1 (300
ng/ml), I-TAC (100 ng/ml), HGF (100 ng/ml), IGF-II (100 ng/ml),
insulin (10 ng/ml), or 0.5% BSA RPMI-1640 (control). After 24 h,
the inserts were removed from the Transwell apparatus. Cells
remaining in the upper chambers were scraped off with cotton wool
and cells that had transmigrated were stained by HEMA 3, according
to the manufacturer’s instructions (Fisher Scientific, Pittsburgh,
PA), and counted either on the lower side of the membranes or on
the bottom of the Transwell inserts.
Adhesion of RMS cells to fibronectin
Cells were made quiescent for 24 h with 0.5% BSA in
RPMI before incubation with SDF-1 (300 ng/ml), I-TAC (100 ng/ml),
HGF (100 ng/ml), IGF-II (100 ng/ml), or insulin (10 ng/ml) for 5
min. Cells were added directly onto the protein-coated wells
(5×103/well) for 5 min. The wells were coated with
fibronectin (10 μg/ml) by incubating overnight at 4°C and
blocked with BSA for 2 h before the experiment. Following
incubation at 37°C, the plates were vigorously washed 3 times and
adherent cells were stained by HEMA 3 and counted under a
microscope.
Fluorescent staining of RMS cells
RMS cells were fixed in 3.5% paraformaldehyde for 20
min, permeabilized by 0.1% Triton X-100, washed in PBS, pre-blocked
with 2% BSA and subsequently stained with antibodies to RasGRF1
(1:200, rabbit polyclonal IgG, Santa Cruz, Inc.), paxillin (1:200,
mouse monoclonal IgG, eBioscience) and phalloidin - Alexa 488
(1:400, Molecular Probes, Eugene, OR). Appropriate secondary Alexa
Fluor 594 mouse anti-rabbit IgG and Alexa Fluor 594 goat anti-mouse
IgG were used (1:400, Molecular Probes). The nuclei were identified
with DAPI (Molecular Probes). The fluorescence images were
collected with the TE-FM Epi-Fluorescence system attached to an
Olympus Inverted Microscope IX81 (Olympus, Center Valley, PA).
Xenografts of human RMS cells into
immunodeficient mice
To evaluate the in vivo metastatic behavior
of three populations of RH30 cells (RH30, RH30 scrambled, and RH30
with knockdown of RasGRF1), cells (5×106 per mouse) were
inoculated into the hind limb muscles of SCID/beige inbred mice.
Six weeks later, the mice were sacrificed for evaluation of the RMS
cells present in blood, bone marrow, liver, and lungs and the
presence of RMS cells (i.e., murine-human chimerism) was evaluated
by the difference in the level of human α-satellite expression. DNA
was amplified in the extracts isolated from bone marrow-, liver-
and lung-derived cells using real-time PCR. Briefly, DNA was
isolated using the QIAamp DNA Mini kit (Qiagen). Detection of human
satellite and murine β-actin DNA levels was conducted by real-time
PCR using an ABI PRISM 7500 Sequence Detection System. A
25-μl reaction mixture containing 12.5 μl SYBR Green
PCR Master Mix, 300 ng DNA template, and forward (5′-ACC ACT CTG
TGT CCT TCG TTG G-3′) and reverse primers (5′-ATC GCG CTC TCA AAA
GGA GTG T-3′ and 5′-AAA CGT CCA CTT GCA GAT TCT AG-3′) for the
α-satellite sequences and forward (5′-GGA TGC AGA AGG AGA TCA
CTG-3′) and reverse primer (5′-CGA TCC ACA CGG AGT ACT TG-3′) for
β-actin was used. The Ct value was determined as before. The number
of human cells present in the murine organs (indicating the degree
of chimerism) was calculated from the standard curve obtained by
mixing different numbers of human cells with a constant number of
murine cells.
Statistical analysis
All results are presented as mean ± standard error
of the mean (SEM). Statistical analysis of the data was performed
using the nonparametric Mann-Whitney test, with p<0.05
considered significant.
Results
RasGRF1 is expressed in human RMS cell
lines and primary tumors
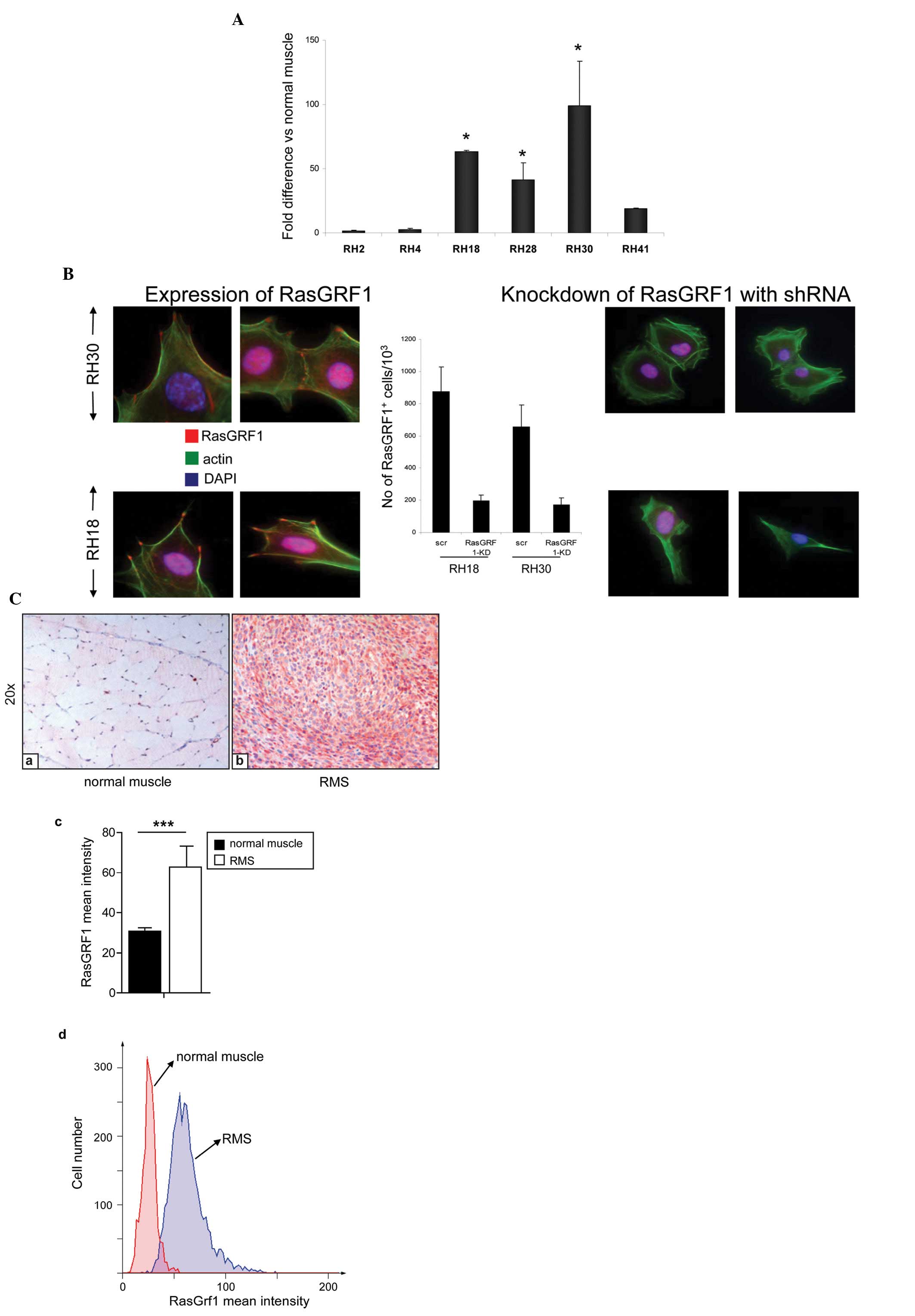
Fig. 1A shows
expression of RasGRF1 mRNA in established human ARMS cell lines
(RH2, RH4, RH18, RH28, RH30 and RH41) compared to RasGRF1 mRNA
expression in normal human skeletal muscle cells. We observed high
(>50 times) RasGRF1 overexpression (at the mRNA level) in 3 and
elevated expression in further 2 ARMS (>10 times) out of 6 cell
lines. RasGRF1 was also upregulated in 3 out of 3 ERMS cell lines
(RD, RH36, SMS-CTR) (data not shown). Interestingly, the ERMS cell
line RD transfected with the PAX3-FKHR trans-gene expressed RasGRF1
at several times the level of wild-type RD cells (data not shown).
By employing immunofluorescence analysis, we observed that RasGRF1
protein is located in the filopodia of RMS cells (Fig. 1B).
We also evaluated expression of RasGRF1 in human
primary ARMS tumor samples and noted pronounced upregulation of its
expression at protein level as compared to normal skeletal muscle
and surrounding tissue from normal patients (Fig. 1C, data not shown).
RasGRF1 controls proliferation of human
RMS cells
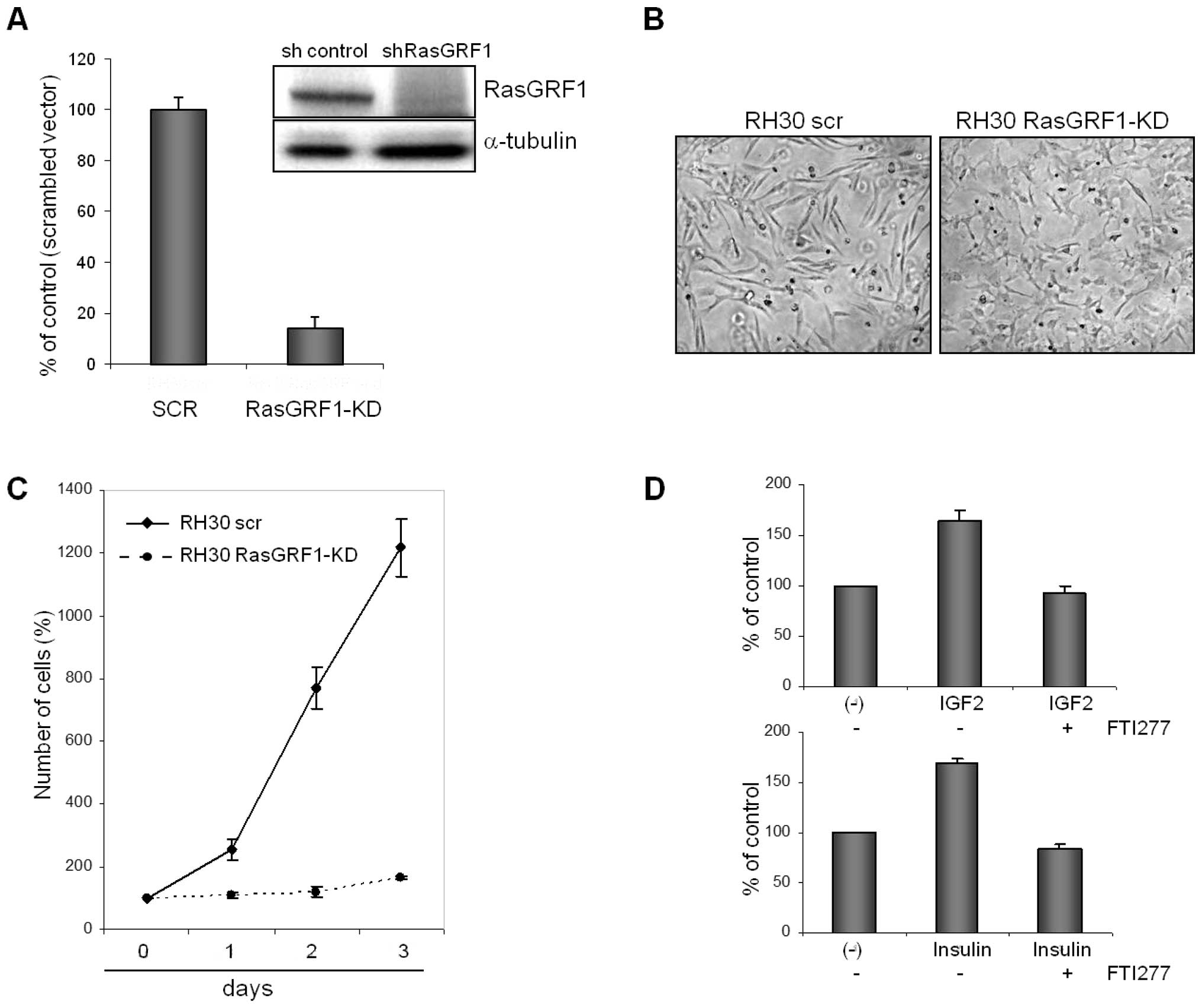
In further experiments, we selected the ARMS cell
line RH30 that expresses RasGRF1 at the highest level (Fig. 1A) and, as we reported previously,
responds robustly to several pro-metastatic chemoattractants such
as SDF-1 (30), HGF/SF (31), and I-TAC (32). Fig.
2A and Fig. 1B show that we
were able to use an shRNA technology to efficiently down-regulate
expression of RasGRF1 in RH30 cells both at the mRNA (Fig. 2A) and protein levels (Fig. 1B). RH30 cells with down-regulated
RasGRF1 changed morphology from a spindle-forming to a more flat
phenotype (Fig. 2B). Most
importantly, down-regulation of RasGRF1 expression in RH30 cells
resulted in a decrease in proliferative potential (Fig. 2C). These cells however, were still
alive and did not undergo apoptosis as evaluated by 0.4% trypan
blue exclusion test and Annexin-V staining.
In parallel, we also knocked down expression of
RasGRF1 in the RH18 cell line, which similarly to RH30, also highly
expresses RasGRF1 and obtained similar results (data not
shown).
Igf-2 and Ins are known factors that stimulate
proliferation of RMS cells (10–12).
Thus, we stimulated RH30 cells by Igf-2 or Ins in the presence or
absence of the Ras-GTPase blocking agent farnesyl transferase
inhibitor (FTI 277). As shown in Fig.
2D, the pro-proliferative effect of Igf-2 and Ins was inhibited
in the presence of FTI 277.
RasGRF1 is involved in intracellular
signaling after stimulation by pro-metastatic chemoattractants
As reported (9–11,
30–33) the pro-metastatic behavior of RMS
cells is influenced by several chemoattractants/growth factors
(i.e., SDF-1, I-TAC, HGF/SF, Igf-2, and Ins). Therefore, in the
next step we evaluated phosphorylation of RasGRF1 in RH30 wild-type
(RH30 wt) and RH30 RasGRF1-kd cells (Fig. 3A). In RH30 wt cells we observed an
increase in both p42/44 MAPK and AKT phosphorylation after
stimulation by SDF-1 and HGF/SF and an increase in p42/44 MAPK
phosphorylation alone after stimulation by Igf-2 and Ins. Since
RH30 cells express very low levels of CXCR7 (32), no activation/phosphorylation of
p42/44 MAPK and/or AKT was observed when the CXCR7 ligand I-TAC was
employed for stimulation. In striking contrast, no phosphorylation
was detected in RH30 RasGRF1-kd cells. Fig. 3B confirms that stimulation by
SDF-1, HGF/SF, Igf-2, and Ins activates phosphorylation of RasGRF1
in RH30 wt but it was not detectable in RH30 RasGRF1-kd cells.
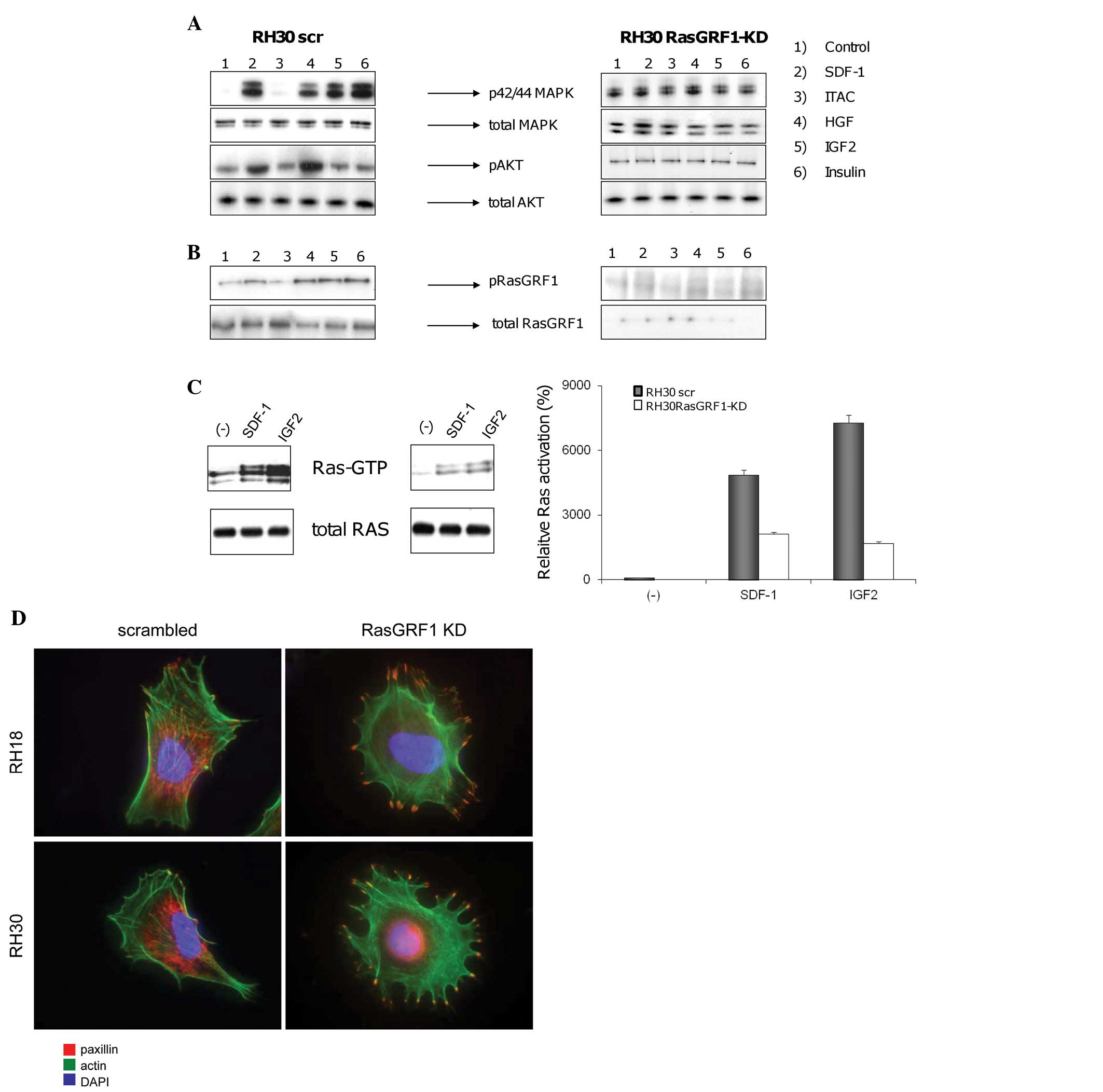 | Figure 3RasGRF1 is involved in chemokine and
growth factor receptor signaling. (A), Effect of RasGRF1
down-regulation on activation of intracellular signaling in ARMS
cells. Phosphorylation of p42/44 MAPK and AKT in RH30-derived cell
lines was stimulated for 5 min by SDF-1 (300 ng/ml), I-TAC (100
ng/ml), HGF (100 ng/ml), IGF-II (100 ng/ml), and insulin (10
ng/ml). The experiment was repeated three times with similar
results. A representative result is shown. (B), RasGRF1
phosphorylation after stimulation with chemokines and growth
factors. RasGRF1 protein phophorylated at Ser929 was detected by
western blot analysis after stimulation for 5 min by SDF-1 (300
ng/ml), I-TAC (100 ng/ml), HGF (100 ng/ml), IGF-II (100 ng/ml), and
insulin (10 ng/ml). (C), Effect of RasGRF1 down-regulation on Ras
activation. A Ras pull-down assay was performed on two RH30-derived
cell lines (RH30scr and RH30 RasGRF1-kd). The cells were stimulated
for 5 min with SDF-1 (300 ng/ml) or IGF-II (100 ng/ml). Ras-GTP was
precipitated by Raf-1 RBD agarose conjugate and detected by Ras
antibody clone RAS10 (Millipore). The same antibody was used to
detect total Ras protein (p21 H-, K- and N-Ras). Western blots were
analyzed by densitometry (right side). The experiment was repeated
three times with similar results. A representative result is shown.
(D), Effect of RasGRF1 down-regulation on paxillin expression and
actin cytoskeleton. Staining of paxillin and actin was performed on
RH30scr, RH30 RasGRF1-kd, RH18scr and RH18 RasGRF-kd cell lines.
The experiment was repeated three times and representative results
are shown. |
These data in toto demonstrate that RasGRF1
is required for signaling from the SDF-1 receptor (CXCR4), the HGF
receptor (c-met), the Igf-2 receptor (IGF-2R) and the Ins receptor
(INS-R). The Ras GTP pull-down assay data shown in Fig. 3C confirm that Ras is activated in
response to SDF-1 and Igf-2 in RH30 wt cells, but as expected, is
activated at only very low levels in RH30 RasGRF1-kd cells.
To address the defect in migration of RasGRF1-kd
cells we evaluated by confocal microscope the distribution of
paxillin, that is involved in interaction between β-integrins on
cell surface and kinases, tructural proteins and regulators of
actin organization in cytoplasm. As predicted from their less
migratory phenotype, RasGRF1-kd cells show a high accumulation of
paxillin in filopodia and more adherent phenotype as compared to
control cells (Fig. 3D).
RasGRF1 is involved in migration but not
adhesion of RMS cells
Next we evaluated the responsiveness of RH30 wt and
RH30 RasGRF1-kd cells to selected chemoattractants in chemotaxis
(Fig. 4A) and adhesion assays
(Fig. 4B). We observed that
down-regulation of RasGRF1 in RH30 cells resulted in inhibition of
responsiveness of these cells to chemotactic gradients of SDF-1,
HGF/SF, Igf-2, and insulin. However, this down-regulation did not
significantly decrease the adhesive potential of these cells in
response to the same factors. In control experiments, inhibition of
chemotaxis was also observed when we blocked Ras-GTPase by
employing farnesyl transferase inhibitor (FTI 277) (data not
shown).
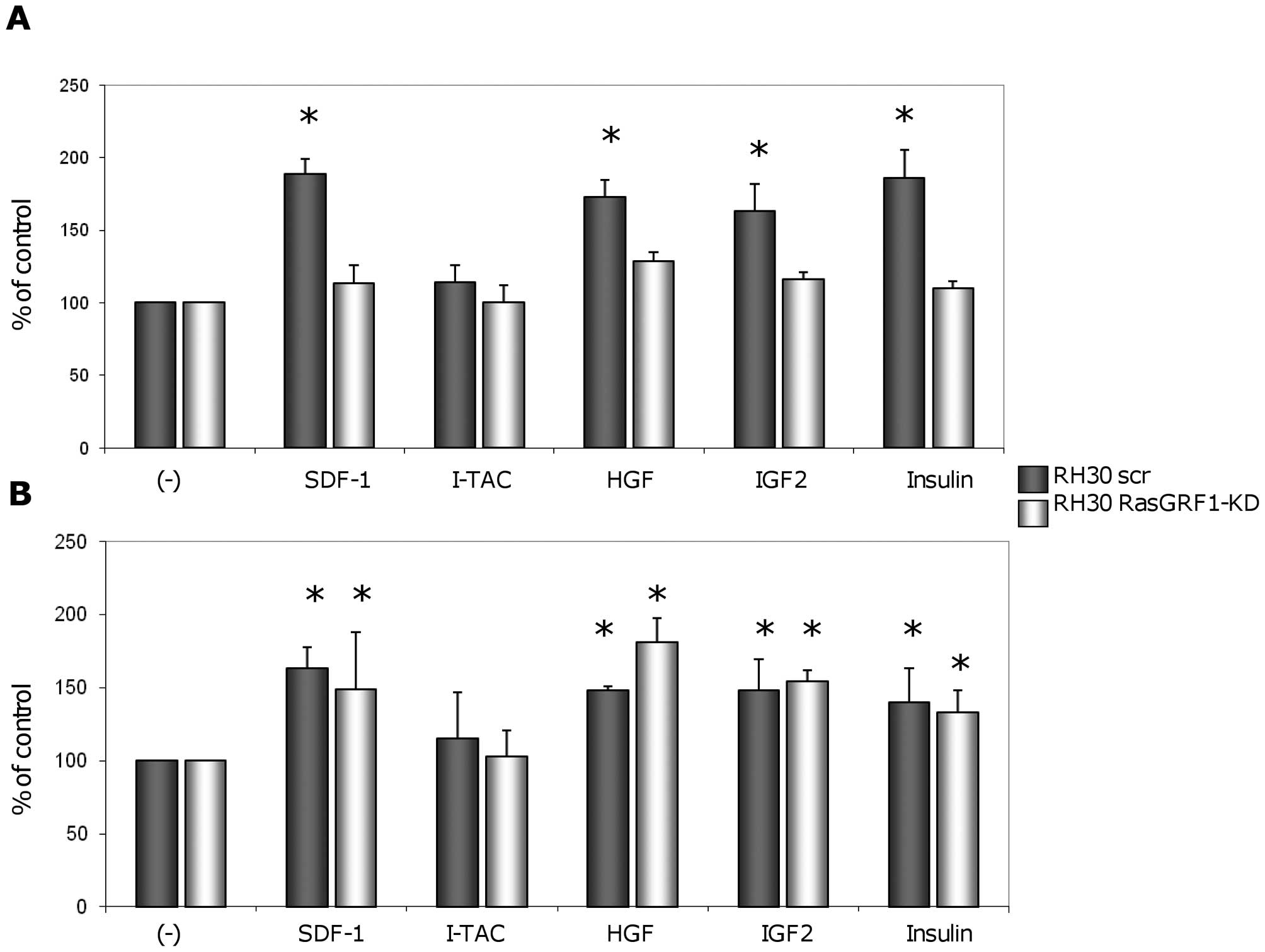 | Figure 4Effect of RasGRF1 knockdown on
migration and adhesion of ARMS cells. (A), ARMS cell chemotaxis
after down-regulating RasGRF1 expression. Chemotaxis of
RH30-derived cells across Transwell membranes covered with gelatin
in response to SDF-1 (300 ng/ml), I-TAC (100 ng/ml), HGF (100
ng/ml), IGF-II (100 ng/ml), and insulin (10 ng/ml) gradients. Black
bars show chemotaxis of control RH30scr cells, while white bars
represent chemotaxis of RH30 cells with down-regulated expression
of RasGRF1. Data from 4 separate experiments are pooled together.
*p<0.05 compared to unstimulated controls (−). (B),
ARMS cell adhesion after down-regulating RasGRF1 expression.
Adhesion of human RMS cells to fibronectin after stimulation by
SDF-1 (300 ng/ml), I-TAC (100 ng/ml), HGF (100 ng/ml), IGF-II (100
ng/ml), and insulin (10 ng/ml). Data from 3 separate experiments
are pooled together. *p<0.05 as compared to
unstimulated controls (−). |
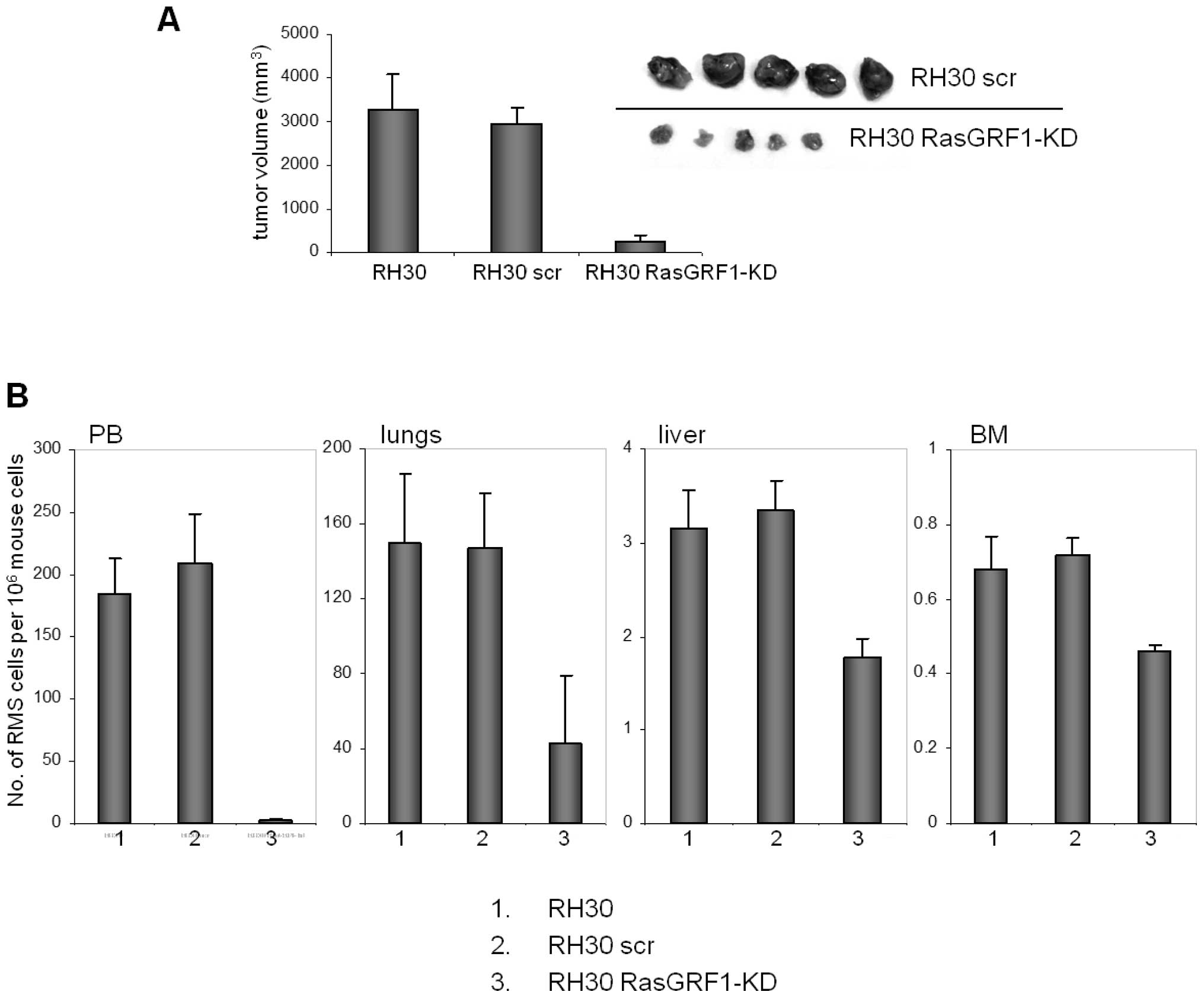
Effect of RasGRF1 down-regulation on in
vivo tumor growth of RH30 cells
Finally, we employed SCID/beige mice to study tumor
formation by RH30 wt and RH30 RasGRF1-kd cells (Fig. 5). We observed that after
down-regulation of RasGRF1, RH30 cells formed significantly smaller
tumors following inoculation into skeletal muscles of
immunodeficient SCID/beige mice (Fig.
5A).
Furthermore, 6 weeks after inoculation of RMS cells,
we observed a much lower number of RMS cells in peripheral blood,
lungs, liver, and bone marrow of mice inoculated with RasGRF1-kd
RH30 cells (Fig. 5B).
Discussion
RMS is the most common soft tissue sarcoma in
children that, as recently postulated, originates from mutated
primitive mesodermal/mesenchymal stem cells (ARMS) or skeletal
muscle satellite cells (ERMS) (8).
Our recent research on VSELs, which are deposited during
development in tissues of young individuals and whose number
rapidly declines with the age, allowed us to present the hypothesis
that VSELs are a ‘missing link’ that would reconcile the embryonic
rest/germ line origin of cancer postulated 150 years ago with
contemporary theories of cancer development (34). In support of this, the analysis of
epigenetic changes in VSELs identified unique methylation patterns
of DMRs in some imprinted genes (Igf2-H19, KCNQ1 and RasGRF1) that,
on the one hand, explain the dormant state of VSELs residing in
adult tissues (27) but on the
other hand, explain the reverse pattern of expression of these
genes reported in RMS seen for example in Beckwith-Wiedemann
syndrome patients (35–38). Since as recently demonstrated VSELs
are at the top of the hierarchy of the mesenchymal stem cell
lineage (39), changes in the
epigenetic state of DMR in some of the imprinted genes in VSELs or
VSELs-derived mesenchymal stem cells could potentially trigger RMS
development.
In research leading to the current paper we became
interested in events downstream from activated receptors and
focused on RasGRF1, which is a GEF for the Ras superfamily of
GTPases and is paternally imprinted in mice (40–42).
While Ras proteins regulate various signaling pathways controlling
cell growth, differentiation, and survival, RasGRF1 was initially
described as highly expressed in brain tissue, while playing a role
in learning and memory. The full-length RasGRF1 protein contains
several domains: a pleckstrin homology domain, a coiled-coil
region, a calmodulin-dependent activation domain, the ilimaquinon
motif, a DBL homology domain, and a CDC25 domain (43,44).
Alternative forms of RasGRF1, ranging in size from approximately 50
to 140 kDa, have been identified and overexpression of larger forms
has been reported to be crucial for transformation of NIH 3T3 cells
(45). Interestingly, the p75
isoform has been reported to be a more effective GEF for H-Ras
(46).
It has been reported that RasGRF1 knockout mice are
smaller than normal littermates and display defects in memory
consolidation associated with different areas of the brain, as well
as defects in β-cell development and glucose homeostasis (47). It has been demonstrated that
RasGRF1 is a c-Jun-regulated gene necessary for promoting
non-adherent growth of c-Myc- or c-Jun-transduced fibroblasts and
not much attention has been paid to a potential role of RasGRF1 in
tumorigenesis, despite this protein having been observed to be
expressed in several tumor types (48).
As demonstrated in this study, RasGRF1, compared to
normal skeletal muscles, is overexpressed in the majority of RMS
cell lines, which was subsequently confirmed by immunohistochemical
detection in patient samples. However, since we compared RasGRF1
expression in proliferating ARMS cells to normal skeletal muscles,
further studies are needed to see if this phenomenon is a result of
malignant phenotype of ARMS cells or rather depends on
proliferative status of myogenic cells.
Moreover, we observed that RasGRF1 becomes
phosphorylated/activated after stimulation with prometastatic
factors, such as SDF-1 and HGF/SF, which suggests its involvement
in signaling of the CXCR4 and c-met receptors, respectively. We
also confirmed our previous observations that in RH30 cells, SDF-1
and HGF/SF activate p42/44 MAPK and AKT, which are involved in cell
migration (30,31). The potential involvement of RasGRF1
in RMS cell migration was subsequently confirmed by confocal
microscopy observations that this GEF localizes within cell
filopodia. More importantly, we noticed that knockdown of RasGRF1
in ARMS cells abolished their chemotactic responsiveness to several
prometastatic factors. This inhibition correlated with a lack of
activation of p42/44 MAPK and AKT, which suggests that RasGRF1 acts
downstream of, for example, the CXCR4 and c-met receptors involved
in cell migration.
Interestingly, the knockdown of RasGRF1 did not
affect adhesion of ARMS cells. Compared to wild-type cells, ARMS
RasGRF1-kd cells showed normal adhesion and microscopic evaluation
of these cells revealed that they change from a spindle-forming to
a more flat morphology. Interestingly, we noted in RasGRF1-kd cells
accumulation in filopodia of paxillin that is involved in
interaction between β-integrins on cell surface and kinases,
structural proteins and regulators of actin organization in
cytoplasm, which supports our data that down-regulation of RasGRF1
inhibits RMS cells migration but does not affect their
adhesion.
It has been reported that insulin family factors
strongly stimulate proliferation of RMS cells, and Igf-2 is an
autocrine factor interacting with both Igf-1R and Ins-R. In this
study we report that knockdown of RasGRF1 by shRNA and inhibition
of Ras by FTI277 results in inhibition of ARMS cell proliferation.
More importantly, RasGRF1-kd cells inoculated in immunodeficient
Beige-SCID mice formed significantly smaller tumors. In addition,
we observed a significantly lower number of circulating ARMS cells
in peripheral blood in these animals compared to mice bearing
tumors formed by control RasGRF1-scr or wt cells. Of note, compared
to wt cells, RasGRF1-kd cells did not respond by chemotaxis to
Igf-2 or an Ins gradient, which correlated with a lack of
activation of p42/44 MAPK and AKT in these cells by these
factors.
Overall, our data suggest that RasGRF1 is required
for Ras-mediated ARMS proliferation. The involvement of RasGRF1 in
insulin factor-mediated cell proliferation is supported by recent
observations that both RasGRF1 knockout mice as well as bimaternal
mice, which in the first 10 weeks after birth display defective
RasGRF1 expression, have reduced body size (47). Furthermore, as reported, Igf-1 did
not stimulate proliferation of β-cells from RasGRF1-deficient mice
and did not activate p42/44 MAPK and AKT in these cells. All these
observations could be explained by involvement of RasGRF1 in
insulin/insulin-like growth factor signaling, and our signal
transduction data lend support to this hypothesis. However, we are
aware that since we compared RasGRF1 expression in proliferating
ARMS cells to normal non-dividing skeletal muscles, further studies
are needed to see if this phenomenon is the result of malignant
phenotype of ARMS cells or rather depends on cell
proliferation.
Since RasGRF1 can act also as GEF for Rac, further
studies will answer which of these RasGRF1 functions are related to
Rac activation (14). Similar
studies are required to shed more light on the role of
RasGRF1-interacting partner proteins that were recently identified
by large-scale proteomic analysis, including ribosomal and
RNA-binding proteins, cytoskeletal proteins, and some other
proteins involved in vesicular trafficking (15,49,50).
In summary, our data for the first time demonstrate
a role for RasGRF1 in signaling from CXCR4, c-met, Igf-1R, and
Ins-R receptors, which is crucial for ARMS migration, metastasis,
and growth. We conclude that RasGRF1, which plays an important role
in ARMS pathogenesis, is a new potential target to develop
efficient, small, blocking molecules to inhibit RMS growth.
Acknowledgements
This study was supported by NIH grant
R01 CA106281-01, NIH R01 DK074720, the Henry M. & Stella M.
Hoenig Endowment and European Union structural funds Innovative
Economy Operational Program POIG 01.02-00-109/09 to MZR and NIH
Grant no. P20RR018733 from the National Center for Research
Resources to M.K., FNP ‘Homing PLUS’ programme cofinanced from
European Union, Regional Development Fund to M.T., the Joanna
McAfee Childhood Cancer Foundation and the Alveolar
Rhabdomyosarcoma Research Fund (to F.G.B.), and by the Austrian
Science Fund (FWF P-18478-B12) and the GEN-AU project
‘Inflammobiota’ [Austrian Ministry of Science and Research (BM:WF)]
to L.K.
References
|
1
|
Barr FG, Galili N, Holick J, Biegel JA,
Rovera G and Emanuel BS: Rearrangement of the PAX3 paired box gene
in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat
Genet. 3:113–117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collins MH, Zhao H, Womer RB and Barr FG:
Proliferative and apoptotic differences between alveolar
rhabdomyosarcoma subtypes: a comparative study of tumors containing
PAX3-FKHR or PAX7-FKHR gene fusions. Med Pediatr Oncol. 37:83–89.
2001. View
Article : Google Scholar
|
|
3
|
Hazelton BJ, Houghton JA, Parham DM, et
al: Characterization of cell lines derived from xenografts of
childhood rhabdomyosarcoma. Cancer Res. 47:4501–4507.
1987.PubMed/NCBI
|
|
4
|
Kelly KM, Womer RB and Barr FG: PAX3-FKHR
and PAX7-FKHR gene fusions in rhabdomyosarcoma. J Pediatr Hematol
Oncol. 20:517–518. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sandberg AA, Stone JF, Czarnecki L and
Cohen JD: Hematologic masquerade of rhabdomyosarcoma. Am J Hematol.
68:51–57. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharp R, Recio JA, Jhappan C, et al:
Synergism between INK4a/ARF inactivation and aberrant HGF/SF
signaling in rhabdomyosarcomagenesis. Nat Med. 8:1276–1280. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gordon T, McManus A, Anderson J, et al:
Cytogenetic abnormalities in 42 rhabdomyosarcoma: a United Kingdom
Cancer Cytogenetics Group Study. Med Pediatr Oncol. 36:259–267.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Charytonowicz E, Cordon-Cardo C,
Matushansky I and Ziman M: Alveolar rhabdomyosarcoma: is the cell
of origin a mesenchymal stem cell? Cancer Lett. 279:126–136. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hahn H, Wojnowski L, Specht K, et al:
Patched target Igf2 is indispensable for the formation of
medulloblastoma and rhabdomyosarcoma. J Biol Chem. 275:28341–28344.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Makawita S, Ho M, Durbin AD, Thorner PS,
Malkin D and Somers GR: Expression of insulin-like growth factor
pathway proteins in rhabdomyosarcoma: IGF-2 expression is
associated with translocation-negative tumors. Pediatr Dev Pathol.
12:127–135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rikhof B, De Jong S, Suurmeijer AJ, Meijer
C and van der Graaf WT: The insulin-like growth factor system and
sarcomas. J Pathol. 217:469–482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang W, Kumar P, Epstein J, Helman L,
Moore JV and Kumar S: Insulin-like growth factor II and PAX3-FKHR
cooperate in the oncogenesis of rhabdomyosarcoma. Cancer Res.
58:4426–4433. 1998.PubMed/NCBI
|
|
13
|
Naini S, Etheridge KT, Adam SJ, et al:
Defining the cooperative genetic changes that temporally drive
alveolar rhabdomyosarcoma. Cancer Res. 68:9583–9588. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Innocenti M, Zippel R, Brambilla R and
Sturani E: CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling
pathways. FEBS Lett. 460:357–362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lavagni P, Indrigo M, Colombo G, et al:
Identification of novel RasGRF1 interacting partners by large-scale
proteomic analysis. J Mol Neurosci. 37:212–224. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rossman KL, Der CJ and Sondek J: GEF means
go: turning on RHO GTPases with guanine nucleotide-exchange
factors. Nat Rev Mol Cell Biol. 6:167–180. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Langenau DM, Keefe MD, Storer NY, et al:
Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes
Dev. 21:1382–1395. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shome D, Honavar SG, Reddy VA and
Vemuganti GK: Orbital embryonal rhabdomyosarcoma in association
with neurofibromatosis type 1. Ophthal Plast Reconstr Surg.
23:147–148. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang P, Grufferman S, Khoury MJ, et al:
Association of childhood rhabdomyosarcoma with neurofibromatosis
type I and birth defects. Genet Epidemiol. 12:467–474. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jung A, Bechthold S, Pfluger T, Renner C
and Ehrt O: Orbital rhabdomyosarcoma in Noonan syndrome. J Pediatr
Hematol Oncol. 25:330–332. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khan S, McDowell H, Upadhyaya M and Fryer
A: Vaginal rhabdomyosarcoma in a patient with Noonan syndrome. J
Med Genet. 32:743–745. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gripp KW, Scott CI Jr, Nicholson L, et al:
Five additional Costello syndrome patients with rhabdomyosarcoma:
proposal for a tumor screening protocol. Am J Med Genet. 108:80–87.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feingold M: Costello syndrome and
rhabdomyosarcoma. J Med Genet. 36:582–583. 1999.PubMed/NCBI
|
|
24
|
O’Neal JP, Ramdas J, Wood WE and
Pellitteri PK: Parameningeal rhabdomyosarcoma in a patient with
Costello syndrome. J Pediatr Hematol Oncol. 26:389–392. 2004.
|
|
25
|
Tidyman WE and Rauen KA: The RASopathies:
developmental syndromes of Ras/MAPK pathway dysregulation. Curr
Opin Genet Dev. 19:230–236. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hettmer S and Wagers AJ: Muscling in:
uncovering the origins of rhabdomyosarcoma. Nat Med. 16:171–173.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shin DM, Zuba-Surma EK, Wu W, et al: Novel
epigenetic mechanisms that control pluripotency and quiescence of
adult bone marrow-derived Oct4(+) very small embryonic-like stem
cells. Leukemia. 23:2042–2051. 2009.PubMed/NCBI
|
|
28
|
Stratton MR, Fisher C, Gusterson BA and
Cooper CS: Detection of point mutations in N-ras and K-ras genes of
human embryonal rhabdomyosarcomas using oligonucleotide probes and
the polymerase chain reaction. Cancer Res. 49:6324–6327. 1989.
|
|
29
|
Ren YX, Finckenstein FG, Abdueva DA, et
al: Mouse mesenchymal stem cells expressing PAX-FKHR form alveolar
rhabdomyosarcomas by cooperating with secondary mutations. Cancer
Res. 68:6587–6597. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Libura J, Drukala J, Majka M, et al:
CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and
regulates locomotion, chemotaxis, and adhesion. Blood.
100:2597–2606. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jankowski K, Kucia M, Wysoczynski M, et
al: Both hepatocyte growth factor (HGF) and stromal-derived
factor-1 regulate the metastatic behavior of human rhabdomyosarcoma
cells, but only HGF enhances their resistance to radiochemotherapy.
Cancer Res. 63:7926–7935. 2003.
|
|
32
|
Grymula K, Tarnowski M, Wysoczynski M, et
al: Overlapping and distinct role of CXCR7-SDF-1/ITAC and
CXCR4-SDF-1 axes in regulating metastatic behavior of human
rhabdomyosarcomas. Int J Cancer. 127:2554–2568. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tarnowski M, Grymula K, Reca R, et al:
Regulation of expression of stromal-derived factor-1 receptors:
CXCR4 and CXCR7 in human rhabdomyosarcomas. Mol Cancer Res. 8:1–14.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ratajczak MZ, Shin DM, Liu R, et al:
Epiblast/germ line hypothesis of cancer development revisited:
lesson from the presence of Oct-4+ cells in adult
tissues. Stem Cell Rev. 6:307–316. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Casola S, Pedone PV, Cavazzana AO, et al:
Expression and parental imprinting of the H19 gene in human
rhabdomyosarcoma. Oncogene. 14:1503–1510. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Anderson J, Gordon A, McManus A, Shipley J
and Pritchard-Jones K: Disruption of imprinted genes at chromosome
region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia.
1:340–348. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scrable H, Cavenee W, Ghavimi F, Lovell M,
Morgan K and Sapienza C: A model for embryonal rhabdomyosarcoma
tumorigenesis that involves genome imprinting. Proc Natl Acad Sci
USA. 86:7480–7484. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhan S, Shapiro DN and Helman LJ:
Activation of an imprinted allele of the insulin-like growth factor
II gene implicated in rhabdomyosarcoma. J Clin Invest. 94:445–448.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Taichman RS, Wang Z, Shiozawa Y, et al:
Prospective identification and skeletal localization of cells
capable of multilineage differentiation in vivo. Stem Cells Dev.
19:1557–1570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De la Puente A, Hall J, Wu YZ, et al:
Structural characterization of Rasgrf1 and a novel linked imprinted
locus. Gene. 291:287–297. 2002.PubMed/NCBI
|
|
41
|
Yoon B, Herman H, Hu B, et al: Rasgrf1
imprinting is regulated by a CTCF-dependent methylation-sensitive
enhancer blocker. Mol Cell Biol. 25:11184–11190. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoon BJ, Herman H, Sikora A, Smith LT,
Plass C and Soloway PD: Regulation of DNA methylation of Rasgrf1.
Nat Genet. 30:92–96. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Martegani E, Vanoni M, Zippel R, et al:
Cloning by functional complementation of a mouse cDNA encoding a
homologue of CDC25, a Saccharomyces cerevisiae RAS activator. EMBO
J. 11:2151–2157. 1992.PubMed/NCBI
|
|
44
|
Buchsbaum R, Telliez JB, Goonesekera S and
Feig LA: The N-terminal pleckstrin, coiled-coil, and IQ domains of
the exchange factor Ras-GRF act cooperatively to facilitate
activation by calcium. Mol Cell Biol. 16:4888–4896. 1996.PubMed/NCBI
|
|
45
|
Chevallier-Multon MC, Schweighoffer F,
Barlat I, et al: Saccharomyces cerevisiae CDC25 (1028–1589) is a
guanine nucleotide releasing factor for mammalian ras proteins and
is oncogenic in NIH3T3 cells. J Biol Chem. 268:11113–11118.
1993.PubMed/NCBI
|
|
46
|
Leaner VD, Donninger H, Ellis CA, Clark GJ
and Birrer MJ: p75-Ras-GRF1 is a c-Jun/AP-1 target protein: its up
regulation results in increased Ras activity and is necessary for
c-Jun-induced nonadherent growth of Rat1a cells. Mol Cell Biol.
25:3324–3337. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Font de Mora J, Esteban LM, Burks DJ, et
al: Ras-GRF1 signaling is required for normal beta-cell development
and glucose homeostasis. EMBO J. 22:3039–3049. 2003.PubMed/NCBI
|
|
48
|
Guerrero C, Rojas JM, Chedid M, et al:
Expression of alternative forms of Ras exchange factors GRF and
SOS1 in different human tissues and cell lines. Oncogene.
12:1097–1107. 1996.PubMed/NCBI
|
|
49
|
Forlani G, Baldassa S, Lavagni P, Sturani
E and Zippel R: The guanine nucleotide exchange factor RasGRF1
directly binds microtubules via DHPH2-mediated interaction. FEBS J.
273:2127–2138. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Arozarena I, Matallanas D, Berciano MT, et
al: Activation of H-Ras in the endoplasmic reticulum by the RasGRF
family guanine nucleotide exchange factors. Mol Cell Biol.
24:1516–1530. 2004. View Article : Google Scholar : PubMed/NCBI
|