Introduction
Hedgehog (HH) signaling is important in development
and carcinogenesis because it controls cell fate and proliferation
(1,2). Three mammalian HH ligands [sonic
(sHH), desert, and indian] are lipid-modified secreted proteins
(1,2). The HH receptor Patched 1 (Ptch1)
inhibits this pathway by locking Smoothened (Smo) in an inactive
conformation. After HH ligand treatment, Ptch1 releases Smo
inhibition, which augments Gli family member (Gli1, Gli2, and Gli3)
expression (1,2). Gli1 and Gli2 typically exert
stimulatory while Gli3 has inhibitory effects (1). Activating the HH pathway affects
expression of Ptch1, cyclin D1, cyclin E, insulin-like growth
factor 2 (IGF2), insulin-like growth factor binding protein 6
(IGFBP6), GILZ and other species (2–5).
HH pathway deregulation occurs in many tumors
including lung (6), breast
(7), and pancreatic (8) cancers. Ptch1 mutations occur in the
Gorlin syndrome-associated cancers basal cell carcinoma (BCC) and
medulloblastoma (9–12). Smo inhibition can chemoprevent
invasive BCC (13). Cyclopamine, a
naturally-occurring HH antagonist, binds to Smo and inhibits HH
signaling (14). Other Smo
inhibitors exist with antineoplastic effects in vitro and in
clinical trials for patients with BCC or medulloblastoma (9–12).
The HH pathway regulates growth of small cell lung cancer (SCLC)
and non-small cell lung cancer (NSCLC) (6,15).
HH pathway members are abundantly expressed in the premalignant and
malignant lungs of cyclin E-expressing transgenic mice (16). Resistance to Smo inhibitors occurs
with acquired Smo mutations (17,18).
This study uncovered growth inhibitory responses to
Smo inhibition in diverse cancer cells using a robotic-based
platform with a genetic database. In this database Ptch1 and Smo
sequences were available with information about expression of
species associated with HH pathway activation. Basal expression of
these species in cancer cells was hypothesized to indicate growth
dependence of these cells on the HH pathway. It was hypothesized
that cancer cells expressing these species would respond to a Smo
inhibitor.
Multiple Smo inhibitors were studied in lung cancer
because the HH pathway is active in subsets of these cancers. Both
murine and human lung cancer cell lines exist. Cyclin E-driven
transgenic and transplantable murine lung cancer models that
spontaneously activated the HH pathway were available for study as
was a paired human normal-malignant lung tissue array with an
associated clinical database. The presented findings implicate use
of Smo inhibitors for lung and other cancers when a gene profile
indicative of HH pathway dependence is expressed in the cancer
cells.
Materials and methods
Cell culture
ED-1 and ED-2 murine lung cancer lines, C-10 murine
immortalized lung epithelial cells, BEAS-2B human immortalized
bronchial epithelial cells, and human lung cancer cell lines (A549,
HOP-62, H-522, U-1752, NCI-H1730, and NCI-H2122) were each cultured
in RPMI-1640 medium with 10% fetal bovine serum (FBS) and 1%
antibiotic and antimycotic solution at 37°C in 5% CO2 in
a humidified incubator, as before (15,16,19–21).
Cell lines were obtained from and authenticated (using genotypic
and phenotypic assays) by ATCC except for murine ED-1 and ED-2 lung
cancer cell lines that were previously described and authenticated
(19,21).
Chemicals
Cyclopamine (LC Laboratories, Wobrun, MA) and
tomatidine (Sigma-Aldrich, St. Louis, MO) were purchased as were
recombinant mouse sHH (R&D Systems, Minneapolis, MN) and FBS
(Gemini Bioproducts, Inc, Calabasas, CA). The Smo inhibitor MK-4101
(22) was provided by Merck. The
SANT-1 Smo inhibitor (15) was
purchased (Tocris Bioscience, Ellisville, MO) as was the SAG Smo
agonist (EMD Millipore, Billerica, MA).
Repression of HH pathway members
Cells were independently treated with the Smo
inhibitors: cyclopamine, SANT-1 and MK-4101. In vivo Smo
inhibition was achieved in mouse lung cancer models with
cyclopamine (intraperitoneal injections, 40 mg/kg) treatments or
with short hairpin RNA (shRNA)-mediated Smo knock-down in ED-1
cells. Individual small interfering RNA (siRNA)-mediated or
shRNA-mediated repression of Gli1, Gli2, or Gli3 was achieved.
High-throughput proliferation assays
Cyclopamine growth effects were investigated in 705
human cancer cell lines using a high-throughput screen (19,23,24).
Cells were treated with cyclopamine at 10 μM (and lower dosages) in
media with 5% FBS and were assayed at 72 h with quantification by
the SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Means of triplicate cyclopamine treatment experiments were compared
to vehicle controls, using optimized methods (19,23,24).
Smo inhibitor responses
The HH pathway affects expression of Ptch1, cyclin
D1, cyclin E, IGF2, IGFBP6, GILZ, Gli family members, and other
species (2–5). The cor.test function (25) of R (26) compared cyclopamine-dependent growth
responses to expressed species. Expression values were from U133
Plus 2.0 Affymetrix arrays and are publically available (27). The data set consisted of 490
samples corresponding to 164 unique cell lines that were typically
examined in triplicate. Correlations were done: a) using all
samples and b) using samples with a cyclopamine growth inhibitory
response ≤0.75 at the 10 μM dosage, corresponding to 110 samples
(in triplicate). Cyclopamine-mediated growth response was compared
with mutation data available from the Sanger database (28).
Ptch1 and smo sequence analyses
Sequencing of the coding regions of Ptch1 was
performed as before (29). For
murine cell lines, murine-specific primers were used to sequence
homologous domains of Ptch1. To assess for Smo mutations in murine
and human cell lines, prior mutations in human Smo were searched
for (A324T, V404M, D473H, E518K, W535L, and T640A) and
corresponding regions of murine Smo were sequenced.
Transient transfection, proliferation and
apoptosis assays
Logarithmically growing ED-1 (3×104 and
4.5×103), ED-2 (8×104), C-10
(1×105), BEAS-2B (3×105), A549
(5×104), HOP-62 (5×104), H-522
(3×105), U1752 (5×104), NCI-H1703
(4.5×105), and NCI-H2122 (6×105) cells were
individually plated onto each well of 6-well tissue culture plates
(BD Bioscience, San Jose, CA) 24 h before transfection or drug
treatments. Three replicate experiments were performed in
triplicate. Logarithmically growing cells were assayed using the
CellTiter-Glo assay (Promega, Madison, WI) (21). Apoptosis was scored with the
Caspase-Glo 3/7 Assay System (Promega).
SiRNA transfections were with Oligofectamine reagent
(Invitrogen, Carlsbad, CA). SiRNAs targeting Gli1, Gli2, Gli3, Smo,
IGFBP6 or a RISC-free control were each purchased (Dharmacon,
Lafayette, CO). Two different siRNAs independently targeted Gli1,
Gli2, Gli3, Smo, or IGFBP6 sequences: 5′-GGUUGGAACUUCUGUGAUG-3′
(Gli1.1), 5′-GAGCAGGCCUCCGUUGUA-3′ (Gli1.2),
5′-GGGAGAAGAAGGAGUUCGU-3′ (Gli2.1), 5′-GGUUUGUGGUUGAGCGGAA-3′
(Gli2.2), 5′-CCAUCGGUGGAAAAGCGU-3′ (Gli3.1),
5′-GAAACGCAAUCACUAUGCA-3′ (Gli3.2), 5′-AGAACCCGCUGUUCACCGA-3′
(Smo1.1), 5′-GCAUCUGUUUUGUAGGCUA-3′ (Smo1.2),
5′-CAUCGAGGCUUCUACCGGA-3′ (IGFBP6-1), and 5′-CAACAGAGGAAUCCAGGCA-3′
(IGFBP6-2). Transient transfections were with FuGENE 6 reagent
(Roche Applied Science, Indianapolis, IN). Reporter assays were
performed after transfection of a GLIBS-Luciferase reporter
construct or TK-Luciferase control plasmid (19). The Dual-Luciferase reporter assay
system (Promega) was used in three replicate experiments, each
performed in triplicate.
Stable knock-down
ED-1 cells stably expressing green fluorescent
protein (GFP) were individually infected with lentivirus expressing
shRNAs targeting Smo: TRCN0000026245 (Smo1) and TRCN0000026295
(Smo2); IGFBP6: TRCN0000114766 (IGFBP6.1) and TRCN0000114768
(IGFBP6.2); GILZ: TRCN0000085743 (GILZ.1) and TRCN0000085746
(GILZ.2), or a scrambled control shRNA (RHS4080) (Open Biosystems,
Huntsville, AL). Selection was with puromycin (5 μg/ml).
Knock-downs were determined using semi-quantitative real-time
reverse transcription (RT) polymerase chain reaction (PCR) assays.
Cells with the most robust Smo knock-down versus controls were
selected for tail vein injection experiments into syngeneic mice
(19,20,30).
Real-time RT-PCR assays
Total RNA was isolated using the RNeasy kit (Qiagen,
Valencia, CA). RT was with the High Capacity cDNA RT kit (Applied
Biosystems, Foster City, CA) and a Peltier Thermal Cycler (MJ
Research, Waltham, MA). Semi-quantitative real-time RT-PCR assays
were with SYBR Green PCR Mastermix (Applied Biosystems) and the
7500 Fast real-time PCR system (Applied Biosystems). Human IGFBP6
and β-actin qPCR assays were performed with TaqMan Universal PCR
Master Mix and the manufacturer’s described assays (Applied
Biosystems). Three replicate experiments were performed. Primers
were: murine Gli1 forward: 5′-CCAAGCCAACTTTATGTCAGGG-3′, and
reverse: 5′-AGCCCGCTTCTTTGTTAATTTGA-3′; murine Gli2 forward:
5′-CAACGCCTACTCTCCCAGAC-3′, and reverse:
5′-GAGCCTTGATGTACTGTACCAC-3′; murine Gli3 forward:
5′-CACAGCTCTACGGCGACTG-3′, and reverse:
5′-CTGCATAGTGATTGCGTTTCTTC-3′; murine Smo forward:
5′-GAGCGTAGCTTCCGGGACTA-3′, and reverse:
5′-CTGGGCCGATTCTTGATCTCA-3′; murine IGFBP6 forward:
5′-TGCTAATGCTGTTGTTCGCTG-3′, and reverse:
5′-CACGGTTGTCCCTCTCTCCT-3′; murine GILZ forward:
5′-ACCACCTGATGTACGCTGTG-3′, and reverse:
5′-TCTGCTCCTTTAGGACCTCCA-3′; murine glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) forward: 5′-AGGTCGGTGTGAACGGATTTG-3′, and
reverse: 5′-TGTAGACCATGTAGTTGAGGTCA-3′; human Gli1 forward:
5′-GGCACCATGAGCCCATCTC-3′, and reverse:
5′-ATCACCTTCCAAGGGTTCCTC-3′; human Gli2 forward:
5′-GGTGAAGCCTCCACCCTTTC-3′, and reverse:
5′-TGCATGTAGTTTACCCTGGGG-3′; human Gli3 forward:
5′-CTCCACGACCACTGAAAAGAAA-3′, and reverse:
5′-TCTCTGTGATAAGTCTGTCCAGG-3′; human Smo forward:
5′-GGCAACAGCATTGCAGTGAAG-3′, and reverse:
5′-GAGGAGAGACACACGAGCCT-3′; and human GAPDH forward:
5′-ATGGGGAAGGTGAAGGTCG-3′, and reverse:
5′-GGGGTCATTGATGGCAACAATA-3′.
Clonal growth assays
Two hundred logarithmically growing ED-1 cells were
plated for triplicate replicate clonal growth assays, as before
(20). Colonies were stained with
Diff Quick (IMEB Inc, San Marcos, CA) and were counted with the
Oxford Optronix Col Count counter (Oxford Optronix, Oxford, UK)
(31).
Normal-malignant lung tissue array
The New Hampshire State Cancer Registry and the
Dartmouth-Hitchcock Medical Center Tumor Registry have been
described (20). Analyses of
normal versus malignant lung tissue arrays were by a pathologist
(Vincent A. Memoli) unaware of findings from the associated
clinical database. Signed consents were obtained; studies were
reviewed and approved by Dartmouth’s Institutional Review Board
(IRB) for human subjects. A paired normal-malignant lung tissue
microarray was constructed (20).
Statistical analysis
Results were expressed as means ± standard
deviations. Results of all independent experiments were pooled to
assess for statistical significance. Z-test and two-sided t-tests
were used for statistical analyses with significance considered for
values of P<0.05.
Cyclopamine treatments of transgenic
mice
Three 9-month-old female mice expressing transgenic
wild-type human cyclin E were each treated daily (intraperitoneal)
for 5 consecutive days with cyclopamine (40 mg/kg) or vehicle (45%
hydroxypropyl-ß-cyclodextrin, HBS) with a total of six mice
studied. Mice were examined using an Institutional Animal Care and
Use Committee (IACUC)-approved protocol. Tissues were
formalin-fixed, paraffin-embedded and sectioned for histopathology
(20). Hematoxylin and eosin
staining as well as Ki-67, cyclin D1, and cyclin E immunostaining
were performed as before (16,19).
Analyses were by a pathologist (Vincent A. Memoli) unaware whether
harvested tissues were from cyclopamine or vehicle-treated
mice.
Murine syngeneic lung cancer
transplantation assays
ED-1 cells were transduced with a GFP expression
vector and transductants were sorted (15,16)
with 8×105 cells injected into tail veins of each female
FVB mouse (8-week old). Ten mice per arm were used and replicate
experiments were performed. To investigate cyclopamine
antineoplastic effects, 10 mice were each intraperitoneally treated
daily for 14 days with cyclopamine (40 mg/kg); 10 additional mice
were each treated with vehicle (45% HBS). Treatments began 2 weeks
after tail-vein injections because lung tumors formed by then (data
not shown). Ten syngeneic FVB mice were each injected with ED-1
cells that were treated with either MK-4101 (10 μM) or vehicle as
part of an IACUC-approved protocol. Mice were sacrificed 28 days
post-tail vein injections. Replicate experiments were performed.
Lung tissues were formalin-fixed, paraffin-embedded and sectioned
for histopathology (16,19,20).
A rabbit polyclonal anti-GFP antibody (product ab290) (Abcam,
Cambridge, MA) was used to identify lung tumors. Hematoxylin
counterstaining was used.
Results
Growth effects of a Smo inhibitor were
studied using a robotic-based platform linked to a genetic database
(19,23,24)
A total of 705 different human cancer lines were
examined for responses to the Smo inhibitor cyclopamine (10 μM).
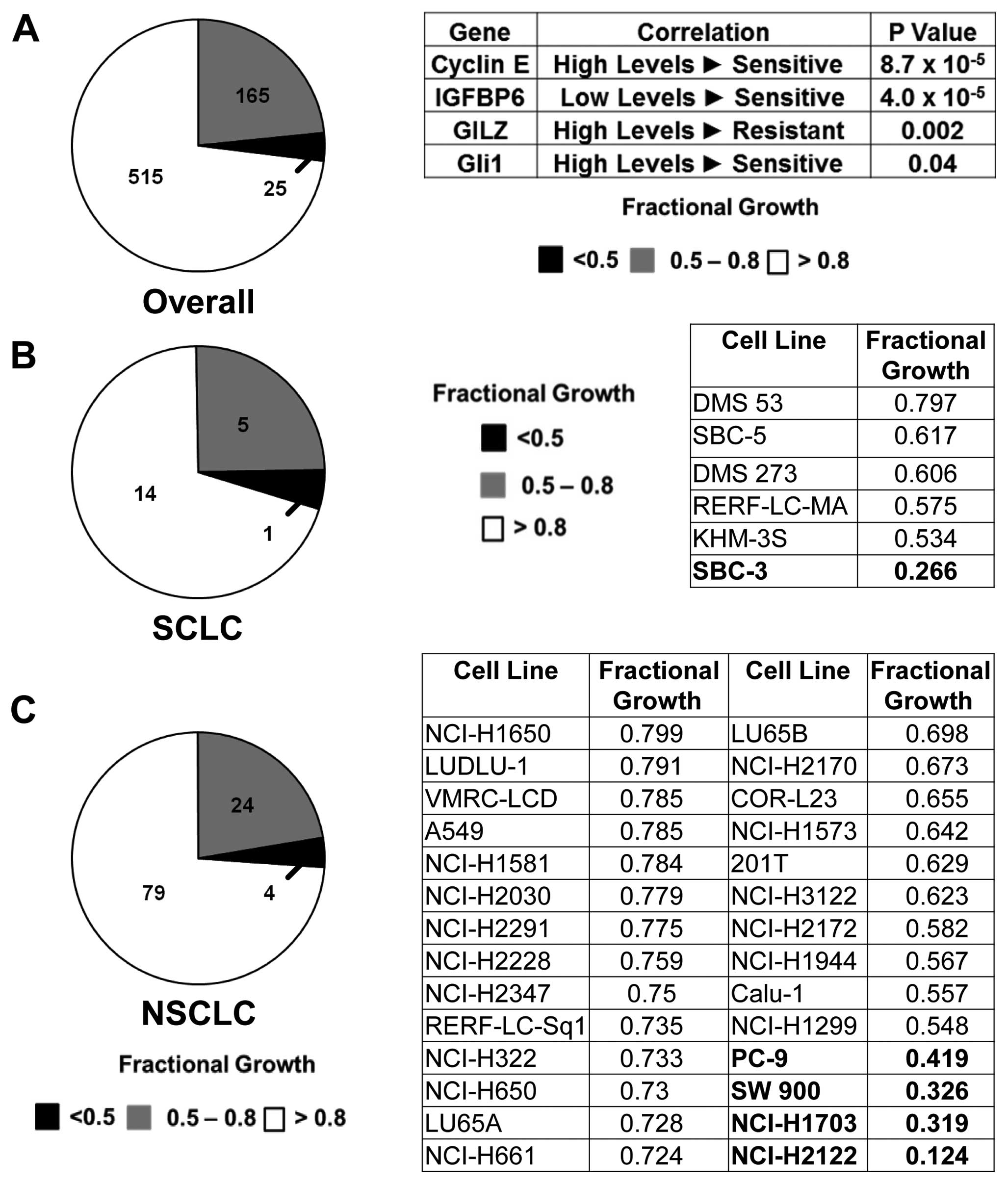
Proliferation was reduced in a subset of cancer cell lines after 72
h of treatment (Fig. 1A, left
panel). Of these lines, 353 were displayed in the Sanger database.
No Ptch1 or Smo mutations were detected (3 lines had Smo
amplifications that did not confer cyclopamine hypersensitivity).
Data from the Sanger database showed the distribution of somatic
mutations in Ptch1 for 860 unique cancer cell lines. Six had
variant sequences of uncertain functional consequences. For the Smo
gene, only 6 out of 866 different cancer cells had sequence
variants of uncertain functional consequence. There were 152 lung
cancer cell lines analyzed. None of these lines had Ptch1 or Smo
mutations.
The HH pathway can affect expression of the Gli
family members, Ptch1, cyclin D1, cyclin E, IGF2, IGFBP6, or GILZ
(1–5). Cyclopamine-dependent growth responses
were compared to these expressed species using the described
database. Most highly significant associations occurred for high
cyclin E (P=0.000009) and low IGFBP6 (P=0.000004) levels (Fig. 1A). Less significant associations
were detected for IGF2 (P=0.005), cyclin D1 (P=0.024), Gli1
(P=0.04), and Gli2 (P=0.05). High GILZ levels were linked to
reduced cyclopamine response (P= 0.002).
Twenty lines were of SCLC and 107 were of NSCLC
origins. Subsets of SCLC (Fig. 1B)
and NSCLC (Fig. 1C) lines
responded to cyclopamine with substantial growth inhibition. NSCLC
responses occurred independently of mutations for Ptch1 or Smo
(Table IA). These responses were
also independent of ras, epidermal growth factor receptor
(EGFR), or p53 mutations (Table IB). K-RAS mutations in NSCLC
are associated with resistance to EGFR-tyrosine kinase inhibitors
(TKIs) (32–35). Growth inhibition in the most
cyclopamine-responsive NSCLC cells lines (NCI-H1703 and NCI-H2122)
were independently confirmed in Fig.
2.
 | Table I.The (A) Ptch1 and Smo mutation status
and (B) ras, p53, and activating EGFR mutation status of the 20
most cyclopamine-sensitive NSCLC cell lines examined in this
study. |
Table I.
The (A) Ptch1 and Smo mutation status
and (B) ras, p53, and activating EGFR mutation status of the 20
most cyclopamine-sensitive NSCLC cell lines examined in this
study.
| A, Ptch1 and Smo
mutation status |
|---|
| Cell line | Ptch1 status | Smo status |
|---|
| NCI-H2122 | Wild-type | Wild-type |
| NCI-H1703 | Wild-type | Wild-type |
| SW900 | Wild-type | Wild-type |
| PC-9 | Not known | Not known |
| NCI-H1299 | Wild-type | Wild-type |
| Calu-1 | Wild-type | Wild-type |
| NCI-H1944 | Not known | Not known |
| NCI-H2172 | Not known | Not known |
| NCI-H3122 | Not known | Not known |
| 201T | Not known | Not known |
| NCI-H1573 | Wild-type | Wild-type |
| COR-L23 | Wild-type | Wild-type |
| NCI-H2170 | Wild-type | Wild-type |
| LU65B | Not known | Not known |
| NCI-H611 | Wild-type | Wild-type |
| LU65A | Not known | Not known |
| NCI-H650 | Wild-type | Wild-type |
| NCI-H322 | Wild-type | Wild-type |
| RERF-LC-Sq1 | Not known | Not known |
| NCI-H2347 | Wild-type | Wild-type |
| B, ras, p53, and
activating EGFR mutation status |
|---|
| Cell line | Ras status | EGFR status | p53 status |
|---|
| NCI-H2122 | Mutant (K-RAS) | Wild-type | Mutant |
| NCI-H1703 | Wild-type | Wild-type | Mutant |
| SW900 | Mutant (K-RAS) | Wild-type | Mutant |
| PC-9 | Wild-type | Mutant | Mutant |
| NCI-H1299 | Mutant (N-RAS) | Wild-type | Wild-type |
| Calu-1 | Mutant (K-RAS) | Wild-type | Wild-type |
| NCI-H1944 | Mutant (K-RAS) | Not known | Wild-type |
| NCI-H2172 | Not known | Not known | Not known |
| NCI-H3122 | Not known | Wild-type | Not known |
| 201T | Wild-type | Wild-type | Not known |
| NCI-H1573 | Mutant (K-RAS) | Wild-type | Mutant |
| COR-L23 | Mutant (K-RAS) | Wild-type | Wild-type |
| NCI-H2170 | Wild-type | Wild-type | Mutant |
| LU65B | Not known | Not known | Mutant |
| NCI-H611 | Wild-type | Wild-type | Mutant |
| LU65A | Not known | Not known | Mutant |
| NCI-H650 | Mutant (K-RAS) | Wild-type | Mutant |
| NCI-H322 | Wild-type | Wild-type | Mutant |
| RERF-LC-Sq1 | Not known | Wild-type | Not known |
| NCI-H2347 | Mutant (N-RAS) | Wild-type | Wild-type |
Studies were next conducted in lung cancer because
an autocrine HH pathway is reported in lung cancer (36). HH signaling is important for growth
of NSCLC and SCLC (6,15). Other reasons for studying lung
cancer included the finding that increased cyclin E expression was
associated with cyclopamine response (Fig. 1A) and cyclin E transgenic mice
recapitulated key features of human lung cancer biology (16,30).
Gain or loss of expression was achieved for the most
significantly associated candidate regulators of cyclopamine
response (cyclin E and IGFBP6) or resistance (GILZ). Gli1 is an
established HH pathway regulator in lung cancer (15). To determine whether cyclin E
affected cyclopamine response, cyclin E was overexpressed in murine
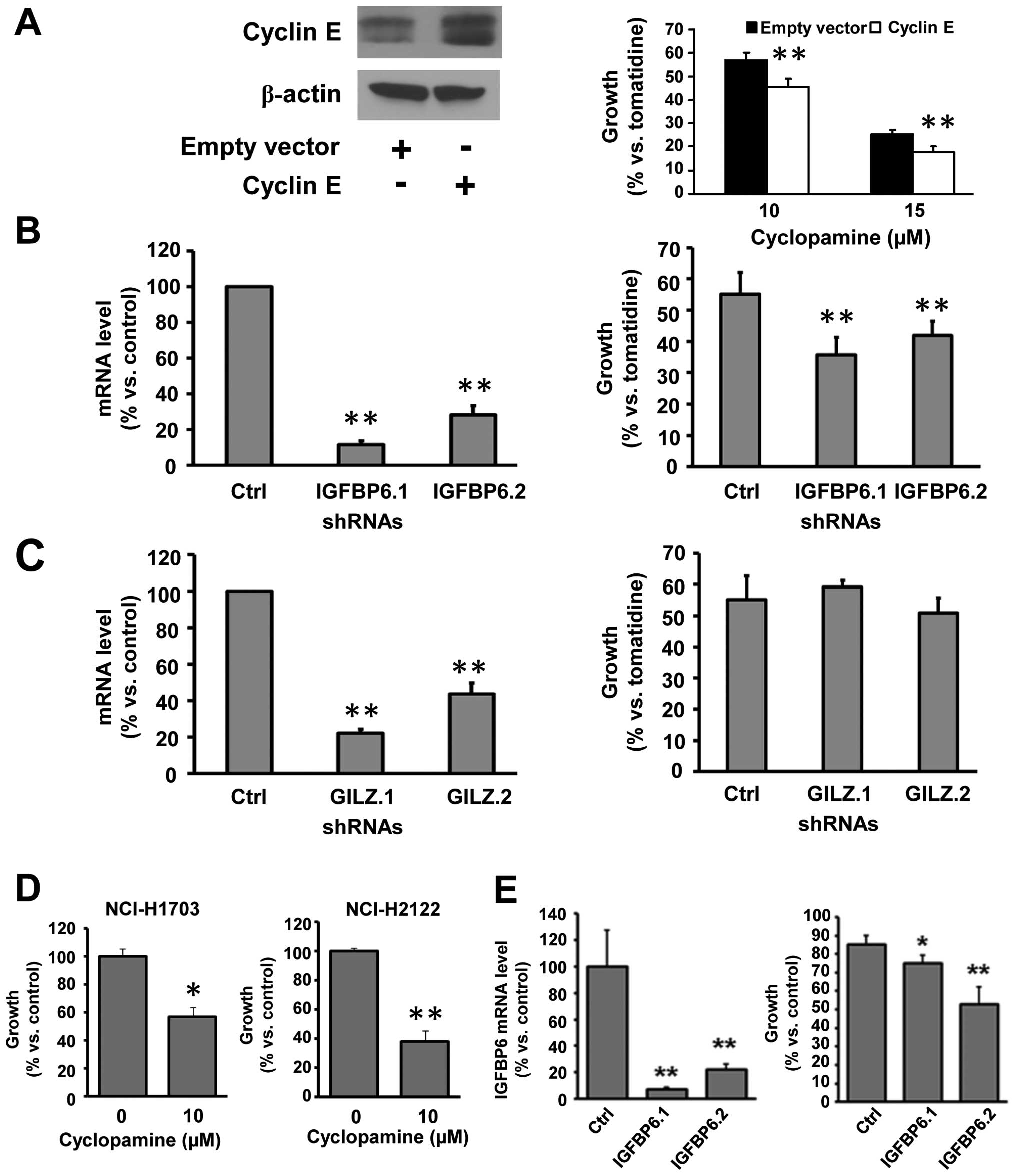
immortalized C-10 lung epithelial cells (Fig. 2A, left panel shows a 2.26-fold
increase in cyclin E protein versus actin expression) and this
significantly (P<0.01) increased cyclopamine-mediated growth
inhibition versus the inactive isomer tomatidine (Fig. 2A, right panel). Similar results
were obtained using a second Smo inhibitor, SANT-1 (data not
shown), which was consistent with prior studies (15).
Two shRNAs (Fig.
2B, left panel) were selected to reduce IGFBP6 expression
independently in ED-1 lung cancer cells derived from a transgenic
mouse that expressed wild-type cyclin E (30). Individual knock-down with these
shRNAs significantly (P<0.01) increased Smo-mediated growth
inhibition in this murine lung cancer cell line versus an inactive
shRNA control (Fig. 2B, right
panel). Thus, gain of cyclin E or loss of IGFBP6 expression
regulated response to a Smo inhibitor. In contrast, two different
shRNAs independently repressed GILZ expression (Fig. 2C, left panel), but these did not
appreciably affect cyclopamine-mediated growth suppression at 72 h
(Fig. 2C, right panel). Findings
were confirmed in human NSCLC cells engineered with IGFBP6
knock-down (Fig. 2D and E).
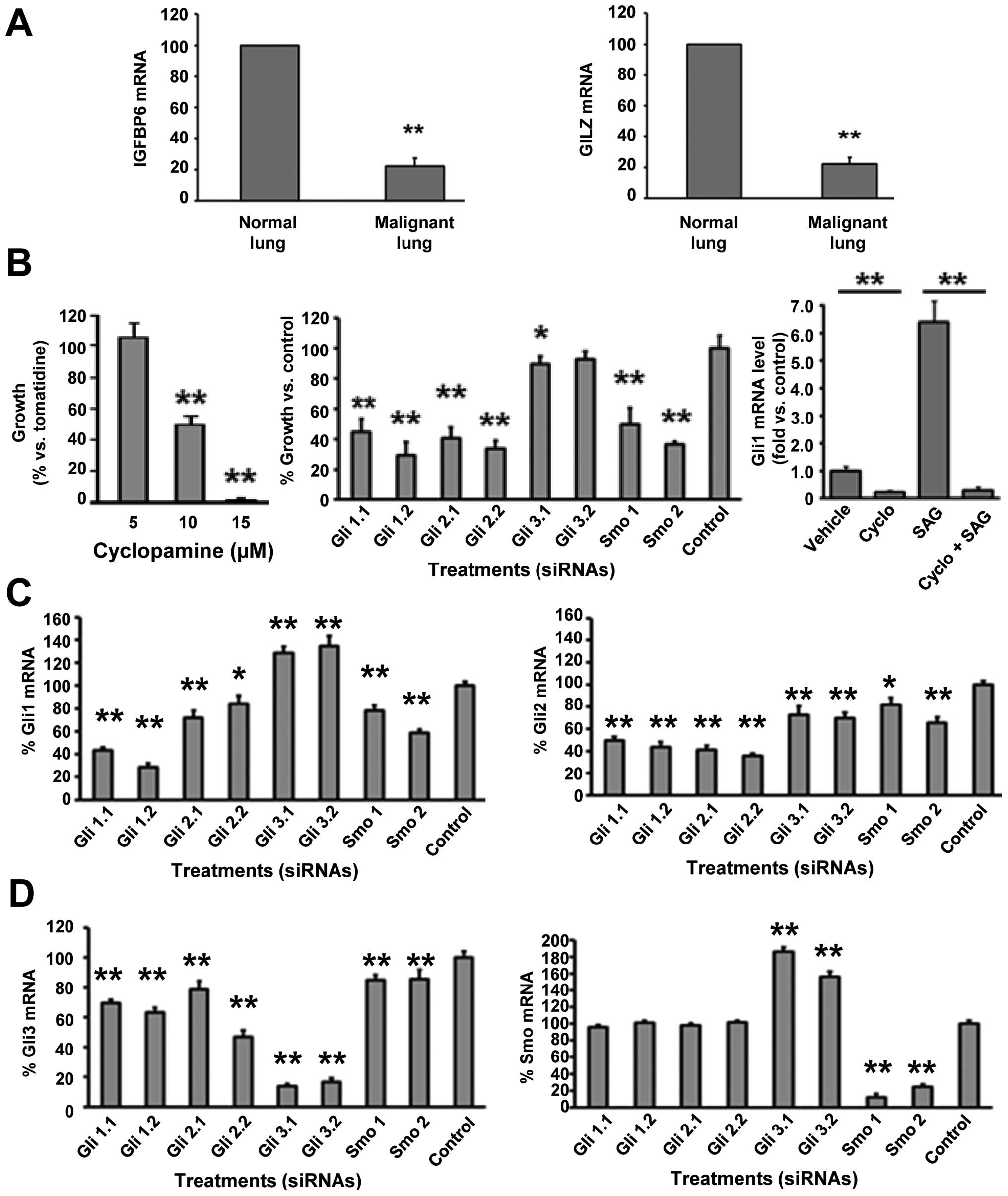
Lung cancers arising in transgenic mice from
increased cyclin E expression in the lung exhibit high levels of
Gli1 (16,30). These transgenic lung cancers are
also shown to express low levels of IGFBP6 and GILZ versus the
adjacent normal lung (Fig. 3A).
Whether cell lines derived from these murine lung cancers responded
to a Smo inhibitor was independently studied in ED-1 and ED-2 lung
cancer cells derived respectively from transgenic mice whose lung
tumors expressed wild-type (ED-1) or degradation-resistant (ED-2)
cyclin E species (30).
Effects of Smo inhibition were explored by targeting
Smo in ED-1 cells. Gli1, Gli2, Gli3, and Smo were independently
targeted using two different siRNAs engineered to repress each of
these species. Independent knock-down of Gli1, Gli2, and Smo each
significantly inhibited ED-1 cell growth (as did cyclopamine
treatment), but Gli3 knock-down did not appreciably affect growth
(Fig. 3B, left and middle panels).
As expected, the Smo agonist SAG augmented Gli1 expression;
cyclopamine co-treatment prevented this (Fig. 3B, right panel). Compensatory
changes in expressed HH pathway members were found after these
different knock-downs in Fig. 3C and
D. Smo knock-down substantially decreased lung cancer cell
growth.
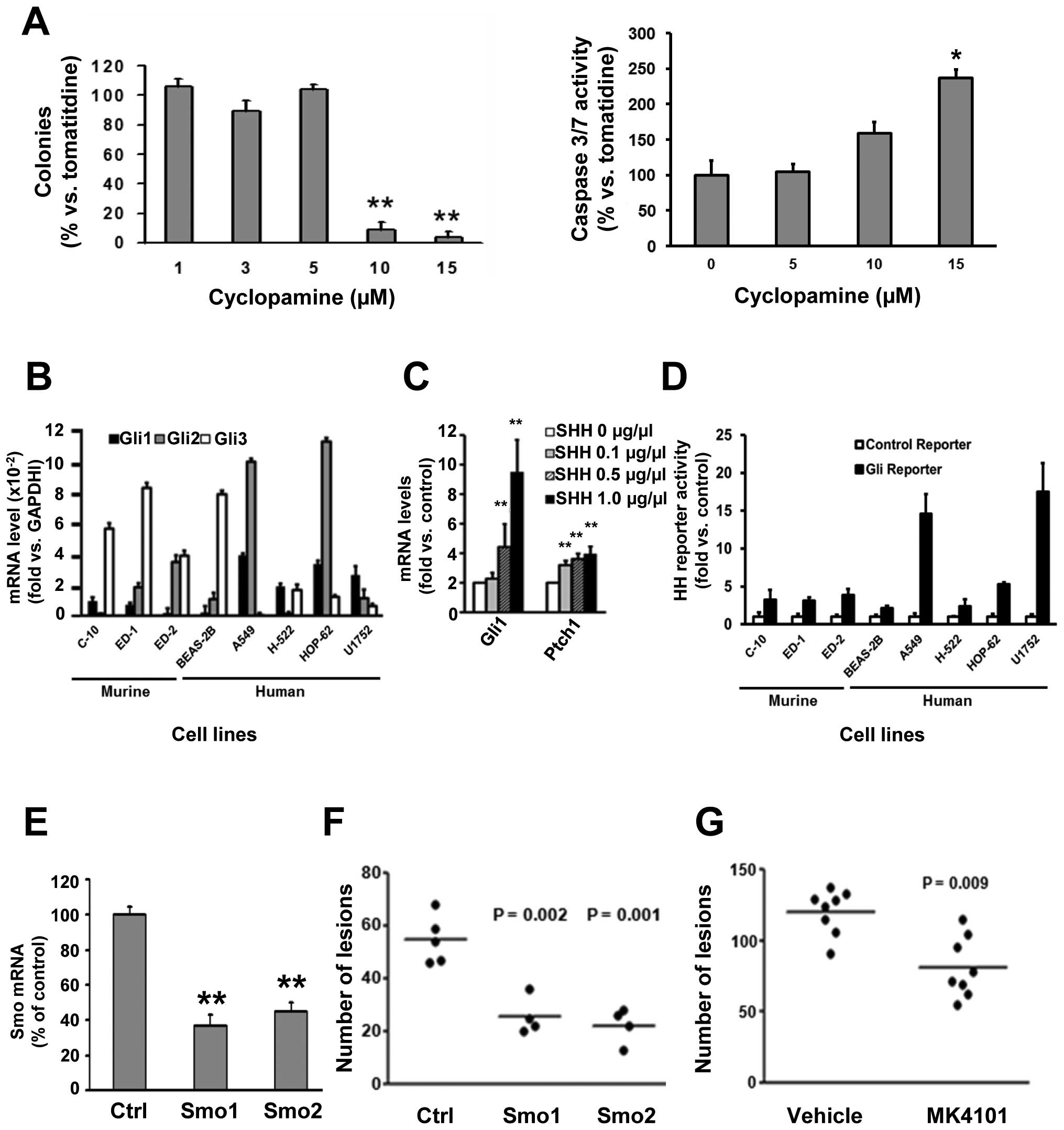
Cyclopamine treatment also significantly (P<0.01)
decreased ED-1 clonal growth while increasing apoptosis (Fig. 4A). ED-1 and ED-2 cells expressed
multiple HH pathway components as did the examined murine and human
immortalized pulmonary epithelial or cancer cell lines (Fig. 4B). Genomic DNA from each of these
cell lines was sequenced for the entire coding region of Ptch1 and
for Smo mutations in regions that conferred Smo inhibitor
resistance (A324T, V404M, D473H, E518K, W535L, and T640A). No
mutations were detected (data not shown). Human lung cancer cell
lines studied were those in which responses to Smo inhibition were
already reported (15,36). To establish whether the HH pathway
was functional in ED-1 cells, these cells were treated with
recombinant sHH at different dosages in low serum (0.5%) containing
medium. HH pathway activity in these cells was confirmed; sHH
treatment significantly (P<0.01) increased mRNA expression for
Gli1 and Ptch (Fig. 4C). Although
ED-1 cells arose from transgenic lung cancers that activated the HH
pathway (16), these cells had
relatively low basal Gli1 and Gli2 mRNA levels (Fig. 4B) and low basal Gli1-reporter
activity (Fig. 4D). This is
consistent with differences reported between ex vivo versus
in vivo HH pathway dependence of lung cancer cells (37). It was therefore not unexpected to
detect prominent Gli1 immunostaining of lung tumors arising from
ED-1 cells after their tail-vein injections into syngeneic FVB mice
(data not shown).
To explore independently Smo inhibitor effects on
lung cancer cells, ED-1 cells were engineered to have Smo
knockdown. This was done using two independent shRNAs versus an
inactive control shRNA (Fig. 4E).
This conferred significant repression (Smo1 shRNA P=0.002 and Smo2
shRNA P=0.001) of transplanted lung cancers (Fig. 4F). Ex vivo treatment of ED-1
cells with another Smo inhibitor MK-4101 significantly reduced ED-1
colony formation (P<0.01, data not shown) and tumorigenicity
(P=0.009) after tail-vein injections into syngeneic mice of these
versus control cells (Fig.
4G).
Cyclopamine responses were examined in cyclin E
transgenic mice because their lung cancers expressed a gene profile
that was indicative of dependence on HH signaling.
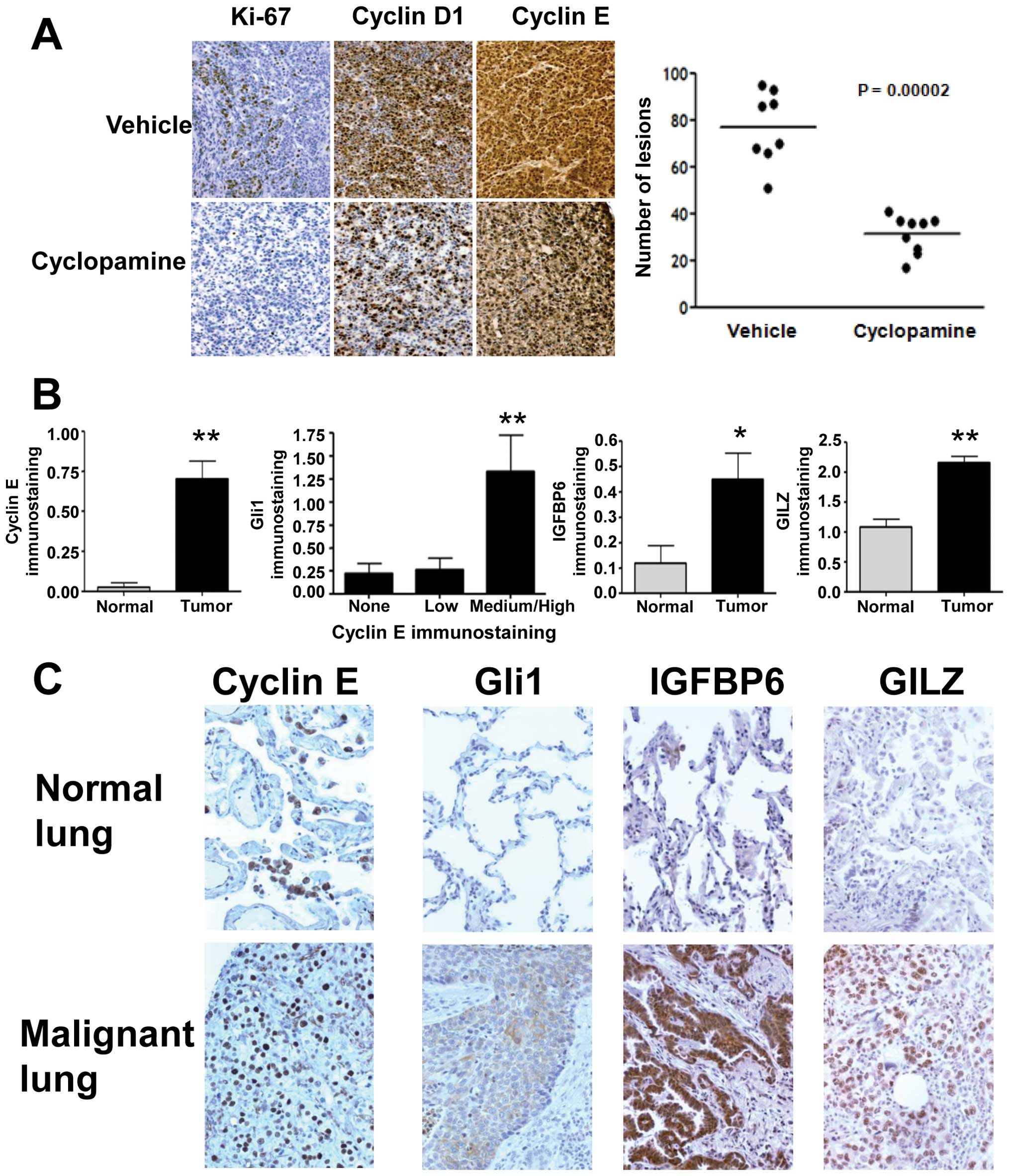
Cyclopamine-treatment decreased immunohistochemical expression of
the proliferation markers Ki-67, cyclin D1 and cyclin E in the
neoplastic versus normal lungs of treated versus age and
sex-matched control mice (Fig. 5A,
left panel). This result suggested that Smo inhibition would also
reduce lung tumor formation in the transplantation model. ED-1
cells were injected into the tail veins of syngeneic mice.
Cyclopamine treatment of mice began after lung tumors were
histologically present (data not shown). This significantly
(P=0.00002) reduced lung cancers in cyclopamine versus
vehicle-treated mice (Fig. 5A,
right panel).
To learn whether a profile indicative of HH pathway
dependence was expressed in human lung cancers, a normal-malignant
lung tissue array (20) was
studied. Immunohistochemical expression profiles revealed
significant differences in the malignant versus normal lung for
cyclin E (P<0.01), IGFBP6 (P<0.01), and GILZ (P<0.01) in
Fig. 5B. Lung cancers with high
cyclin E levels expressed significantly (P<0.01) higher levels
of Gli1 than did cases with reduced cyclin E levels (Fig. 5B). Cases with high cyclin E and low
IGFBP6 levels had increased (P<0.0001) Gli1 immunohistochemical
expression. Representative results are presented (Fig. 5C).
Discussion
Smo inhibitors are active against Gorlin
syndrome-associated BCC or medulloblastoma where Ptch mutations
occur (9–13). Smo mutations confer resistance to
Smo inhibitors (17,18). This study comprehensively
interrogated 705 epithelial cancer cell lines for growth response
to the Smo inhibitor cyclopamine. Findings were compared with
expressed HH pathway-regulated species using a linked genetic
database. Ptch and Smo mutations that respectively conferred Smo
inhibitor response or resistance were undetected. Rare variant
sequences were found, but their functional impact was not
established. Because HH pathway activation occurs in lung cancers,
findings were validated using different Smo inhibitors in human
lung cancer cell lines, transgenic and transplantable murine lung
cancer models, and paired normal-malignant lung tissue arrays.
Ptch1 or Smo mutations were undetected in examined murine and human
immortalized lung epithelial and cancer cell lines.
Smo inhibitor growth response was most significantly
associated with high cyclin E (P= 0.000009), low IGFBP6
(P=0.000004), and high Gli1 (P=0.04) levels; high GILZ levels were
associated with reduced response (P= 0.002) in Fig. 1. This profile implicated a basal
dependence on the HH pathway for the growth of these cancer cells.
This possibility was validated in murine and human lung cancer cell
lines and transgenic as well as transplantable murine lung cancer
models in Figs. 1–5. Functional consequences were shown
using several pharmacological and genetic Smo inhibitors.
Differential expression of the same species was
observed in a malignant versus normal human lung tissue array
(Fig. 5). A profile indicative of
Smo inhibitor response was also observed in murine lung cancers
(Fig. 3). While high IGFBP6 levels
were present in some lung tumors, it is notable that lung cancers
with both high cyclin E and reduced IGFBP6 expression significantly
(P<0.0001) increased Gli1 expression. This pattern was one
indicating response to a Smo inhibitor. This points out the
potential clinical need to discern profiles of HH pathway-regulated
species in human tumors to learn which are likely to be responsive
to a Smo inhibitor. Future clinical trials that explore activity of
Smo inhibitors should also determine these profiles to uncover
possible HH pathway dependence.
Several Smo inhibitors conferred similar effects.
Stromal effects are engaged to confer some of these anti-neoplastic
effects. This could account for differences between in vitro
and in vivo effects of Smo inhibition (37,38).
The findings presented in Fig. 1
could explain why trials with Smo inhibitors might underestimate
clinical anti-tumor effects of Smo inhibition. Only some cancers
express a gene profile indicating possible HH pathway dependence.
Notably, autocrine HH pathway signaling occurs in lung cancers
(36). The findings reported here
are consistent with this prior study.
These findings have implications for combination
cancer therapy. Responses to Smo inhibition occurred whether or not
RAS or p53 (or Ptch1 or Smo) mutations
were present in cancer cells (Table
I). In clinical lung cancers, K-RAS mutations confer
resistance to EGFR-TKIs (32,34,35).
Yet, clinical trials revealed activity against lung cancers having
K-RAS mutations when an EGFR-TKI was combined with a
rexinoid (33). This clinical
activity was associated with reduced cyclin D1 expression in
post-treatment lung cancer biopsies (33). In the present study, in vivo
responses to Smo inhibition were linked to cyclin D1 repression
(Fig. 5). Adding a Smo inhibitor
to a regimen that targets cyclin D1 for repression might enhance
clinical anti-tumor activity. Smo responses were also associated
with cyclin E expression (Figs. 1
and 5). Targeting the cyclin
E-cdk2 complex exerted anti-tumor responses despite presence of
K-RAS mutations by inducing anaphase catastrophe (19,39).
Combining a cdk2 antagonist with a Smo inhibitor might augment
anti-neoplastic activity.
High Smo inhibitor dosages were associated with
these anti-neoplastic effects. A similar dose-response relationship
for HH inhibition was observed in different tumor contexts and this
might depend on expressed drug transporters (40,41).
It is notable that the findings displayed in Fig. 3B (right panel) using a Smo agonist
argue against off-target effects of cyclopamine. The reduction of
lung tumors after Smo inhibition in the transplantation model
reported here is notable since these lung cancers did not have
Ptch1 or Smo mutations. Smo inhibitors might treat or prevent other
cancers that lack these mutations.
Taken together, findings presented here indicate a
Smo inhibitor should be considered in cancers that lack Smo or
Ptch1 mutations. This is especially the case when the tumors
express a gene profile indicating basal activation of the HH
pathway. This could implicate a dependence on the HH pathway for
growth or survival of the same tumors. Future clinical research
should explore the translational consequences of these findings for
cancer therapy and prevention.
Acknowledgements
This study was supported by National
Institutes of Health (NIH) and National Cancer Institute (NCI)
grants R01-CA087546 (E.D.), R01-CA111422 (E.D.), R03-CA132166
(E.D.), a Samuel Waxman Cancer Research Foundation award (E.D.), a
research grant from Merck (E.D.) and an American Cancer Society
Clinical Research Professorship (E.D.) supported by a gift from the
FM Kirby Foundation. A.M.B. was supported by an NIH National
Research Service Award (T32-CA009658). We thank Dr Jason Sparkowski
(Merck) for providing MK-4101.
References
|
1.
|
PW InghamAP McMahonHedgehog signaling in
animal development: paradigms and principlesGenes
Dev1530593087200110.1101/gad.93860111731473
|
|
2.
|
M Pasca di MaglianoM HebrokHedgehog
signaling in cancer formation and maintenanceNat Rev
Cancer3903911200314737121
|
|
3.
|
H ZhuH-W LoThe human glioma-associated
oncogene homolog 1 (GLI1) family of transcription factors in gene
regulation and diseasesCurr
Genomics11238245201010.2174/13892021079123310821119888
|
|
4.
|
WJ IngramCA WickingSM GrimmondAR ForrestBJ
WainwrightNovel genes regulated by sonic hedgehog in pluripotent
mesenchymal
cellsOncogene2181968205200210.1038/sj.onc.120597512444557
|
|
5.
|
M Duman-ScheelL WengS XinW DuHedgehog
regulates cell growth and proliferation by inducing cyclin D and
cyclin ENature417299304200210.1038/417299a12015606
|
|
6.
|
DN WatkinsDM BermanSG BurkholderB WangPA
BeachySB BaylinHedgehog signaling within airway epithelial
progenitors and in small-cell lung
cancerNature422313317200310.1038/nature0149312629553
|
|
7.
|
M KuboM NakamuraA TasakiHedgehog signaling
pathway is a new therapeutic target for patients with breast
cancerCancer
Res6460716074200410.1158/0008-5472.CAN-04-041615342389
|
|
8.
|
SP ThayerMP di MaglianoPW HeiserHedgehog
is an early and late mediator of pancreatic cancer
tumorigenesisNature425851856200310.1038/nature0200914520413
|
|
9.
|
CM RudinCL HannJ LaterraTreatment of
medulloblastoma with hedgehog pathway inhibitor GDC-0449N Engl J
Med36111731178200910.1056/NEJMoa090290319726761
|
|
10.
|
I CaroJA LowThe role of the hedgehog
signaling pathway in the development of basal cell carcinoma and
opportunities for treatmentClin Cancer
Res1633353339201010.1158/1078-0432.CCR-09-257020439455
|
|
11.
|
DD Von HoffPM LoRussoCM RudinInhibition of
the hedgehog pathway in advanced basal-cell carcinomaN Engl J
Med36111641172200919726763
|
|
12.
|
JA LowFJ de SauvageClinical experience
with hedgehog pathway inhibitorsJ Clin
Oncol2853215326201010.1200/JCO.2010.27.994321041712
|
|
13.
|
JY YangAA MarghoobEmerging treatments and
signaling pathway inhibitorsSemin Cutan Med Surg31S14S182011
|
|
14.
|
JK ChenJ TaipaleMK CooperPA
BeachyInhibition of hedgehog signaling by direct binding of
cyclopamine to smoothenedGenes
Dev1627432748200210.1101/gad.102530212414725
|
|
15.
|
Z YuanJA GoetzS SinghFrequent requirement
of hedgehog signaling in non-small cell lung
carcinomaOncogene2610461055200710.1038/sj.onc.120986016909105
|
|
16.
|
Y MaS FieringC BlackTransgenic cyclin E
triggers dysplasia and multiple pulmonary adenocarcinomasProc Natl
Acad Sci USA10440894094200710.1073/pnas.060653710417360482
|
|
17.
|
GJP DijkgraafB AlickeL WeinmannSmall
molecule inhibition of GDC-0449 refractory smoothened mutants and
downstream mechanisms of drug resistanceCancer
Res71435444201110.1158/0008-5472.CAN-10-287621123452
|
|
18.
|
RL YauchGJP DijkgraafB AlickeSmoothened
mutation confers resistance to a hedgehog pathway inhibitor in
medulloblastomaScience326572574200910.1126/science.117938619726788
|
|
19.
|
F GalimbertiSL ThompsonX LiuTargeting the
cyclin E-cdk-2 complex represses lung cancer growth by triggering
anaphase catastropheClin Cancer
Res16109120201010.1158/1078-0432.CCR-09-215120028770
|
|
20.
|
X LiuLF SempereH OuyangMicroRNA-31
functions as an oncogenic microRNA in mouse and human lung cancer
cells by repressing specific tumor suppressorsJ Clin
Invest12012981309201010.1172/JCI3956620237410
|
|
21.
|
X LiuLF SempereF GalimbertiUncovering
growth-suppressive microRNAs in lung cancerClin Cancer
Res1511771183200910.1158/1078-0432.CCR-08-135519228723
|
|
22.
|
S GuptaN TakebeP LorussoTargeting the
hedgehog pathway in cancerTher Adv Med
Oncol2237250201010.1177/1758834010366430
|
|
23.
|
U McDermottSV SharmaL DowellIdentification
of genotype-correlated sensitivity to selective kinase inhibitors
by using high-throughput tumor cell line profilingProc Natl Acad
Sci USA1041993619941200710.1073/pnas.070749810418077425
|
|
24.
|
U McDermottSV SharmaJ
SettlemanHigh-throughput lung cancer cell line screening for
genotype-correlated sensitivity to an EGFR kinase inhibitorMethods
Enzymol438331341200810.1016/S0076-6879(07)38023-318413259
|
|
25.
|
R: Test for Association/Correlation
Between Paired Sampleshttp://stat.ethz.ch/R-manual/R-patched/library/stats/html/cor.test.html
|
|
26.
|
rproject.orghttp://www.rproject.org/
|
|
27.
|
GSK Cancer Cell Line Genomic Profiling
Datahttps://cabig.nci.nih.gov/caArray_GSKdata/
|
|
28.
|
S BamfordE DawsonS ForbesThe COSMIC
(catalogue of somatic mutations in cancer) database and websiteBr J
Cancer91355358200415188009
|
|
29.
|
E LindströmT ShimokawaR ToftgårdPG
ZaphiropoulosPTCH mutations: distribution and analysesHum
Mutat27215219200616419085
|
|
30.
|
SJ FreemantleE DmitrovskyCyclin E
transgenic mice: discovery tools for lung cancer biology, therapy,
and preventionCancer Prev
Res315131518201010.1158/1940-6207.CAPR-10-029721149327
|
|
31.
|
Q FengD SekulaY GuoUBE1L causes lung
cancer growth suppression by targeting cyclin D1Mol Cancer
Ther737803788200810.1158/1535-7163.MCT-08-075319074853
|
|
32.
|
E MassarelliM Varella-GarciaX TangKRAS
mutation is an important predictor of resistance to therapy with
epidermal growth factor receptor tyrosine kinase inhibitors in
non-small-cell lung cancerClin Cancer
Res1328902896200710.1158/1078-0432.CCR-06-304317504988
|
|
33.
|
KH DragnevT MaJ CyrusBexarotene plus
erlotinib suppress lung carcinogenesis independent of KRAS
mutations in two clinical trials and transgenic modelsCancer Prev
Res4818828201110.1158/1940-6207.CAPR-10-037621636548
|
|
34.
|
H LinardouIJ DahabrehD
KanaloupitiAssessment of somatic k-RAS mutations as a mechanism
associated with resistance to EGFR-targeted agents: a systematic
review and meta-analysis of studies in advanced non-small-cell lung
cancer and metastatic colorectal cancerLancet
Oncol9962972200810.1016/S1470-2045(08)70206-718804418
|
|
35.
|
W PaoTY WangGJ RielyKRAS mutations and
primary resistance of lung adenocarcinomas to gefitinib or
erlotinibPLoS
Med2e17e17200510.1371/journal.pmed.002001715696205
|
|
36.
|
S SinghZ WangD Liang FeiHedgehog-producing
cancer cells respond to and require autocrine hedgehog
activityCancer
Res7144544463201110.1158/0008-5472.CAN-10-231321565978
|
|
37.
|
J VestergaardMW PedersenN PedersenHedgehog
signaling in small-cell lung cancer: frequent in vivo but a rare
event in vitroLung
Cancer52281290200610.1016/j.lungcan.2005.12.01416616798
|
|
38.
|
JK ChenJ TaipaleKE YoungT MaitiPA
BeachySmall molecule modulation of smoothened activityProc Natl
Acad Sci USA991407114076200210.1073/pnas.18254289912391318
|
|
39.
|
F GalimbertiSL ThompsonS RaviDA ComptonE
DmitrovskyAnaphase catastrophe is a target for cancer therapyClin
Cancer Res1712181222201110.1158/1078-0432.CCR-10-117821288923
|
|
40.
|
J Sims-MourtadaJG IzzoJ AjaniKSC ChaoSonic
hedgehog promotes multiple drug resistance by regulation of drug
transportOncogene2656745679200710.1038/sj.onc.121035617353904
|
|
41.
|
T TangJY TangD LiTargeting superficial or
nodular basal cell carcinoma with topically formulated small
molecule inhibitor of smoothenedClin Cancer
Res1733783387201110.1158/1078-0432.CCR-10-337021558397
|