Introduction
PECAM-1, also known as CD31, is a 130-kDa
glycoprotein member of the immunoglobulin (Ig)-superfamily of type
I transmembrane cell adhesion molecules, which is expressed widely
on hematopoietic cells as well as on endothelial cells (1,2).
PECAM-1 has a 118 amino-acid cytoplasmic tail that contains 2
immunoreceptor tyrosine-based inhibition motifs (ITIMs) that
encompass Y663 and Y686 of human PECAM-1. The ITIMs become tyrosine
phosphorylated mainly by the Src family tyrosine kinases in
response to various stimuli and recruit several SH2-domain
containing signaling molecules, including the protein-tyrosine
phosphatase SHP2 and the Src family tyrosine kinases. By coupling
with these and various other signaling molecules, PECAM-1 is
implicated in modulation of intracellular signaling mechanisms
regulating a variety of cellular events, including integrin
activation, chemotaxis, apoptosis and cell adhesion (1–4).
Recent studies on PECAM-1 deficient mice have further revealed that
it plays a regulatory role in SDF-1-induced migration of
hematopoietic stem cells and megakaryocytes to the bone marrow
vascular niche (5,6). The hematopoietic cytokine IL-3 has
been shown to induce tyrosine phosphorylation of PECAM-1 in
hematopoietic cells (7). However,
its significance in the signal transduction mechanisms by which
this hematopoietic cytokine regulates proliferation and apoptosis
of cells is still unknown. PECAM-1 is also expressed on various
types of leukemias, including acute myeloid leukemia (AML)
(8), acute lymphoblastic leukemia
(ALL) (9), and chronic lymphocytic
leukemia (CLL) (10,11), and has been implicated in prognosis
of CLL, although it remains controversial. Thus, PECAM-1 is
expected to play important roles in regulation of hematopoiesis and
in leukemogenesis, possibly through modulation of apoptosis, cell
adhesion and migration, although the molecular mechanisms involved
have not been explored.
The BCR/ABL fusion protein is encoded by the fusion
gene generated by a reciprocal t(9;22) (q34;q11.2) chromosomal
translocation causing the Philadelphia chromosome (Ph), which is
the molecular signature of chronic myeloid leukemia (CML) and is
also observed in 30–40% of ALL (12,13).
BCR/ABL is a tyrosine kinase that is constitutively activated and
confers survival and proliferation advantages on hematopoietic
cells, thus directly contributing to leukemogenesis. CML cells
express the p210 form of BCR/ABL, and Ph-positive (Ph+)
ALL cells mostly express the p190 form, which is generated by the
difference in location of gene fusion. Imatinib, a tyrosine kinase
inhibitor that blocks the catalytic activity of BCR/ABL, has
demonstrated unprecedented efficacy for treatment of CML or
Ph+ ALL (12–14). However, the resistance to imatinib
may develop in significant portions of patients under treatment,
especially in those with CML in advanced stages or with
Ph+ ALL mostly due to the emergence of mutations in the
BCR/ABL kinase domain, including the most prevalent E255K and T315I
mutations. We previously showed that the E255K or T315I mutant
possessed increased in vitro kinase activities as well
increased ability to induce phosphorylation of itself and several
cellular substrates when expressed in COS7 cells or in
hematopoietic BaF3 cells as compared with unmutated (native)
BCR/ABL (15–17). The increases in transformation
abilities for these mutants have also been reported (18,19).
The Src family tyrosine kinases are also activated in
BCR/ABL-dependent or independent ways and may confer imatinib
resistance on these leukemic cells (20–22).
To develop more efficient therapeutic strategies against
Ph+ leukemias, it is very important to gain more
insights into the molecular mechanisms involved in imatinib
resistance of these leukemias.
In the present study, we show that PECAM-1 is
heavily tyrosine phosphorylated on its ITIMs in BCR/ABL-expressing
cells, including primary Ph+ leukemic cells, at least
partly dependent on the BCR/ABL kinase activity. Tyrosine
phosphorylated PECAM-1 is physically associated with the SHP2
tyrosine phosphatase and most likely acted as a major substrate for
SHP2 in these cells. Intriguingly, tyrosine phosphorylation of
PECAM-1 as well as its physical association with SHP2 was enhanced
by the imatinib-resistance E255K or T315I mutation. Moreover,
overexpression of PECAM-1 enhanced cell adhesion and downregulated
imatinib-induced apoptosis on BCR/ABL-expressing hematopoietic
cells. These results suggest that PECAM-1 is involved in
BCR/ABL-mediated signaling and may enhance the anti-apoptotic
effect.
Materials and methods
Cells and reagents
A clone of murine IL-3-dependent BaF3 cells
transfected with a BCR/ABL cDNA under the control of a
tetracycline-inducible promoter, Ton.B210 and the parental control
clone, Ton.BaF, were kindly provided by Dr G. Daley (23). Ton.B210 cells were cultured in 10%
fetal calf serum (FCS) containing RPMI-1640 medium supplemented
either with 10% Wehi3B conditioned medium as the source of IL-3 or
with 1 μg/ml doxycycline, which induces the expression of
BCR/ABL. Ton.B210/E255K or Ton.B210/T315I cells (16), which inducibly express BCR/ABL with
the E255K or T315I mutation, respectively, and 32Dcl3 cells
expressing BCR/ABL, Ton.32Dp210 (17), were described previously. The human
CML cell line K562 was obtained from the Riken Cell Bank (Ibaraki,
Japan). TMD-5 cells, a double Ph+ ALL-derived cell line
expressing the p190 form of BCR/ABL, were kindly provided by Dr S.
Tohda (24). Leukemic blasts were
isolated from patients with CML myeloid crisis, Ph+ ALL,
or Ph+ biphenotypic acute leukemia as described
previously (16). Written informed
consent was provided according to the Declaration of Helsinki, and
the study was approved by the ethics committee of Tokyo Medical and
Dental University. PLAT-A (25),
an amphotropic virus packaging cell line, and 293T (26), a human embryonic kidney cell line,
were kindly provided by Dr T. Kitamura and Dr S. Yamaoka,
respectively, and maintained in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% FCS.
Imatinib was kindly provided by Novartis (Basel,
Switzerland). Dasatinib and sorafenib were purchased from Toronto
Research Chemicals Inc. (Toronto, Canada) and LKT Laboratories (St.
Paul, MN), respectively. Doxycycline and fibronectin were from
Calbiochem (San Diego, CA) and Gibco-BRL (Grand Island, NY),
respectively. DiOC6 was purchased from Invitrogen
(Carlsbad, CA).
Antibodies against PECAM-1 (SC1506), Lyn (SC15),
CrkL (SC319), SHP2 (SC280), BCR (SC885), and phospho-Y694-STAT5
(SC9359) were purchased from Santa Cruz Biotechnology (Santa Cruz,
CA). An anti-phosphotyrosine monoclonal antibody (4G10, 05-321) as
well as antibody against Gab2 (06-967) was purchased from Millipore
(Billerica, MA). Antibodies against phospho-Y416-Src (9359) and
cleaved caspase-3 (9661) were purchased from Cell Signaling
Technology (Beverly, MA). Antibodies against phospho-Y396-Lyn
(1645) and β-actin were purchased from Epitomics Inc. (Burlingame,
CA) and Sigma, respectively.
Expression plasmids
Expression plasmids for BCR/ABL, pcDNA3-BCR/ABL, and
that for the 56-kDa form of Lyn, pXM-LynA, were described
previously (27,28). Retrovirus vectors, pRevTRE and pMIG
(Addgene plasmid 9044), were obtained from Clontech (Palo Alto, CA)
and Addgene (Cambridge, MA), respectively. pMXs-IG (29) was kindly provided by Dr T.
Kitamura. Expression plasmids for wild-type PECAM-1 and its mutant
with Y663F and Y686F mutations in the ITIM motives, PECAM-1-ITIM
(−), in pcDNA3.0 vector were kindly provided by Dr D. Newman
(30,31). The coding regions for wild-type and
mutant PECAM-1 were subcloned from these plasmids into retroviral
vectors pREV-TRE (HindIII/EcoRV), pMIG (EcoRI)
and pXMs-IG (EcoRI) using the restriction enzymes in
parentheses. Expression plasmids for wild-type and
substrate-trapping mutant of SHP2, pIRES2-EGF-SHP2-Wt and -DA
(Addgene plasmids 12283 and 12286) (32), respectively, were obtained from
Addgene. The coding sequences for SHP2-Wt and -DA were excised from
these plasmids using XhoI and SmaI and subcloned into
pREV-TRE to give pREV-SHP2-Wt and -DA. An expression plasmid for
BCR/ABL, pTetP210, was kindly provided by Dr G. Daley (23). The coding sequence for BCR/ABL was
excised from pTetP210 using EcoRI and subcloned into pRxZiN
obtained from the Riken Gene Bank to give pRxP210.
Transfection and infection
For transient expression in 293T cells, cells were
transfected with indicated plasmids using the Lipofectamine reagent
(Invitrogen) according to the manufacturer’s instructions. Cells
were harvested 48 h after transfection for immunoprecipitation and
immunoblotting.
To obtain BaF3 cells constitutively expressing
BCR/ABL, Ton.BaF cells were infected with the recombinant
retrovirus obtained from PLAT-A transfected with pRxP210, as
described previously (33).
Infected cells were cultured in medium lacking IL-3, and a clone
expressing BCR/ABL at a high level was selected by limited dilution
to give Ton.Bp210-8. This cell line was subsequently transduced
again with pRev-PECAM-1, -ITIM (−), or pRevTRE and selected in
medium containing hygromycin. Cells were used for subsequent
experiments after expression of PECAM-1 or PECAM-1-ITIM (−) was
confirmed by immunoblotting. To obtain Ton.B210 cells
overexpressing wild-type SHP2 or the D425A mutant, Ton.B210 cells
were transduced with pRev-SHP2-Wt or -DA, respectively, and
selected in medium containing hygromycin. To obtain Ton.32Dp210 or
K562 cells overexpressing PECAM-1 or PECAM-1-ITIM (−), these cells
were transduced by the retrovirus vectors in pMXs-IG for pMIG, as
described above. GFP-positive cells were sorted by flow cytometry,
and expression of PECAM-1 or its mutant as well as BCR/ABL was
confirmed by immunoblotting.
Immunoprecipitation and
immunoblotting
Cells were lysed and subjected to
immunoprecipitation and immunoblotting as described previously
(34). The results shown are
representative of experiments repeated at least three times.
Analyses of cell viability, apoptosis,
caspase-3 cleavage, and mitochondrial membrane potential
(Δψm)
Cell viability was assessed by counting viable and
non-viable cell numbers by the trypan blue dye exclusion method.
Flow cytometric analysis of cell cycle and apoptosis was performed
as described previously (16).
Flow cytometric analysis of caspase-3 cleavage was performed using
specific antibodies against cleaved caspase-3 as described
previously (17). For analysis of
Δψm, cells were stained with DiOC6
(Invitrogen) and analyzed by flow cytometry as described previously
(16).
Cell adhesion assay
Adhesion assays were performed essentially as
described previously (35,36). In brief, cells were labeled with 5
μM BCECF/AM (Dojindo, Kumamoto, Japan) and plated on wells
coated with 5 μg/ml fibronectin for 30 min at 37°C. Adherent
cells were measured by Cytofluor II fluorescent plate reader
(PerSeptive Biosystems, Foster City, CA). After subtraction of
background cell binding to bovine serum albumin-coated wells, the
percentage of adherent cells was determined by dividing the
fluorescence intensity of the adherent cells by that of the initial
cell input.
Results
PECAM-1 is tyrosine phosphorylated in
primary Ph+ leukemic and TMD-5 cells in a manner
dependent on the BCR/ABL kinase activity
To examine possible involvement of PECAM-1 in
BCR/ABL-mediated signaling, we first examined whether it is
tyrosine phosphorylated in primary Ph+ leukemic cells.
As shown in Fig. 1A, PECAM-1 was
conspicuously phosphorylated on tyrosine in Ph+
biphenotypic acute leukemia or ALL cells, which was mostly
abolished by treatment with the tyrosine kinase inhibitor imatinib
or dasatinib. Essentially the same results were obtained with
primary leukemic cells from another patient with Ph+ ALL
expressing the p190 form of BCR/ABL and a patient with CML in
myeloid blastic crisis expressing the p210 form of BCR/ABL
(Fig. 1B and C, respectively). We
also examined a Ph+ ALL cell line expressing the p190
form of BCR/ABL, TMD-5, and found that PECAM-1 was also tyrosine
phosphorylated in these cells and was dephosphorylated after
treatment with imatinib (Fig. 1D and
E). These results suggest that PECAM-1 is a substrate of both
p190 and p210 forms of BCR/ABL in these leukemic cells.
PECAM-1 is tyrosine phosphorylated partly
through the Src kinases and is a major substrate of SHP2 in
BCR/ABL-expressing cells
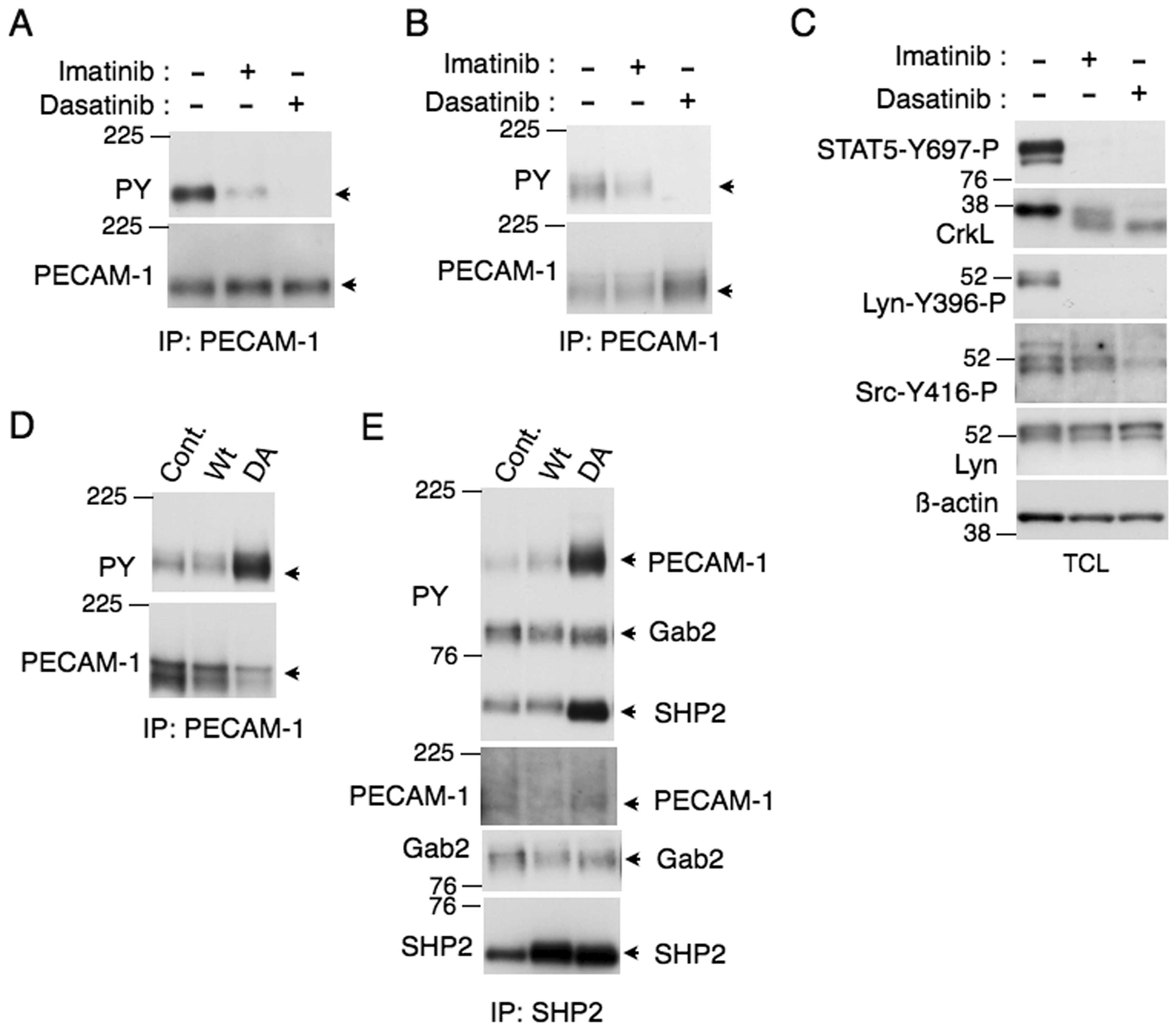
We next examined murine model hematopoietic cell
lines 32Dcl3 and BaF3 engineered to express BCR/ABL. As shown in
Fig. 2A and B, PECAM-1 was
tyrosine phosphorylated also in these cells, which was completely
dephosphorylated by dasatinib. However, imatinib only partially
inhibited tyrosine phosphorylation of PECAM-1 in these cells.
Because dasatinib, but not imatinib, also inhibits the Src family
tyrosine kinases in addition to BCR/ABL, we examined the activation
specific tyrosine phosphorylation of these kinases. As shown in
Fig. 2C, western blot analysis
with an antibody specific for Lyn activation showed that imatinib
drastically inhibited activation of Lyn in BCR/ABL-expressing BaF3
cells. However, examination with an antibody that detects
activation of the various Src family kinases revealed that some of
the activated kinases were resistant to imatinib (Fig. 2C). On the other hand, dasatinib
strongly inhibited activation of the Src family kinases including
Lyn. These data suggest that Lyn is activated by BCR/ABL in these
cells, while some of the other Src family members are
constitutively activated independent of BCR/ABL. We also examined
the well-established BCR/ABL substrates STAT5 and CrkL in these
cells (12). Similar to PECAM-1,
tyrosine phosphorylation of CrkL, which was examined by the
mobility shift assay, was also abrogated by dasatinib but only
partially inhibited by imatinib (Fig.
2C). By contrast, imatinib abrogated tyrosine phosphorylation
of STAT5 in BaF3 cells expressing BCR/ABL. These results suggest
that the tyrosine phospho rylation of PECAM-1 as well as CrkL, but
not that of STAT5, is mediated at least partly by the Src family
kinases activated independent of BCR/ABL in these cells.
We next examined the possibility that tyrosine
phospho rylated PECAM-1 is a substrate for the SHP2 tyrosine
phosphatase in these cells, because SHP2 forms a physical complex
with tyrosine phosphorylated PECAM-1 in various types of cells
(4,37). For this purpose, we overexpressed
wild-type SHP2 or its dominant-negative, substrate-trapping mutant
SHP-2-D425A (7,32,38)
in Ton.B210 cells. As shown in Fig. 2D
and E, overexpression of SHP2-D425A, but not wild-type SHP2,
profoundly enhanced tyrosine phosphorylation of PECAM-1 and SHP2.
Although SHP2 was found to form a complex with PECAM-1 as well as
Gab2 in these cells, tyrosine phosphorylation of PECAM-1, but not
that of Gab2, that was associated with SHP2 was drastically
enhanced by overexpression of SHP2-D425A (Fig. 2E). Tyrosine phosphorylation of SHP2
was also prominently increased in SHP-2-D425A-expressing Ton.B210
cells. These results suggest that PECAM-1 is a major binding
partner and substrate of SHP2 in BCR/ABL-expressing cells.
PECAM-1 is tyrosine phosphorylated on
ITIM by BCR/ABL and Lyn in 293T cells
To examine further the mechanisms of tyrosine
phosphorylation of PECAM-1 induced by BCR/ABL, we next examined it
in transiently transfected 293T cells. As shown in Fig. 3A, co-expression of BCR/ABL induced
tyrosine phosphorylation of wild-type PECAM-1 but not that of
PECAM-1-ITIM (−) with the mutated ITIM motives (Y663F, Y686F), thus
indicating that BCR/ABL induces tyrosine phosphorylation of one or
both of these ITIM motives. In accordance with a previous report
(39), a smaller 120-kDa form of
PECAM-1, which most likely represents a differently glycosylated
form, was unambiguously observed in transfected cells.
Because Lyn was activated by BCR/ABL and the Src
family kinases were implicated in induction of tyrosine
phosphorylation of PECAM-1 in BaF3 cells (Fig. 2B and C), we next examined the
ability of Lyn to phosphorylate PECAM-1. When co-expressed in 293T
cells, Lyn induced a robust tyrosine phosphorylation of PECAM-1,
which was more conspicuously observed than that induced by BCR/ABL
(Fig. 3B and C). Lyn also failed
to induce phosphorylation of PECAM-1-ITIM (−). These results are
consistent with the idea that the Src kinases including Lyn may
partly mediate tyrosine phosphorylation of PECAM-1 on the ITIM
motives in cells expressing BCR/ABL.
Tyrosine phosphorylation of PECAM-1 is
enhanced by the E255K or T315I imatinib-resistant mutation in
BCR/ABL
E255K and T315I are the most predominant mutations
of BCR/ABL causing imatinib resistance in patients and may increase
the kinase activity or change the substrate preferences of BCR/ABL
(15,18). Thus, we next examined tyrosine
phosphorylation of PECAM-1 in cells expressing BCR/ABL harboring
these mutations. As shown in Fig.
4A, PECAM-1 was more prominently tyrosine phosphorylated in
BaF3 cells expressing the E255K or T315I mutant as compared with
cells expressing native BCR/ABL. Moreover, as shown in Fig. 4B, SHP2 physically associated with
PECAM-1 more prominently in cells expressing the E255K or T315I
mutant as compared with cells expressing native BCR/ABL, while
these mutants had less significant effects on complex formation
between SHP2 and Gab2. Tyrosine phosphorylation of STAT5 was also
enhanced, though not as significantly as that of PECAM-1, in cells
expressing the E255K or T315I mutant (Fig. 4C).
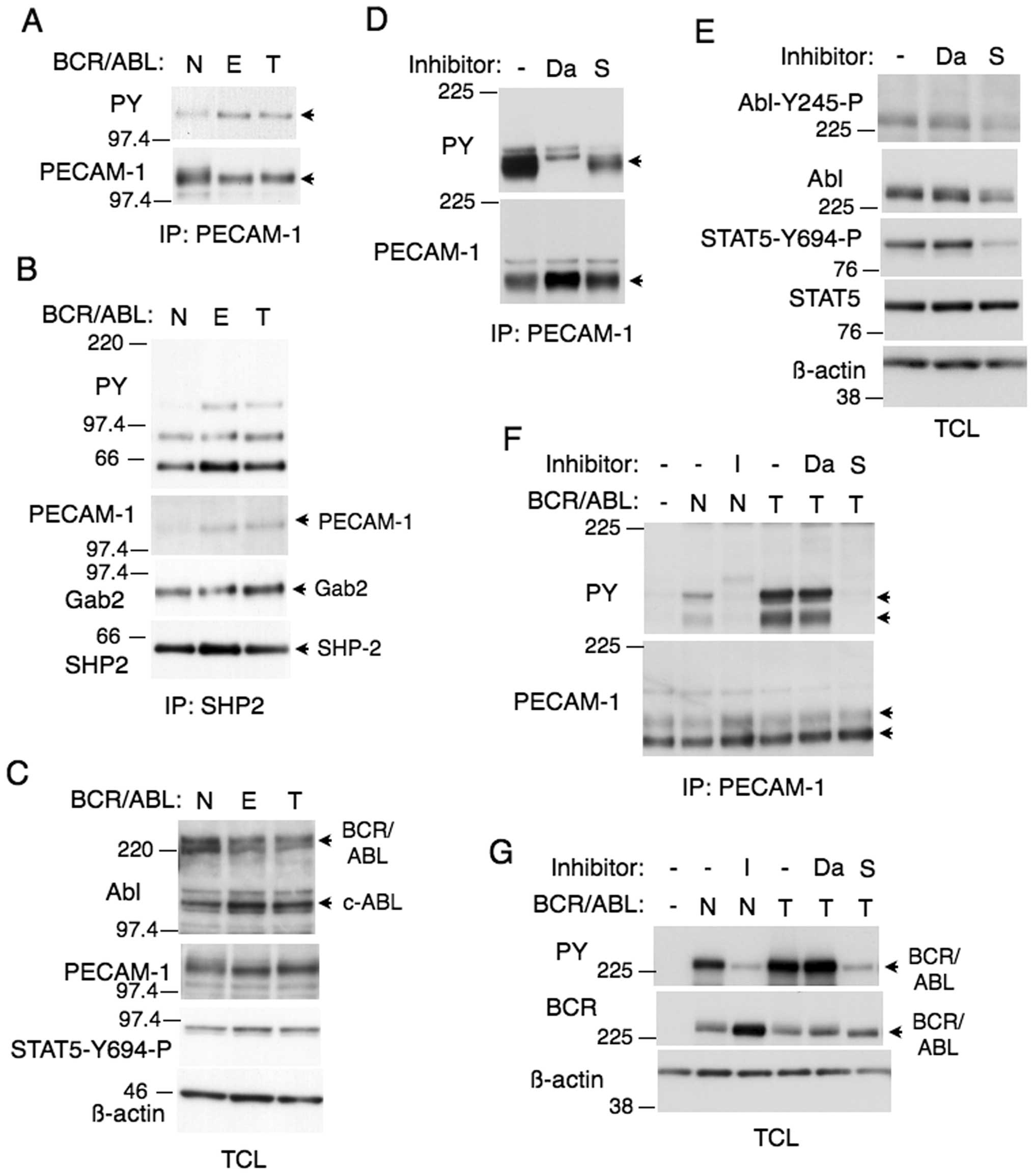 | Figure 4.PECAM-1 is more prominently tyrosine
phosphorylated in cells expressing the E255K or T315I mutant
BCR/ABL. (A, B and C) Ton.B210 (N), Ton.B210/E255K (E), or
Ton.B210/T315I (T) cells, cultured in the presence of doxycycline,
were lysed. Immunoprecipitates with (A) anti-PECAM-1 or (B)
anti-SHP2 as well as (C) total cell lysates (TCL) were subjected to
western blot analysis using indicated antibodies. The position of
PECAM-1 is indicated by an arrowhead. Positions of other proteins
are also indicated. (D and E) Ton.B210/T315I cells were treated
with 0.5 μM dasatinib (Da) or 10 μM sorafenib (S) for
2 h or left untreated as control, as indicated, and analyzed. (F
and G) 293T cells were transiently transfected with plasmids coding
for native BCR/ABL (N) or the T315I mutant (T), as indicated. Cells
were treated with 3 μM imatinib (I), 0.5 μM dasatinib
(Da), or 10 μM sorafenib (S) or left untreated, as
indicated, for 5 h before analysis. |
We next examined the effect of dasatinib on tyrosine
phosphorylation of PECAM-1 in BaF3 cells expressing the T315I
mutant, which is totally resistant to dasatinib as well as imatinib
but sensitive to the multi-kinase inhibitor sorafenib (17,22).
As shown in Fig. 4D, dasatinib or
sorafenib partially inhibited tyrosine phosphorylation of PECAM-1.
It was confirmed that sorafenib, in contrast to dasatinib, showed
inhibitory effect on autophosphorylation of BCR/ABL or
phosphorylation of its substrate STAT5 in these cells (Fig. 4E), thus indicating it partially
inhibited the T315I mutant in these cells. In 293T cells, the T315I
mutant also induced tyrosine phosphorylation of PECAM-1 much more
prominently than native BCR/ABL, which was abolished by sorafenib
but not affected by dasatinib and correlated with
autophosphorylation of BCR/ABL (Fig.
4F and G). The increase in autophosphorylation of the T315I
mutation as compared with native BCR/ABL was not so prominent as
compared with that in PECAM-1 phosphorylation. These results
suggest that the tyrosine phosphorylation of PECAM-1 is mediated
both directly by the T315I BCR/ABL mutant and by the Src family
kinases in BaF3 cells and exclusively by BCR/ABL in 293T cells.
Overexpression of PECAM-1 enhances cell
adhesion and downregulates imatinib-induced apoptosis in
BCR/ABL-expressing cells in an ITIM-independent manner
To examine the cellular effects of PECAM-1 in
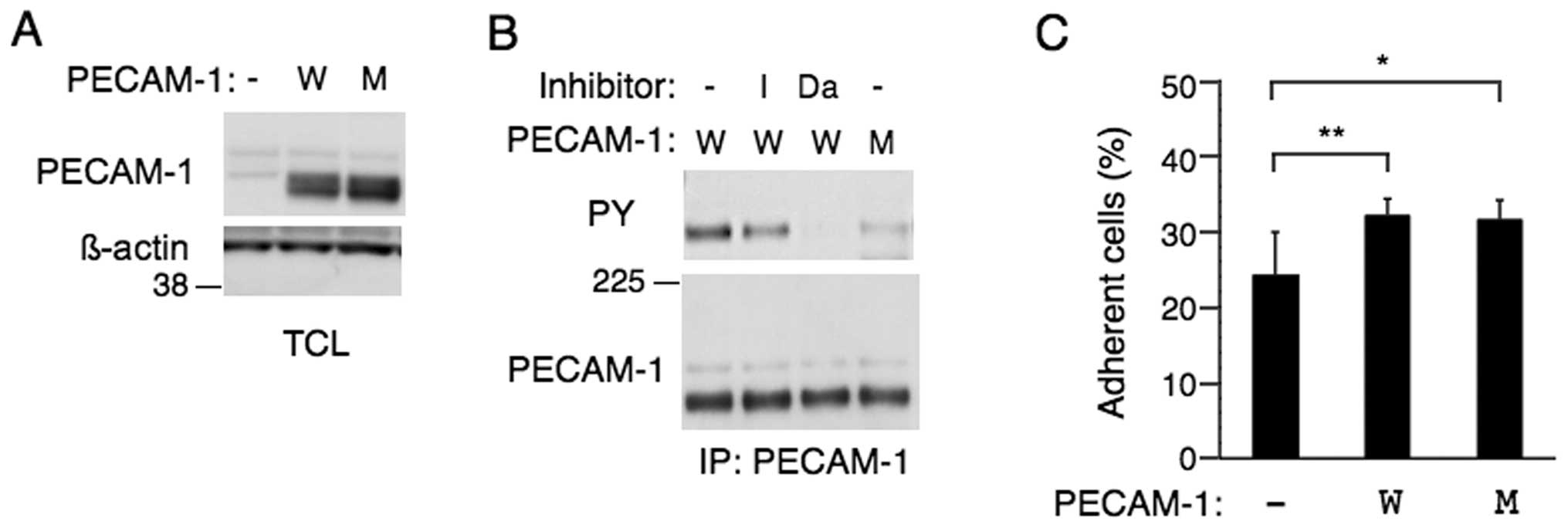
Ph+ leukemic cells, we next examined the human CML K562
cell line, which expresses endogenous PECAM-1 at a barely
detectable level (Fig. 5A). As in
other BCR/ABL-expressing cells, PECAM-1 overexpressed in K562 cells
was tyrosine phosphorylated, which was moderately inhibited or
abolished by imatinib or dasatinib, respectively (Fig. 5B). On the other hand, K562 cells
overexpressing PECAM-1-ITIM (−) showed a very low level of PECAM-1
phosphorylation, thus suggesting that the ITIM motives are mainly
tyrosine phosphorylated also in these Ph+ leukemic
cells.
We first examined the possible effect of PECAM-1 on
cell adhesion. As shown in Fig.
5C, adhesion of K562 to fibronectin-coated plate was enhanced
by overexpression of PECAM-1 or PECAM-1-ITIM (−). These data are in
agreement with the previous reports that PECAM-1 may play a role in
regulation of integrin activation and cell adhesion in various cell
types (3,37).
We next examined the possibility that PECAM-1 may
affect the sensitivity of Ph+ leukemic cells to the
tyrosine kinase inhibitors, because PECAM-1 has been implicated in
prevention of apoptosis in various types of cells (40–44).
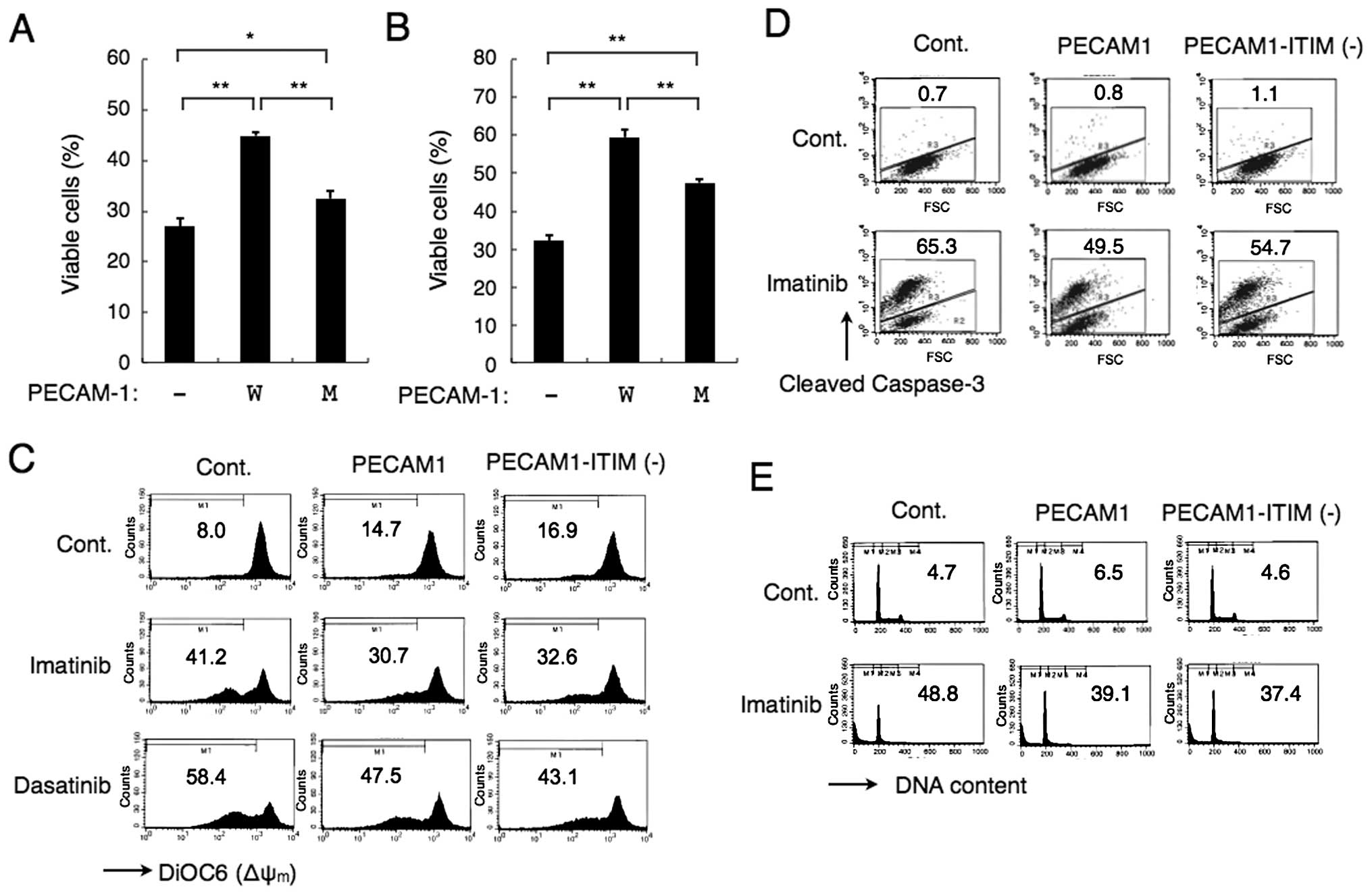
As shown in Fig. 6A and B, the
decline in viability induced by imatinib was downregulated by
overexpression of PECAM-1 or, to a lesser degree, by that of
PECAM-1-ITIM (−) in BCR/ABL-expressing 32D cells or in K562 cells,
respectively. Moreover, PECAM-1 or its mutant was found to
partially prevent depolarization of Δψm in K562 cells
treated with imatinib or dasatinib (Fig. 6C). In accordance with this, PECAM-1
also partially protected K562 cells from imatinib-induced cleavage
of caspase-3 (Fig. 6D). It was
finally confirmed that PECAM-1 or its mutant decreased the number
of K562 cells with sub-G1 DNA content, a hallmark of cells
undergoing apoptosis, after treatment with imatinib (Fig. 6E). Similarly, overexpression of
PECAM-1 or its mutant in 32D cells partially downregulated
depolarization of Δψm, cleavage of caspase-3, and
appearance of cells with sub-G1 DNA content induced by imatinib
(data not shown). Overexpression of PECAM-1 and its mutant showed
comparable anti-apoptotic effects in these repeated experiments in
K562 and 32D cells. These data suggest that PECAM-1 may partially
protect BCR/ABL-expressing leukemic cells treated with the tyrosine
kinase inhibitors from activation of mitochondria-mediated
apoptotic pathway leading to caspase activation and DNA
fragmentation, at least partly, in an ITIM-independent manner.
Discussion
We have found that PECAM-1 was heavily tyrosine
phosphorylated in all the four Ph+ leukemic blast
samples we examined, including those from biphenotypic AL, ALL and
CML-BC patients, the effect was drastically inhibited by imatinib,
thus indicating its phosphorylation was mediated by BCR/ABL
(Fig. 1). Further studies
indicated that tyrosine phosphorylation of PECAM-1 was exclusively
mediated by BCR/ABL in 293T cells but was additionally mediated
through the Src family kinases, including Lyn, that are activated
constitutively or by BCR/ABL in various hematopoietic cell lines,
such as BaF3, 32D and K562 cells (Figs. 2–5). In this regard, it is noteworthy that
BCR/ABL-independent activation of Lyn has been implicated in
development of imatinib-resistance in patients with
mutation-negative BCR/ABL (21).
Although tyrosine phosphorylation of PECAM-1 ITIMs per se
may not be required for the anti-apoptotic effect of PECAM-1 in
imatinib-treated leukemic cells as discussed below, it is tempting
to speculate that interaction of Lyn with PECAM-1 might be involved
in acquisition of resistance in these cases, which needs to be
addressed in future studies. Previous studies have shown that
PECAM-1 is expressed in various types of leukemic cells and
implicated its expression in development of the central nervous
system involvement of ALL (9),
emigration of AML cells from the bone marrow by transendothelial
migration (8), and in
determination of prognosis of CLL (10). However, the tyrosine
phosphorylation status of PECAM-1 has not been examined in these
leukemic cells. Thus, its examination in various leukemic cells may
shed more light on the significance of PECAM-1 in pathogenesis and
prognosis of leukemias.
It is well established that PECAM-1 recruits SHP2
through interaction between its tyrosine phosphorylated ITIMs and
the SH2 domains of the tyrosine phosphatase and activates its
phosphatase activity (3,4,37).
In agreement with this, we observed that tyrosine phosphorylated
PECAM-1 formed a complex with SHP2 in BCR/ABL-expressing
hematopoietic cells (Fig. 4B).
Furthermore, by using the substrate-trapping, loss-of-function
mutant of SHP2, we revealed that PECAM-1 is a major substrate of
SHP2 in these cells, because PECAM-1 represented an SHP2-associated
tyrosine phosphorylated protein that was most significantly
enhanced by introduction of SHP2-D425A in BaF3 cells expressing
BCR/ABL (Fig. 2E). Previously,
Wheadon et al(7) showed
that tyrosine phosphorylation of PECAM-1 and Gab2 that bound SHP2
was significantly increased by overexpression of a
substrate-trapping C459S mutant of SHP2 in BaF3 cells stimulated
with IL-3, thus indicating these proteins are substrates of SHP2 in
these cells. Somewhat different from their results, we found that
tyrosine phosphorylation of Gab2 that bound SHP2 was not
significantly enhanced by overexpression of SHP2-D425A as compared
with that of PECAM-1. Therefore, although BCR/ABL and the IL-3
receptor activate similar signaling events involving SHP2 and Gab2,
PECAM-1 may play a relatively more significant role as an
SHP2-binding substrate as compared with Gab2 in signaling events
downstream of BCR/ABL. Previously, SHP2 was postulated to be
activated by binding with Gab2 and to play a crucial role in
leukemogenesis by BCR/ABL (45–47).
The present study raises a possibility that PECAM-1 may also play
an important role in activation of the SHP2 signaling events
downstream of BCR/ABL.
Intriguingly, the imatinib-resistant BCR/ABL mutants
E255K and T315I showed increased ability as compared with native
BCR/ABL to induce tyrosine phosphorylation of PECAM-1 when
expressed in the murine model hematopoietic cell line BaF3 cells or
in 293T cells (Fig. 4).
Furthermore, the E255K and T315I mutations enhanced the complex
formation between PECAM-1 and SHP2 (Fig. 4B). The enhanced tyrosine
phosphorylation of PECAM-1 by the E255K and T315I mutants may be at
least partly due to their enhanced activities (15–17).
However, as compared with tyrosine phosphorylation of BCR/ABL and
STAT5, that of PECAM-1 was more significantly enhanced by these
mutations (Fig. 4A, C, F and G).
In this regard, it was previously reported that the
imatinib-resistant mutants, including E255K and T315I, exhibited
different patterns of substrate phosphorylation as compared with
native BCR/ABL, thus suggesting altered substrate specificity and
pathway activation (18).
Therefore, it is possible that the E255K and T315I mutants may more
efficiently interact with and phosphorylate PECAM-1 as compared
with native BCR/ABL. Because these mutants are endowed with not
only imatinib resistance but also enhanced transforming activities
(18,19), a possible significance of PECAM-1
in transforming mechanisms for BCR/ABL including these mutants
needs to be addressed in future studies.
The present study revealed that PECAM-1 may enhance
the anti-apoptotic effect of BCR/ABL and partially confer imatinib
resistance on BCR/ABL-expressing cells by inhibiting the
mitochondria-mediated apoptotic mechanisms in a manner at least
partly independent of phosphorylation of ITIMs (Fig. 6). Anti-apoptotic effects of PECAM-1
have been previously reported for several types of cells under
various conditions, including endothelial cells withdrawn from
serum (41,44), hematopoietic cells withdrawn from
GM-CSF (42) and an ALL cell line
treated with UV irradiation or DNA-damaging chemotherapeutic agents
VP16 and AraC (40,43). It was also found that PECAM-1
prevented mitochondria-dependent apoptosis of HEK293T cells induced
by overexpression of Bax (43). In
these studies, involvement of its tyrosine phosphorylation and
binding with SHP2 in anti-apoptotic effects has been controversial
(40,43,44).
Moreover, activation of the PI3K/Akt signaling pathway with
upregulation of anti-apoptotic Bcl-2 and Bcl-Xl expression has been
implicated in some of these reports (41,42,44),
but not in others (40,43). Thus, it is speculated that
anti-apoptotic effects of PECAM-1 are mediated through several
different mechanisms in different types of cells under various
apoptotic stimuli. Intriguingly, PECAM-1 was reported to bind
tyrosine-phosphorylated β-catenin, which was independent of
tyrosine phosphorylation of PECAM-1, and to affect its degradation
through GSK3β-mediated degradation (48). It was also reported that β-catenin
is stabilized through tyrosine phosphorylation by BCR/ABL and may
play an essential role in survival of leukemic stem cells
expressing BCR/ABL (49–51). Further studies are in progress in
our laboratory to address the possible involvement of β-catenin in
PECAM-1-mediated anti-apoptotic mechanisms independent of ITIM
phospho rylation. It is also notable that overexpression of PECAM-1
or PECAM-1-ITIM (−) in K562 cells enhanced adhesion of these cells
to fibronectin (Fig. 5C), because
cell adhesion has been strongly implicated in survival and drug
resistance of leukemic cells (52). Taken together with the previous
report implicating PECAM-1 in migration of hematopoietic cells to
the bone marrow niche (6), where
adhesion as well as soluble factors mediate prosurvival effects
(52), it is possible that PECAM-1
may play a more prominent role in protection of Ph+
leukemic cells from apoptosis in patients treated with imatinib
than that expected from the present study. Further studies are
warranted to address these possibilities and to elucidate the
mechanisms underlying the anti-apoptotic effect of PECAM-1 in
Ph+ leukemic cells.
Acknowledgements
We thank Dr Debra Newman for kind
advice and generous gifts of experimental materials. We also thank
Drs G. Daley, S. Tohda, T. Kitamura, S. Yamaoka and A. Bennett for
kind gifts of experimental materials. This study was supported in
part by grants from Ministry of Education, Culture, Sports, Science
and Technology of Japan (grant nos 21591194, 24591384, 22591030 and
24790966).
References
|
1.
|
Newman PJ: The biology of PECAM-1. J Clin
Invest. 99:3–8. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Privratsky JR, Newman DK and Newman PJ:
PECAM-1: conflicts of interest in inflammation. Life Sci. 87:69–82.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ilan N and Madri JA: PECAM-1: old friend,
new partners. Curr Opin Cell Biol. 15:515–524. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Jackson DE: The unfolding tale of PECAM-1.
FEBS Lett. 540:7–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Dhanjal TS, Pendaries C, Ross EA, et al: A
novel role for PECAM-1 in megakaryocytokinesis and recovery of
platelet counts in thrombocytopenic mice. Blood. 109:4237–4244.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Ross EA, Freeman S, Zhao Y, et al: A novel
role for PECAM-1 (CD31) in regulating haematopoietic progenitor
cell compartmentalization between the peripheral blood and bone
marrow. PLoS One. 3:e23382008. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wheadon H, Edmead C and Welham MJ:
Regulation of interleukin-3-induced substrate phosphorylation and
cell survival by SHP-2 (Src-homology protein tyrosine phosphatase
2). Biochem J. 376:147–157. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Gallay N, Anani L, Lopez A, et al: The
role of platelet/endothelial cell adhesion molecule 1 (CD31) and
CD38 antigens in marrow microenvironmental retention of acute
myelogenous leukemia cells. Cancer Res. 67:8624–8632. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Akers SM, O’Leary HA, Minnear FL, et al:
VE-cadherin and PECAM-1 enhance ALL migration across brain
microvascular endothelial cell monolayers. Exp Hematol. 38:733–743.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ibrahim S, Jilani I, O’Brien S, et al:
Clinical relevance of the expression of the CD31 ligand for CD38 in
patients with B-cell chronic lymphocytic leukemia. Cancer.
97:1914–1919. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Tonino SH, Spijker R, Luijks DM, van Oers
MH and Kater AP: No convincing evidence for a role of CD31-38
interactions in the pathogenesis of chronic lymphocytic leukemia.
Blood. 112:840–843. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Goldman JM and Melo JV: Chronic myeloid
leukemia - advances in biology and new approaches to treatment. N
Engl J Med. 349:1451–1464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wong S and Witte ON: The BCR-ABL story:
bench to bedside and back. Annu Rev Immunol. 22:247–306. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gruber F, Mustjoki S and Porkka K: Impact
of tyrosine kinase inhibitors on patient outcomes in Philadelphia
chromosome-positive acute lymphoblastic leukaemia. Br J Haematol.
145:581–597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yamamoto M, Kurosu T, Kakihana K, Mizuchi
D and Miura O: The two major imatinib resistance mutations E255K
and T315I enhance the activity of BCR/ABL fusion kinase. Biochem
Biophys Res Commun. 319:1272–1275. 2004. View Article : Google Scholar
|
|
16.
|
Kurosu T, Tsuji K, Kida A, Koyama T,
Yamamoto M and Miura O: Rottlerin synergistically enhances
imatinib-induced apoptosis of BCR/ABL-expressing cells through its
mitochondrial uncoupling effect independent of protein kinase
C-delta. Oncogene. 26:2975–2987. 2007. View Article : Google Scholar
|
|
17.
|
Kurosu T, Ohki M, Wu N, Kagechika H and
Miura O: Sorafenib induces apoptosis specifically in cells
expressing BCR/ABL by inhibiting its kinase activity to activate
the intrinsic mitochondrial pathway. Cancer Res. 69:3927–3936.
2009. View Article : Google Scholar
|
|
18.
|
Griswold IJ, MacPartlin M, Bumm T, et al:
Kinase domain mutants of Bcr-Abl exhibit altered transformation
potency, kinase activity, and substrate utilization, irrespective
of sensitivity to imatinib. Mol Cell Biol. 26:6082–6093. 2006.
View Article : Google Scholar
|
|
19.
|
Skaggs BJ, Gorre ME, Ryvkin A, et al:
Phosphorylation of the ATP-binding loop directs oncogenicity of
drug-resistant BCR-ABL mutants. Proc Natl Acad Sci USA.
103:19466–19471. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Danhauser-Riedl S, Warmuth M, Druker BJ,
Emmerich B and Hallek M: Activation of Src kinases p53/56lyn and
p59hck by p210bcr/abl in myeloid cells. Cancer Res. 56:3589–3596.
1996.PubMed/NCBI
|
|
21.
|
Wu J, Meng F, Kong LY, et al: Association
between imatinib-resistant BCR-ABL mutation-negative leukemia and
persistent activation of LYN kinase. J Natl Cancer Inst.
100:926–939. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Quintas-Cardama A, Kantarjian H and Cortes
J: Flying under the radar: the new wave of BCR-ABL inhibitors. Nat
Rev Drug Discov. 6:834–848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Klucher KM, Lopez DV and Daley GQ:
Secondary mutation maintains the transformed state in BaF3 cells
with inducible BCR/ABL expression. Blood. 91:3927–3934.
1998.PubMed/NCBI
|
|
24.
|
Tohda S, Sakashita C, Fukuda T, Murakami N
and Nara N: Establishment of a double Philadelphia
chromosome-positive acute lymphoblastic leukemia-derived cell line,
TMD5: effects of cytokines and differentiation inducers on growth
of the cells. Leuk Res. 23:255–261. 1999. View Article : Google Scholar
|
|
25.
|
Morita S, Kojima T and Kitamura T: Plat-E:
an efficient and stable system for transient packaging of
retroviruses. Gene Ther. 7:1063–1066. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
DuBridge RB, Tang P, Hsia HC, Leong PM,
Miller JH and Calos MP: Analysis of mutation in human cells by
using an Epstein-Barr virus shuttle system. Mol Cell Biol.
7:379–387. 1987.PubMed/NCBI
|
|
27.
|
Mizuchi D, Kurosu T, Kida A, et al:
BCR/ABL activates Rap1 and B-Raf to stimulate the MEK/Erk signaling
pathway in hematopoietic cells. Biochem Biophys Res Commun.
326:645–651. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Chin H, Arai A, Wakao H, Kamiyama R,
Miyasaka N and Miura O: Lyn physically associates with the
erythropoietin receptor and may play a role in activation of the
Stat5 pathway. Blood. 91:3734–3745. 1998.PubMed/NCBI
|
|
29.
|
Kitamura T, Koshino Y, Shibata F, et al:
Retrovirus-mediated gene transfer and expression cloning: powerful
tools in functional genomics. Exp Hematol. 31:1007–1014. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Jackson DE, Kupcho KR and Newman PJ:
Characterization of phosphotyrosine binding motifs in the
cytoplasmic domain of platelet/endothelial cell adhesion molecule-1
(PECAM-1) that are required for the cellular association and
activation of the protein-tyrosine phosphatase, SHP-2. J Biol Chem.
272:24868–24875. 1997. View Article : Google Scholar
|
|
31.
|
Newman DK, Hamilton C and Newman PJ:
Inhibition of antigen-receptor signaling by platelet endothelial
cell adhesion molecule-1 (CD31) requires functional ITIMs, SHP-2,
and p56(lck). Blood. 97:2351–2357. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Kolli S, Zito CI, Mossink MH, Wiemer EA
and Bennett AM: The major vault protein is a novel substrate for
the tyrosine phosphatase SHP-2 and scaffold protein in epidermal
growth factor signaling. J Biol Chem. 279:29374–29385. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Oshikawa G, Nagao T, Wu N, Kurosu T and
Miura O: c-Cbl and Cbl-b ligases mediate
17-allylaminodemethoxygeldanamycin-induced degradation of
autophosphorylated Flt3 kinase with internal tandem duplication
through the ubiquitin proteasome pathway. J Biol Chem.
286:30263–30273. 2011. View Article : Google Scholar
|
|
34.
|
Miura O, Cleveland JL and Ihle JN:
Inactivation of erythropoietin receptor function by point mutations
in a region having homology with other cytokine receptors. Mol Cell
Biol. 13:1788–1795. 1993.PubMed/NCBI
|
|
35.
|
Arai A, Nosaka Y, Kanda E, Yamamoto K,
Miyasaka N and Miura O: Rap1 is activated by erythropoietin or
interleukin-3 and is involved in regulation of beta1
integrin-mediated hematopoietic cell adhesion. J Biol Chem.
276:10453–10462. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Jin A, Kurosu T, Tsuji K, et al: BCR/ABL
and IL-3 activate Rap1 to stimulate the B-Raf/MEK/Erk and Akt
signaling pathways and to regulate proliferation, apoptosis, and
adhesion. Oncogene. 25:4332–4340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Newman PJ and Newman DK: Signal
transduction pathways mediated by PECAM-1: new roles for an old
molecule in platelet and vascular cell biology. Arterioscler Thromb
Vasc Biol. 23:953–964. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Flint AJ, Tiganis T, Barford D and Tonks
NK: Development of ‘substrate-trapping’ mutants to identify
physiological substrates of protein tyrosine phosphatases. Proc
Natl Acad Sci USA. 94:1680–1685. 1997.
|
|
39.
|
Albelda SM, Muller WA, Buck CA and Newman
PJ: Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a
novel vascular cell-cell adhesion molecule. J Cell Biol.
114:1059–1068. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Bergom C, Goel R, Paddock C, et al: The
cell-adhesion and signaling molecule PECAM-1 is a molecular
mediator of resistance to genotoxic chemotherapy. Cancer Biol Ther.
5:1699–1707. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Bird IN, Taylor V, Newton JP, et al:
Homophilic PECAM-1(CD31) interactions prevent endothelial cell
apoptosis but do not support cell spreading or migration. J Cell
Sci. 112:1989–1997. 1999.PubMed/NCBI
|
|
42.
|
Ferrero E, Belloni D, Contini P, et al:
Transendothelial migration leads to protection from
starvation-induced apoptosis in CD34+CD14+
circulating precursors: evidence for PECAM-1 involvement through
Akt/PKB activation. Blood. 101:186–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Gao C, Sun W, Christofidou-Solomidou M, et
al: PECAM-1 functions as a specific and potent inhibitor of
mitochondrial-dependent apoptosis. Blood. 102:169–179. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Limaye V, Li X, Hahn C, et al: Sphingosine
kinase-1 enhances endothelial cell survival through a
PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2
family members. Blood. 105:3169–3177. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Chan G, Kalaitzidis D and Neel BG: The
tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis
Rev. 27:179–192. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Scherr M, Chaturvedi A, Battmer K, et al:
Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2
expression in chronic myeloid leukemia (CML). Blood. 107:3279–3287.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Chen J, Yu WM, Daino H, Broxmeyer HE,
Druker BJ and Qu CK: SHP-2 phosphatase is required for
hematopoietic cell transformation by Bcr-Abl. Blood. 109:778–785.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Biswas P, Canosa S, Schoenfeld D, et al:
PECAM-1 affects GSK-3beta-mediated beta-catenin phosphorylation and
degradation. Am J Pathol. 169:314–324. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Coluccia AM, Vacca A, Dunach M, et al:
Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through
its tyrosine phosphorylation. EMBO J. 26:1456–1466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Hu Y, Chen Y, Douglas L and Li S:
beta-Catenin is essential for survival of leukemic stem cells
insensitive to kinase inhibition in mice with BCR-ABL-induced
chronic myeloid leukemia. Leukemia. 23:109–116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Shang YC, Zhang C, Wang SH, et al:
Activated beta-catenin induces myogenesis and inhibits adipogenesis
in BM-derived mesenchymal stromal cells. Cytotherapy. 9:667–681.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Meads MB, Hazlehurst LA and Dalton WS: The
bone marrow microenvironment as a tumor sanctuary and contributor
to drug resistance. Clin Cancer Res. 14:2519–2526. 2008. View Article : Google Scholar : PubMed/NCBI
|