Introduction
Taxanes, including docetaxel, are widely used for
the treatment of squamous cell carcinoma (SCC) of the head and neck
(SCCHN), suggesting the highest single agent activity (1,2).
They exert their cytotoxic effects by promoting microtubule
assembly and stabilizing microtubule dynamics, thereby inhibiting
cell proliferation (3). Although
the clinical use of taxanes is often limited by their potentially
serious side-effects, such as diarrhea, myelosuppression, mucositis
and peripheral neuropathy, docetaxel has been shown to be
efficacious in the treatment of patients with advanced head and
neck cancer, as well as in neoadjuvant settings (4,5).
However, the molecular mechanisms underlying the growth-inhibitory
effect of docetaxel in SCCHN has not yet been extensively
investigated.
Apoptosis (programmed cell death), plays a pivotal
role in the regulation of various physiological and pathological
conditions, and it is also thought to mediate chemotherapy-induced
cytotoxicity (6). Apoptosis
induced by chemotherapy seems to require, at least partially, the
activation of the caspase cascade (7). To date, 2 major pathways leading to
apoptosis have been identified: an extrinsic (receptor) pathway and
an intrinsic (mitochondrial) pathway (8). The cascade led by caspase-8 (the
extrinsic pathway) is involved in death-receptor-mediated
apoptosis, such as the one triggered by Fas and tumor necrosis
factor (TNF). Upon activation, these receptors recruit the
Fas-associated death domain (FADD), which in turn binds to
procaspase-8, leading to cleavage into its active form (8). The subsequent activation of the
effector caspases, such as caspase-3, occurs either directly or
after an amplification step involving the mitochondria (9). On the other hand, the mitochondrial
(intrinsic) pathway is triggered by cytochrome c release
from the mitochondria. Cytochrome c release into the
cytoplasm leads to the formation of a complex with Apaf-1 that
binds to procaspase-9 via its caspase recruit domain (CARD)
(10). This complex, known as the
apoptosome complex can, in the presence of deoxyadenosine
triphosphate (dATP), activate procaspase-9, which in turn activates
effector caspases including caspase-3 (11). Thus, the cascade of caspase
activation plays an important role in the induction of apoptosis in
cancer cells. Paradoxically, however, chemotherapeutic agents that
promote apoptosis also activate the transcription factor, nuclear
factor-κB (NF-κB) (12), which
suppresses caspase activation by enhancing the expression of
anti-apoptotic proteins, including survivin, a cellular inhibitor
of apoptosis protein (cIAP)-1; cIAP-2, an X-linked inhibitor of
apoptosis protein (XIAP); and B-cell lymphoma 2 (Bcl-2) (12–15).
Since a human oral cancer cell line (B88) exhibited constitutively
activated NF-κB activity in our previous studies (16,17),
we hypothesized that the downregulation of anti-apoptotic proteins
through the suppression of NF-κB activity would be a promising
strategy for the treatment of patients with oral cancer.
A vitamin E constituent may be one such candidate
agent derived from natural sources that can have great potential
for preventing and treating oral cancer. Vitamin E is a general
term representing a family of compounds that is further divided
into 2 subgroups: tocopherols and tocotrienols (18). Although tocopherols and
tocotrienols exist in α, β, γ and δ forms, the two differ
structurally in that tocopherols contain a saturated phytyl chain,
whereas tocotrienols possess an unsaturated side chain. Thus far,
tocopherols have been studied extensively; however, very little is
known about tocotrienols. Previous studies have clearly established
that tocotrienols, but not tocopherols, display potent
antiproliferative and apoptotic activity againt neoplastic mammary
epithelial cells with treatment at low doses that have little or no
effect on normal cell growth and function (19,20).
For instance, studies have shown that γ-tocotrienol, but not
tocophenol, can inhibit both constitutive and inducible NF-κB
activation in various cancer cell lines (21,22).
This activity correlates well with the downregulation of
NF-κB-regulated gene products, such as anti-apoptotic proteins
(22). Therefore, it is considered
that the combined treatment with low doses of docetaxel and
γ-tocotrienol may result in an enhanced therapeutic response in
patients with oral cancer.
In the present study, we report that the
simultaneous treatment of human oral cancer (B88) cells with low
doses of docetaxel and γ-tocotrienol suppresses docetaxel-induced
NF-κB activity, leading to the inhibition of the expression of
anti-apoptotic proteins, which results in the activation of
initiator caspases, caspase-8 and -9, as well as an effector
caspase, caspase-3. We also found that these cells actually entered
apoptosis, as evaluated by the cleavage of poly(ADP-ribose)
polymerase (PARP) and DNA fragmentation.
Materials and methods
Cells and media
A metastatic human oral cancer cell line (B88) was
previously established in our laboratory (23). This cell clone was cultured in DMEM
(Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine
serum (FBS) (Gibco) and 100 mg/ml penicillin-streptomycin (Gibco)
in the presence of 5% CO2 in an incubator at 37°C.
In vitro cell growth assay
Cells (5×103 cells per well) were grown
on 96-well plates (Falcon Labware, Lincoln Park, NJ) in DMEM
supplemented with 10% serum in the presence or absence of docetaxel
(0.5, 1, and 2 nM) (obtained from Sigma, St. Louis, MO) and
γ-tocotrienol (50, 75, and 100 μM) (obtained from Eizai Food &
Chemical Co., Tokyo, Japan, with a purity exceeding 98.7%) alone,
or both for 6 days. Thereafter, 10 μl of 5 mg/ml
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were added to each well and incubated for 4 h. The blue dye taken
up by the cells was dissolved in dimethyl sulfodide (100 μg/ml),
and the absorbance was measured with a Titertek spectrophoto meter
(Flow, Irvine, UK) at 540 nm. All assays were run in
triplicate.
Nuclear and cytosolic extract
preparations
The cells were seeded on 100-mm plastic petri dishes
(Falcon Labware). Twenty-four hours after seeding, the cells were
treated with either docetaxel, γ-tocotrienol, or both for 48 h, and
nuclear extracts were then obtained according to a previously
described method (24). The cells
were washed twice with ice-cold PBS before being resuspended in 400
μl of ice-cold lysis buffer consisting of 10 mM
N-2-hydroxyethylpiperazine-N′-2-ethane sulfonic acid (HEPES) (pH
7.9), 10 mM KCl, 0.1 mM ethylenediaminetetra-acetate (EDTA), 0.1 mM
ethyleneglycolbis(b-aminoethyl ether)-N,N′-tetraacetic acid (EGTA),
0.5 mM dithiothreitol (DTT), 0.5 mg/ml benzamidine and 2 mg/ml
aprotinin for 15 min. Nonidet P-40 was added to a final
concentration of 0.3%, and the lysates were vortexed before being
pelleted in a microfuge. The supernatants of this centrifugation
were designated cytosolic extracts. Each nuclear pellet was
resuspended in 50 μl of extraction buffer consisting of 10 mM HEPES
(pH 7.9), 400 mM NaCl, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM
DTT, 0.5 mM phenylmethylsulfonyl fluoride and 2 mg/ml aprotinin and
then placed on ice for 30 min. The nuclear extracts were pelleted,
and the supernatants were designated nuclear extracts. The protein
concentrations contained in samples were determined by a Bio-Rad
(Hercules, CA) protein assay kit.
Labeling of oligonucleotides and
electrophoretic mobility shift assay (EMSA)
The probe consisted of NF-κB-specific
double-stranded oligonucleotides with the sequence
5′-AGTTGAGGGGACTTTCCCAGGC-3′ containing the κB site from the
κ-light-chain enhancer in B cells. Oligonucleotides were biotin
end-labeled using the biotin 3′ end labeling kit (Pierce Chemical,
Rockford, IL). To extract the labeled probe, 50 μl of
chloroform:isoamylalcohol (24:1) were added to each tube, followed
by centrifugation briefly at 13,000 × g. The top aqueous phase
containing the labeled probe was removed and saved for binding
reactions. Nuclear extract proteins (5 μg) were used for EMSA with
a LightShift Chemiluminescent EMSA kit (Pierce Chemical Co.)
according to the manufacturer’s instructions. In brief, nuclear
extract proteins were preincubated with the binding buffer for 5
min and then incubated with double-stranded, biotin-labeled
consensus oligonucleotides for 15 min at room temperature. The
specificity of the complex was analyzed by incubation with an
excess of unlabeled competitor oligonucleotides (100-fold molar
excess of labeled probe). Samples were run on 6% polyacrylamide
gels. Subsequently, the DNA was transferred onto a positive nylon
membrane, UV cross-linked, probed with streptavidin-HP conjugate,
and incubated with the substrate of the enhanced chemiluminescence
kit. The membrane was then exposed to X-ray film (Amersham
Hyperfilm™ ECL, GE Healthcare Ltd., Chalfont St. Giles, UK).
Western blot analysis of inhibitor of κB
(IκB)-α, survivin, cIAP-1, cIAP-2, X-linked inhibitor of apoptosis
protein (XIAP), Bcl-2, caspase-8, caspase-9, caspase-3, Apaf-1,
cytochrome c, PARP and β-actin
Cytosolic extracts (20 μg) were subjected to
electrophoresis on 10% SDS-polyacrylamide gels, then transferred
onto nylon membranes. The membranes were blocked with 3% bovine
serum albumin and incubated with each of the following antibodies
(all from Cell Signaling Technology, Beverly, MA, USA): anti-IκB-α,
anti-survivin, anti-cIAP-1, anti-cIAP-2, anti-XIAP, anti-Bcl-2,
anti-caspase-8, anti-caspase-9, anti-caspase-3, anti-Apaf-1,
anti-cytochrome c, anti-PARP and anti-β-actin. In the case
of the detection of cytochrome c, possible mitochondrial
contamination from the same sample was monitored using an antibody
against mitochondrial cytochrome c oxidase subunit IV
(Molecular Probes, Eugene, OR). After intervening rinses with PBS,
the antibody was detected using a chemiluminescence western blot
analysis kit (Amersham, Tokyo, Japan) according to the
manufacturer’s instructions.
Assessment of apoptosis induced by
docetaxel and γ-tocotrienol
Cells that sloughed off the surface of the dish were
collected and fixed with 1% paraformaldehyde (pH 7.4) for 10 min at
room temperature. The apoptotic nuclear morphology was examined by
Hoechst 33258 staining as described previously (25). Briefly, fixed cells plated on a
glass slide were stained with 5 μg/ml of Hoechst 33258 (Hoechst
Japan, Tokyo, Japan) for 30 min at room temperature, washed with
PBS, and mounted with 80% glycerol in PBS for fluorescence
microscopy.
Results
Cell growth inhibition by docetaxel and
γ-tocotrienol
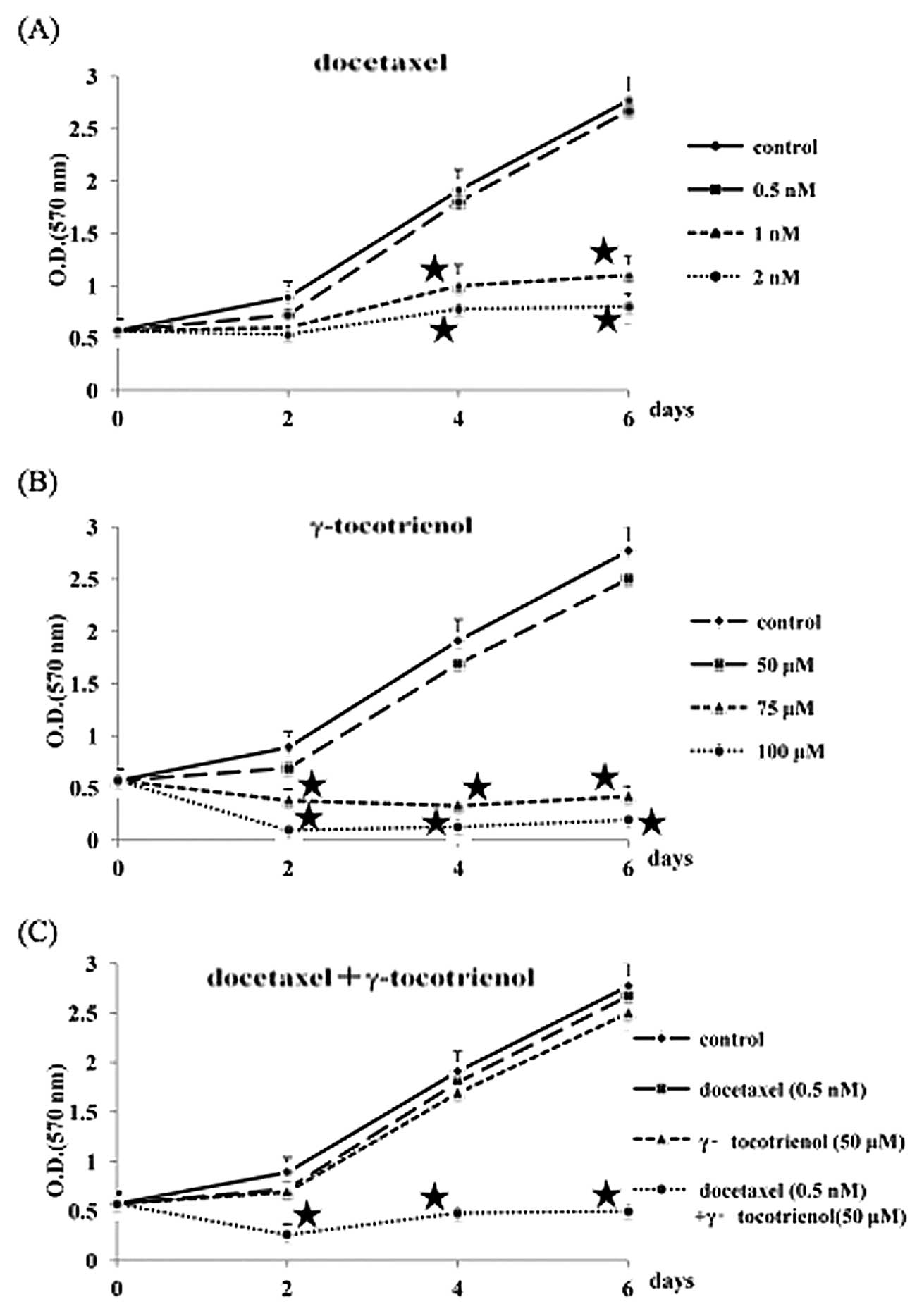
The growth inhibitory response of B88 cells to
docetaxel and γ-tocotrienol was investigated by MTT assay for 6
days. As shown in Fig. 1 (A and
B), when the number of untreated cells measured at day 6 was
determined to be 100, the growth inhibitory ratios in the 0.5, 1,
and 2 nM docetaxel groups were 4, 60, and 71%, respectively, and
those in the 50, 75, and 100 μM γ-tocotrienol groups were 10, 85,
and 93%, respectively. Therefore, we used docetaxel and
γ-tocotrienol at the respective concentrations of 0.5 nM and 50 μM,
neither of which significantly inhibited B88 cell growth when
compared to the growth of the control cells. Whether or not
γ-tocotrienol can potentiate the effect of docetaxel against B88
cells was also examined. As can be observed in Fig. 1C, the doses of docetaxel (0.5 nM)
and γ-tocotrienol (50 μM) that minimally inhibited growth when used
alone exhibited an enhanced suppression of cell growth when used in
combination.
γ-tocotrienol inhibition of constitutive
and docetaxel-induced NF-κB activation in B88 cells
We then examined the mechanisms by which
γ-tocotrienol potentiates the effects of docetaxel in B88 cells.
Since various agents, including anticancer agents, have been shown
to activate NF-κB through diverse pathways, we investigated the
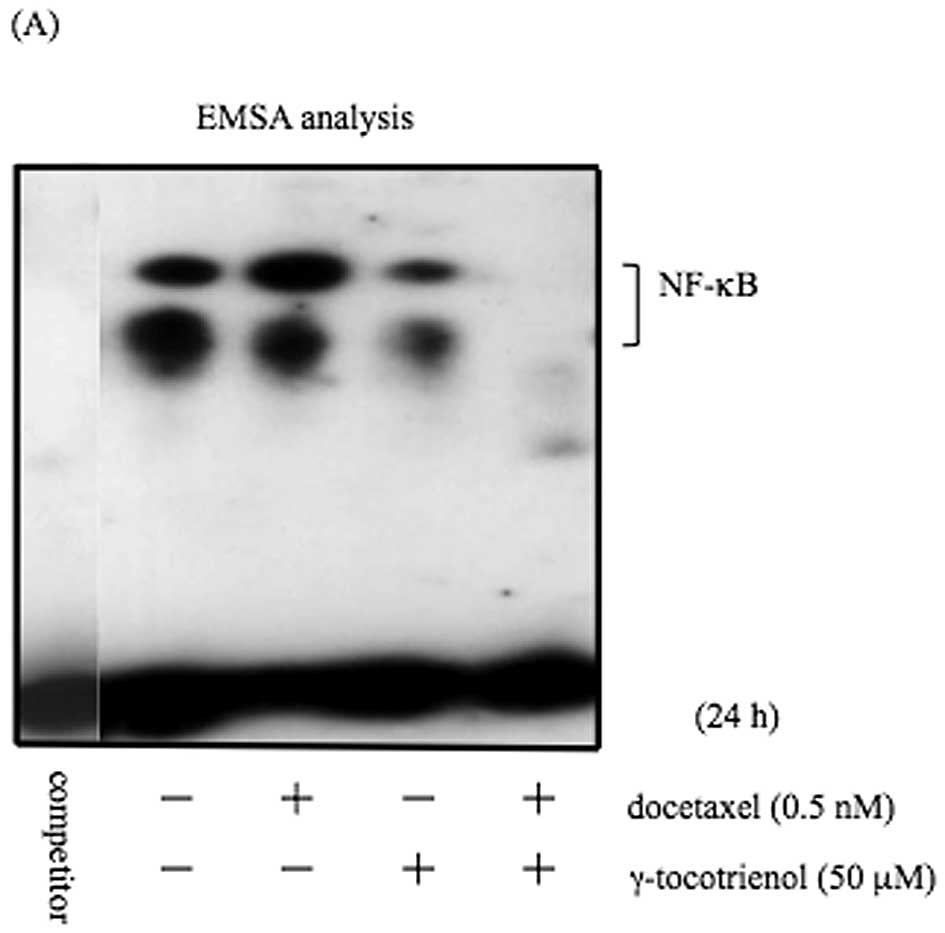
effect of γ-tocotrienol on docetaxel-induced NF-κB activation. EMSA
analysis demonstrated that γ-tocotrienol suppressed the
constitutively active and docetaxel-induced NF-κB activation
(Fig. 2A). The specific binding of
NF-κB to DNA was abrogated with an excess of unlabeled probe,
indicating the NF-κB activity contained in the cells. In addition,
time-course analysis of the effects of these agents on NF-κB
activity in the B88 cells revealed that docetaxel alone stimulated
the nuclear p65 expression at 12, 24, and 48 h after treatment.
However, docetaxel inversely suppressed cytoplasmic IκB-α protein
expression (Fig. 2B). As shown in
Fig. 2C, when B88 cells were
treated with γ-tocotrienol alone, nuclear p65 expression was
significantly inhibited from 12 to 24 h after treatment. Of note,
this docetaxel-induced expression of nuclear p65 was suppressed by
the combined treatment with γ-tocotrienol in B88 cells, whereas
cytoplasmic IκB-α protein expression was conversely enhanced
(Fig. 2D). These results suggest
that γ-tocotrienol is a potent modulator of both constitutive and
inducible NF-κB activation in oral cancer cells.
Effects of docetaxel and γ-tocotrienol
treatment on the expression of anti-apoptotic proteins
Since NF-κB regulates the expression of several
anti-apoptotic proteins, including survivin, cIAP-1, cIAP-2, XIAP,
and Bcl-2 (13–15,26–28),
we examined the expression levels of these anti-apoptotic proteins
by treatment with docetaxel, γ-tocotrienol, or both. As shown in
Fig. 3, although docetaxel induced
the expression of these anti-apoptotic proteins in a time-dependent
manner (Fig. 3A), the inhibitory
effects on the expression of anti-apoptotic proteins were observed
in the γ-tocotrienol-treated cells (Fig. 3B). However, the simultaneous
treatment of cells with docetaxel and γ-tocotrienol led to the
downregulation of the expression of these anti-apoptotic proteins
in a time-dependent manner, in agreement with the degree of
inhibition of NF-κB activity (Fig.
3C).
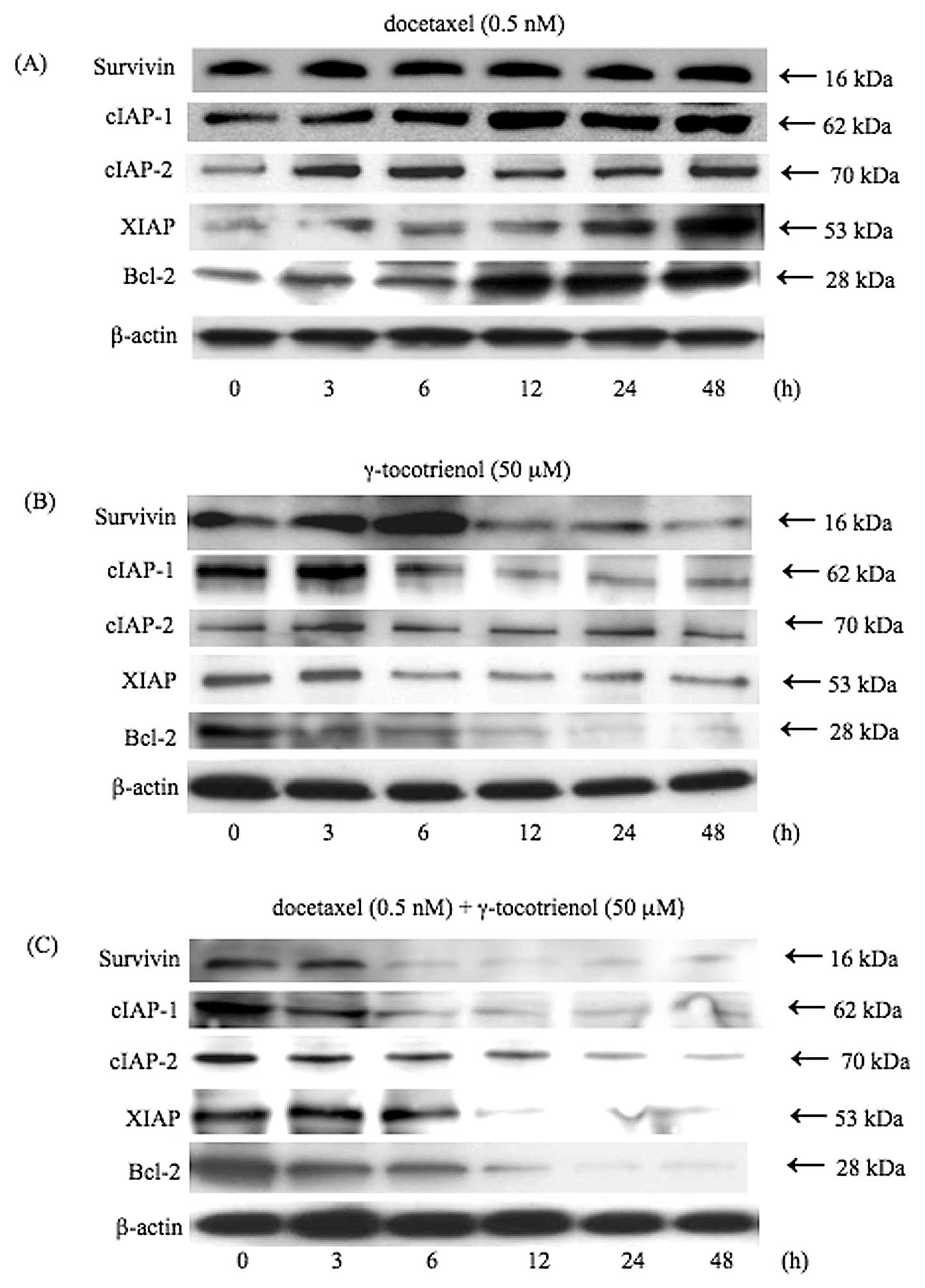 | Figure 3.γ-tocotrienol inhibition of the
anti-apoptotic gene products, survivin, cIAP-1, cIAP-2, XIAP and
Bcl-2. Cells were left untreated or incubated with either (A)
docetaxel (0.5 nM), (B) γ-tocotrienol (50 μM), or (C) both agents
for 48 h. Whole-cell extracts were prepared and were analyzed by
western blot analysis using antibodies against survivin, cIAP-1,
cIAP-2, XIAP, Bcl-2 and β-actin as indicated. Although docetaxel
stimulated the expression of these anti-apoptotic proteins,
γ-tocotrienol slightly suppressed the expression of these proteins.
On the other hand, simultaneous treatment of cells with both agents
significantly inhibited the expression of anti-apoptotic proteins.
As a loading control for the protein samples, the expression of
β-actin is demonstrated. Results are representative of 3
independent experiments. |
Processing of caspases-3, -8, and -9 by
docetaxel and γ-tocotrienol
To understand the mechanisms involved in the
enhanced cytotoxicity of the combined treatment with docetaxel and
γ-tocotrienol, we analyzed the apoptotic cascades, including the
processing of procaspases-3, -8 and -9, the release of cytochrome
c from the mitochondria, and the induction of Apaf-1 by
using western blot analysis. Since we have previously shown that in
the apoptotic pathway, caspase-3 is a downstream caspase
responsible for the cleavage of important substrates such as PARP
in B88 cells (16), we first
examined the processing of caspase-3 by docetaxel and
γ-tocotrienol. As shown in Fig.
4A, when administered simultaneously, both agents converted
procaspase-3 with a molecular weight of 35 kDa into active
caspase-3 with a molecular weight of 17 kDa. To analyze the
processing of procaspase-8, which stimulates the release of
cytochrome c from the mitochondria (29), cytosolic extracts obtained from the
treated cells were subjected to western blot analysis. As shown in
Fig. 4B, procaspase-8 with a
molecular weight of 57 kDa and 54 kDa was cleaved to active species
with a molecular weight of 10 kDa. To further examine the
processing of procaspase-9 mediated by cytochrome c,
cytosolic extracts were subjected to western blot analysis. As
shown in Fig. 4C, when B88 cells
were treated with both agents, procaspase-9 with a molecular weight
of 47 kDa was converted to an active form of caspase-9 with a
molecular weight of 37 kDa.
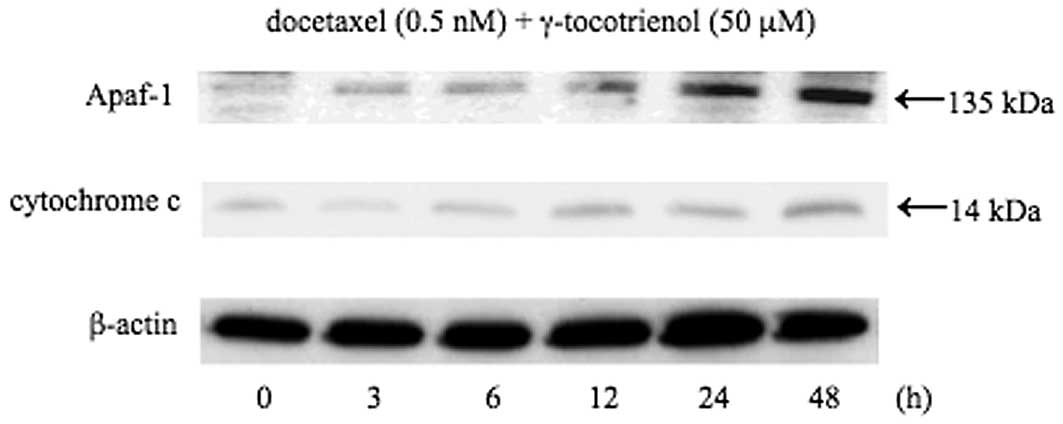
Induction of Apaf-1 and cytochrome c
release by docetaxel and γ-tocotrienol
The initiation of mitochondria-mediated apoptosis
requires the release of cytochrome c into the cytoplasm,
where a complex (apoptosome) is formed with procaspase-9, Apaf-1
and dATP (10). The release of
cytochrome c into the cytoplasm is thought to be the
limiting factor in caspase-9 activation and the subsequent
activation of caspase-3 (10).
Thus, we assessed cytochrome c release into the cytoplasm in
response to docetaxel and γ-tocotrienol in B88 cells. The results
shown in Fig. 5 demonstrate that
simultaneous treatment with these agents induced the release of
cytochrome c into the cytoplasm in a time-dependent manner.
Apaf-1, another component of the apoptosome complex, was also
augmented in the agent-treated B88 cells.
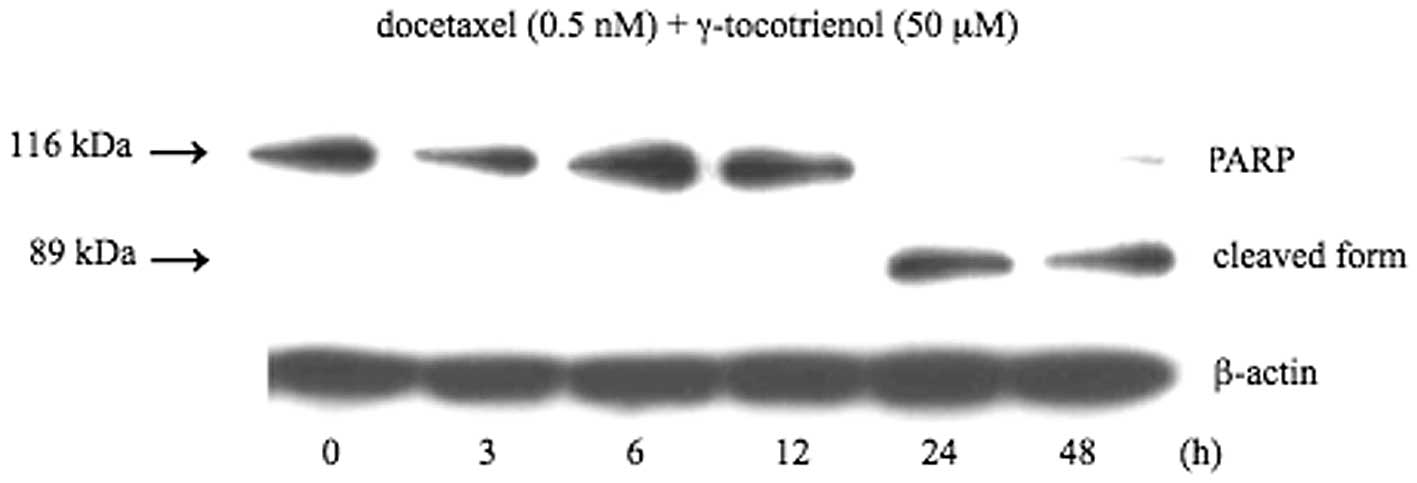
Cleavage of PARP by docetaxel and
γ-tocotrienol
PARP is a 116-kDa nuclear enzyme that detects and
binds DNA strand breaks produced by various apoptotic stimuli. It
is known as a substrate for caspase-3. Caspase-3 mediated PARP
cleavage into a molecular weight of 89 kDa is considered a hallmark
of apoptosis (30). Therefore, we
investigated the cleavage of PARP by both agents. As shown in
Fig. 6, increased cleavage of PARP
was observed at 24 and 48 h following treatment with both
agents.
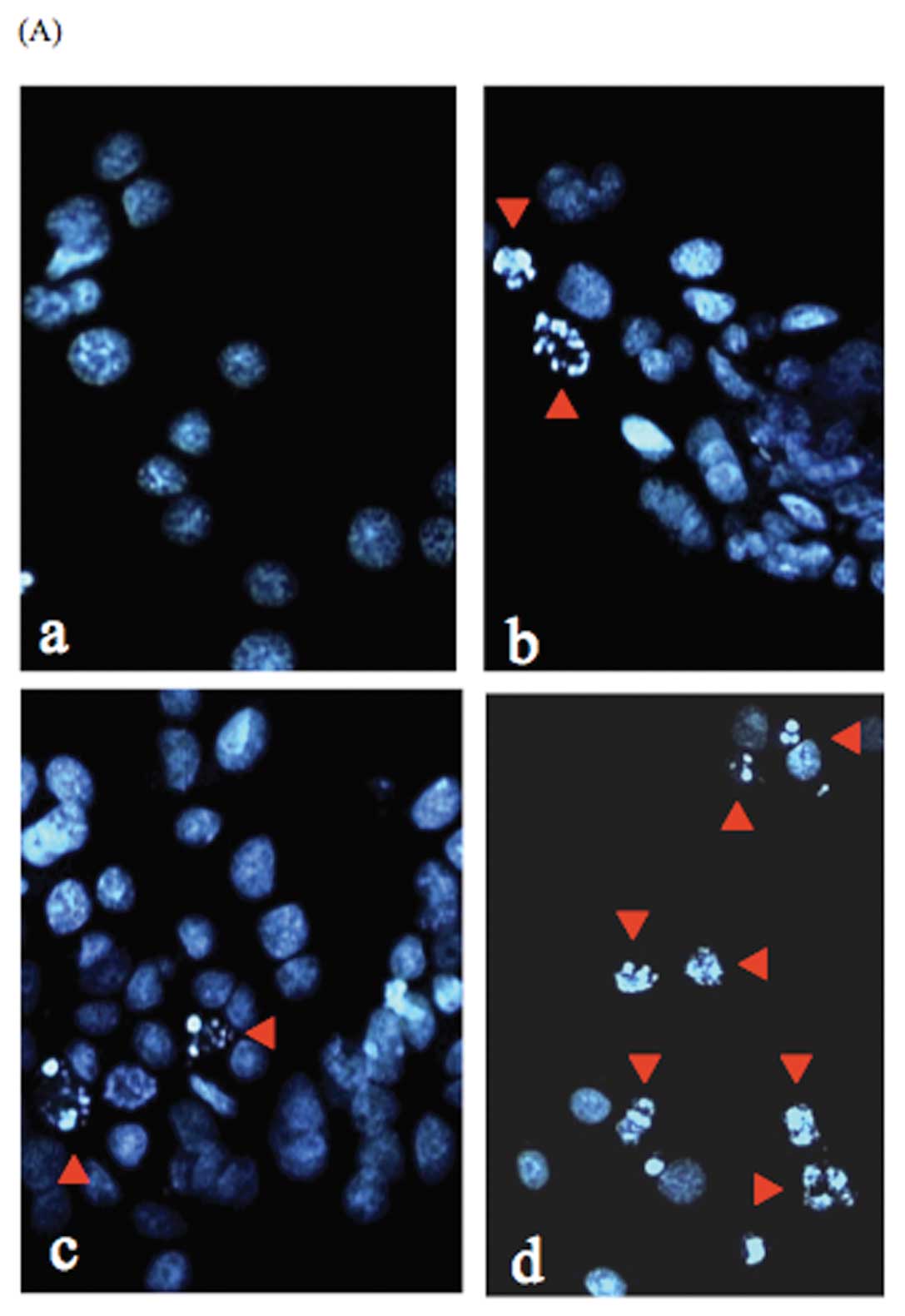
Induction of apoptosis in B88 cells
treated with docetaxel and γ-tocotrienol
A photomicrograph of Hoechst 33258-stained B88
nuclei, from floating cells obtained by treatment with docetaxel,
γ-tocotrienol, or both for 24 h, exhibited chromatin condensation
and nuclear fragmentation typical of apoptosis (Fig. 7A). The percentages of apoptotic
cells were 3.0% in the untreated control cells (Fig. 7B), 7.9% in the docetaxel-treated
cells (Fig. 7B), 8.2% in the
γ-tocotrienol-treated cells (Fig.
7B), and 55.1% in the cells treated with both agents (Fig. 7B).
Discussion
Although chemotherapy and radiation therapy have
proven to be highly effective in eradicating early oral SCC, the
successful management of advanced (stage III and IV) oral SCC
remains disappointing in the majority of cases (31). The poor clinical response in
patients with advanced oral SCC has prompted the investigation of
molecular targets for chemotherapy and radiotherapy in the
treatment of oral cancer. Based on these considerations, we have
previously analyzed the possible molecules responsible for the
proliferation, invasion and metastasis of cancer cells, and have
shown that human head and neck cells in oral squamous and salivary
gland carcinomas have a significantly augmented NF-κB activity as
compared to their normal counterparts (23,32),
suggesting that NF-κB may be an important therapeutic target for
the improvement of conventional chemotherapy in oral SCC. Thus,
novel agents that are non-toxic, and can significantly inhibit
constitutive and inducible NF-κB activation, as well as
NF-κB-regulated gene products could remarkably augment
chemotherapeutic drug-induced apoptosis. Therefore, the objective
of the present study was to investigate whether or not
γ-tocotrienol, a component of vitamin E, can enhance the antitumor
activity of docetaxel against human oral cancer cells. The results
showed that γ-tocotrienol suppressed the proliferation of human
oral cancer cells, potentiated docetaxel-induced apoptosis, and
inhibited constitutively active and inducible NF-κB activation, as
well as the expression of NF-κB-regulated gene products. Our
results are in partial agreement with those from other studies
(33,34), reporting that γ-tocotrienol
suppresses the proliferation of gastric and pancreatic cancer
cells.
Specifically, we found for the first time that a low
dose of γ-tocotrienol, when used in combination with docetaxel at a
non-cytotoxic dose, is highly effective in inducing apoptosis in
oral cancer cells. This is a very important finding as, although
γ-tocotrienol has previously been suggested to induce apoptosis in
gastric and pancreatic cancer cells (33,34.), its effect in
combination with chemotherapeutic agents, such as docetaxel has not
been previously investigated in human oral cancer. In this study,
we demonstrate that this effect may be exerted through the
downregulation of NF-κB-mediated survival proteins, such as
survivin, Bcl-2, cIAP-1, cIAP-2 and XIAP by γ-tocotrienol.
Moreover, since we have previously shown that constitutive and
inducible NF-κB expressed in human oral cancer cells has been
linked with chemoresistance (17),
the suppression of NF-κB activity by γ-tocotrienol may account for
the enhanced chemosensitivity of oral cancer cells to
docetaxel.
In the present study, we report that even a
non-cytotoxic dose of docetaxel can induce NF-κB DNA-binding
activity in oral cancer cells, thus raising the possibility that
cytotoxic doses of docetaxel used in the clinical setting could
stimulate, to a greater degree, NF-κB activity in cancer cells.
This possibility may significantly limit the antitumor activity of
docetaxel in patients with oral cancer. On the other hand,
γ-tocotrienol decreased NF-κB-binding activity in B88 cells. It is
noteworthy that γ-tocotrienol alone had minimal effects on cell
viability, whereas the combination with docetaxel increased
antitumor activity with respect to chemotherapy alone in oral
cancer cells. Although the effects of γ-tocotrienol in tumor cells
are not yet fully understood, its abilities to induce cell cycle
arrest (35), activate p53
(36), activate caspase-8
(21), suppress adhesion molecules
(37), downregulate c-Myc and
telomerase (38) and inhibit
angiogenesis (39) have been
suggested. Since the NF-κB pathway plays a critical role in
tumorigenesis, chemosensitization, apoptosis, cell adhesion, the
expression of c-Myc and human telomerase reverse transcriptase and
cell cycle arrest (40),
γ-tocotrienol must therefore modulate these pathways and molecules
by suppressing NF-κB activity.
Based on the fact that the activation of caspase-3,
an executioner caspase in the apoptotic pathway, leads to the
cleavage of PARP in B88 cells (16), analysis of the conversion of
procaspase-3 to caspase-3 by docetaxel and γ-tocotrienol is indeed
important in any evaluation of apoptotic cell death. Consistent
with this premise, the degree of growth suppression by these agents
correlated well with the activation status of caspase-3 in B88
cells, indicating that caspase-3 targets cellular proteins, such as
PARP and DNA fragmentation factor (41), for proteolytic cleavage resulting
in cell death (42).
In conclusion, the results of the present study
indicate that γ-tocotrienol potentiates the anticancer activity of
docetaxel by downregulating the expression of NF-κB and
NF-κB-regulated gene products, leading to the inhibition of
proliferation. A number of clinical trials with tocotrienols in
patients with pancreatic, prostate and breast cancer are already in
progress. Based on our present results, well-designed animal and
clinical studies are required for the potential translation of our
preclinical findings in patients with oral cancer.
Acknowledgements
This study was supported by a
Grant-in-Aid from the Ministry of Education, Culture, Sports,
Science, and Technology of Japan.
References
|
1.
|
Forastiere AA and Urba SG: Experimental
therapeutic approaches for recurrent head and neck cancer. Cancer
Treat Res. 74:263–281. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Colevas AD and Posner MR: Docetaxel in
head and neck cancer: a review. Am J Clin Oncol. 21:482–486. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Eisenhauer EA and Vermorken JB: The
taxoids. Comparative clinical pharmacology and therapeutic
potential. Drugs. 55:5–30. 1998.PubMed/NCBI
|
|
4.
|
Schoffski P, Catimel G, Planting AS, et
al: Docetaxel and cisplatin: an active regimen in patients with
locally advanced, recurrent or metastatic squamous cell carcinoma
of the head and neck. Results of a Phase II study of the EORTC
Early Clinical Studies Group. Ann Oncol. 10:119–122. 1999.
View Article : Google Scholar
|
|
5.
|
Couteau C, Chouaki N, Leyvraz S, et al: A
Phase II study of docetaxel in patients with metastatic squamous
cell carcinoma of the head and neck. Br J Cancer. 81:457–462. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Vaux DL and Korsmeyer SJ: Cell death in
development. Cell. 96:245–254. 1999. View Article : Google Scholar
|
|
7.
|
Fulda S, Susin S, Kroemer G and Debatin
KM: Molecular ordering of apoptosis induced by anticancer drugs in
neuroblastoma cells. Cancer Res. 58:4453–4460. 1998.PubMed/NCBI
|
|
8.
|
Ferreira CG, Span SW, Peters GJ, Kruyt FAE
and Giaccone G: Chemotherapy triggers apoptosis in a
caspase-8-dependent and mitochondria-controlled manner in the
non-small cell lung cancer cell line NCI-H460. Cancer Res.
60:7133–7141. 2000.PubMed/NCBI
|
|
9.
|
Scaffidi C, Fulda S, Srinivasan A, et al:
Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17:1675–1687.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Li P, Nijhawan D, Budihardjo I, et al:
Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9
complex initiates an apoptotic protease cascade. Cell. 91:479–489.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Srinivasula SM, Ahmad M, Fernades-Alnemri
T and Alnemri ES: Autoactivation of procaspase-9 by Apaf-1-mediated
oligomerization. Mol Cell. 1:949–957. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Baldwin AS Jr: Control of oncogenesis and
cancer therapy resistance by the transcription factor NF-κB. J Clin
Invest. 107:241–246. 2001.
|
|
13.
|
Chu Z-L, McKinsey TA, Liu L, Gentry JJ,
Malim MH and Ballard DW: Suppression of tumor necrosis
factor-induced cell death by inhibitor of apoptosis c-IAP2 is under
NF-κB control. Proc Natl Acad Sci USA. 94:10057–10062.
1997.PubMed/NCBI
|
|
14.
|
Zhu L, Fukuda S, Cordis G, Das DK and
Maulik N: Anti-apoptotic protein survival plays a significant role
in tubular morphogenesis of human coronary arteriolar endothelial
cells by hypoxic preconditioning. FEBS Lett. 508:369–374. 2001.
View Article : Google Scholar
|
|
15.
|
Kreuz S, Siegmund D, Scheurich P and
Wajant H: NF-κB inducers upregulate cFLIP, a
cycloheximide-sensitive inhibitor of death receptor signaling. Mol
Cell Biol. 21:3964–3973. 2001.
|
|
16.
|
Azuma M, Tamatani T, Ashida Y, Takashima
R, Harada K and Sato M: Cisplatin induces apoptosis in oral
squamous carcinoma cells by the mitochondria-mediated but not the
NF-κB-suppressed pathway. Oral Oncol. 39:282–289. 2003.PubMed/NCBI
|
|
17.
|
Tamatani T, Azuma M, Ashida Y, et al:
Enhanced radio-sensitization and chemosensitization in
NF-κB-suppressed human oral cancer cells via the inhibition of
γ-irradiation- and 5-FU-induced production of IL-6 and IL-8. Int J
Cancer. 108:912–921. 2004.PubMed/NCBI
|
|
18.
|
Wada S: Cancer preventive effects of
vitamin E. Curr Pharm Biotechnol. 13:156–164. 2012. View Article : Google Scholar
|
|
19.
|
McIntyre BS, Briski KP, Gapor A and
Sylvester PW: Antiproliferative and apoptotic effects of
tocopherols and tocotrienols on preneoplastic and neoplastic mouse
mammary epithelial cells. Proc Soc Exp Biol Med. 224:292–301. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
McIntyre BS, Briski KP, Tirmenstein MA,
Fariss MW, Gapor A and Sylvester PW: Antiproliferative and
apoptotic effects of tocopherols and tocotrienols on normal mouse
mammary epithelial cells. Lipids. 35:171–180. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Shah SJ and Sylvester PW: γ-Tocotrienol
inhibits neoplastic mammary epithelial cell proliferation by
decreasing Akt and nuclear factor κB activity. Exp Biol Med
(Maywood). 230:235–241. 2005.
|
|
22.
|
Ahn KS, Sethi G, Krishnan K and Aggarwal
BB: γ-Tocotrienol inhibits nuclear factor-κB signaling pathway
through inhibition of receptor-interacting protein and TAK1 leading
to suppression of antiapoptotic gene products and potentiation of
apoptosis. J Biol Chem. 282:809–820. 2007.
|
|
23.
|
Tamatani T, Azuma M, Aota K, Yamashita T,
Bando T and Sato M: Enhanced IκB kinase activity is responsible for
the augmented activity of NF-κB in human head and neck carcinoma
cells. Cancer Lett. 171:165–172. 2001.
|
|
24.
|
Imbert V, Rupec RA, Livolsi A, et al:
Tyrosine phosphorylation of IκB-α activates NF-κB without
proteolytic degradation. Cell. 85:787–798. 1996.
|
|
25.
|
Kluck RM, Bossy-Wetzel E, Green DR and
Newmeyer DD: The release of cytochrome c from mitochondria: a
primary site for Bcl-2 regulation of apoptosis. Science.
275:1132–1136. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
You M, Ku P-T, Hrdlickova R and Bose HR
Jr: ch-IAP1, a member of the inhibitor-of-apoptosis protein family,
is a mediator of the antiapoptotic activity of the v-rel
oncoprotein. Mol Cell Biol. 17:7328–7341. 1997.PubMed/NCBI
|
|
27.
|
Catz SD and Johnson JL: Transcriptional
regulation of bcl-2 by nuclear factor κB and its significance in
prostate cancer. Oncogene. 20:7342–7351. 2001.
|
|
28.
|
Grumont RJ, Rourke IJ and Gerondakis S:
Rel-dependent induction of A1 transcription is required to protect
B cells from antigen receptor ligation-induced apoptosis. Genes
Dev. 13:400–411. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kuwana T, Smith JJ, Muzio M, Dixit V,
Newmeyer DD and Kornbluth S: Apoptosis induction by caspase-8 is
amplified through the mitochondrial release of cytochrome c. J Biol
Chem. 273:16589–16594. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ding Z, Yang X, Pater A and Tang SC:
Resistance to apoptosis is correlated with the reduced caspase-3
activation and enhanced expression of antiapoptotic proteins in
human cervical multidrug-resistant cells. Biochem Biophys Res
Commun. 270:415–420. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Clark JR and Frei E III: Chemotherapy for
head and neck cancer: progress and controversy in the management of
patients with MO disease. Semin Oncol. 16:44–57. 1989.PubMed/NCBI
|
|
32.
|
Azuma M, Motegi K, Aota K, Yamashita T,
Yoshida H and Sato M: TGF-β1 inhibits NF-κB activity through
induction of IκB-α expression in human salivary gland cells: a
possible mechanism of growth suppression by TGF-β1. Exp Cell Res.
250:213–211. 1999.
|
|
33.
|
Kunnumakkara AB, Sung B, Ravindran J, et
al: γ-Tocotrienol inhibits pancreatic tumors and sensitizes them to
gemcitabine treatment by modulating the inflammatory
microenvironment. Cancer Res. 70:8695–8705. 2010.
|
|
34.
|
Manu KA, Shanmugam MK, Ramachandran L, et
al: First evidence that γ-tocotrienol inhibits the growth of human
gastric cancer and chemosensitizes it to capecitabine in a
xenograft mouse model through the modulation of NF-κB pathway. Clin
Cancer Res. 18:2220–2229. 2012.
|
|
35.
|
Wada S, Satomi Y, Murakoshi M, Noguchi N,
Yoshikawa T and Nishino H: Tumor suppressive effects of tocotrienol
in vivo and in vitro. Cancer Lett. 229:181–191. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Agarwal MK, Agarwal ML, Athar M and Gupta
S: Tocotrienol-rich fraction of palm oil activates p53, modulates
Bax/Bcl2 ratio and induces apoptosis independent of cell cycle
association. Cell Cycle. 3:205–211. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Naito Y, Shimozawa M, Kuroda M, et al:
Tocotrienols reduce 25-hydroxylcholesterol-induced
monocyte-endothelial cell interaction by inhibiting the surface
expression of adhesion molecules. Atherosclerosis. 180:19–25. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Eitsuka T, Nakagawa K and Miyazawa T:
Down-regulation of telomerase activity in DLD-1 human colorectal
adenocarcinoma cells by tocotrienol. Biochem Biophys Res Commun.
348:170–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Miyazawa T, Inokuchi H, Hirokane H,
Tsuzuki T, Nakagawa K and Igarashi M: Anti-angiogenic potential of
tocotrienol in vitro. Biochemistry (Mosc). 69:67–69. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Aggarwal BB: Nuclear factor-kappa B: the
enemy within. Cancer Cell. 6:203–208. 2004.
|
|
41.
|
Liu X, Zou H, Slaughter C and Wang X: DFF,
a heterodimeric protein that functions downstream of caspase-3 to
trigger DNA fragmentation during apoptosis. Cell. 89:175–184. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Shearwin-Whyatt L, Baliga B, Doumanis J
and Kumar S: Chimeric caspase molecules with potent cell killing
activity in apoptosis-resistant cells. Biochem Biophys Res Commun.
282:1114–1119. 2001. View Article : Google Scholar : PubMed/NCBI
|