Introduction
Lung cancer is the leading cause of cancer mortality
worldwide. Although remarkable developments in various cancer
treatments have been made, the overall 5-year relative survival
rate of patients with lung cancer remains less than 20% in most
countries (1–3). Moreover, in the advanced stages of
lung cancer, only palliative therapies (chemotherapy and/or
radiotherapy) are given as the standard of care (4). Therefore, new therapeutic agents to
improve the prognosis of lung cancer are urgently needed. Various
novel therapeutic strategies are currently under investigation
because the clinical use of cytotoxic drugs is limited due to
intrinsic or acquired resistance and toxicity (5). In addition, there has been a paradigm
shift in cancer therapeutics from the use of conventional cytotoxic
drugs to the use of variable molecular-targeted therapeutics
including gefitinib in lung cancer or trastuzumab in breast cancer
(6,7). Some candidate molecules are expressed
in specific cancer cells and methods of targeting these alterations
have been developed. For example, several monoclonal antibodies or
small molecules that can inhibit the growth and proliferation of
specific cancers are now available (8,9). A
better understanding of the molecular mechanisms of targeted drug
action has shed light on the treatment of lung cancer, and novel
agents that target specific intracellular pathways related to the
distinctive properties of cancer cells continue to be
developed.
Epidermal growth factor receptor (EGFR) is
occasionally mutated in non-small cell lung cancer and
heterogeneity in treatment response could result from differences
in EGFR mutation status (10,11).
The A549 tumor-cell line with wild-type EGFR, derived from a human
alveolar epithelial cell carcinoma, has been studied in
vitro to evaluate lung cancer behavior (12). HCC827 cells are lung adenocarcinoma
cells with an activating mutation in the EGFR tyrosine kinase
domain (13). In the present
study, we evaluated a new cell surface molecule expressed on both
A549 and HCC827 cells to consider the different response dependent
on EGFR mutation status.
Centrocyte/centroblast marker 1 (CM1) is a new
putative germinal center marker defined by a monoclonal antibody
developed against concanavalin-A-activated peripheral blood
mononuclear cells (PBMCs). It was originally reported that several
cancer cell lines, such as Raji, Ramos and IM-9, which originate
from human B cells, express CM1 molecules on their cell membranes
(14). Moreover, the expression of
CM1 is induced during transformation of B cells by Epstein-Barr
virus infection. Most importantly, ligation of CM1-induced
apoptosis of CM1+ cells (15,16).
These studies suggest that CM1 may be expressed on other cancer
cells including lung cancer and serve as a potential target in
CM1+ cancer cells. In this study, we investigated the
expression and role of CM1 molecules in both A549 and HCC827 lung
cancer cells.
Materials and methods
Cell preparation and culture
A549 and HCC827 cells were obtained from the
American Type Culture Collection (ATCC, Rockville, MD, USA). These
cells were grown and maintained in RPMI-1640 medium (HyClone,
Logan, UT, USA) containing 2 mM L-glutamine, 10 U/ml penicillin,
100 μg/ml streptomycin and 10% heat-inactivated fetal bovine
serum (HyClone) at 37°C in a 5% CO2 incubator.
Flow cytometry
The cells were washed twice with phosphate-buffered
saline (PBS) and incubated with either fluorescein isothiocyanate
(FITC)-conjugated mouse anti-human CM1 antibody (a gift from Dr
W.J. Lee, Seoul National University, Seoul, Korea), mouse
anti-human Fas antibody (BD Pharmingen, San Jose, CA, USA) or mouse
anti-human FasL antibody (BD Pharmingen) for 30 min on ice. The
cells incubated with anti-Fas and anti-FasL antibodies were further
stained with FITC-conjugated goat anti-mouse IgG antibody (Sigma,
St. Louis, MO, USA) for 30 min on ice and washed twice with PBS.
MOPC (IgG1) was purchased from Sigma-Aldrich (St. Louis, MO, USA)
as the isotype control antibody. A FACSCalibur (BD Pharmingen) flow
cytometry was used for analysis.
Confocal microscopy
To detect surface molecules, cells were incubated
with FITC-conjugated anti-CM1 antibody (mouse IgG1) and,
to detect intracellular molecules, cells were first treated with
permeabilization buffer (0.1% saponin in PBS) before incubation
with various antibodies. Cells were then incubated with primary
antibody against cytochrome c (mouse IgG2b, Santa
Cruz Biotechnology, Santa Cruz, CA, USA), AIF (mouse
IgG2b, Santa Cruz Biotechnology) or endoG (mouse IgG2b,
Santa Cruz Biotechnology). Cells were then washed thrice with PBS,
and incubated with FITC-conjugated goat anti-mouse IgG antibody
(Sigma-Aldrich) for 30 min. The nucleus was stained with propidium
iodide (PI, BD Pharmingen) at RT for 10 min. After being washed
thrice with PBS, cells were mounted onto microscopic slides under
coverslips using fluorescent mounting medium (DakoCytomation,
Glostrup, Denmark). Fluorescent cells were examined by Confocal
Laser-Scanning microscopy (510 META, Carl Zeiss, Jena, Germany) at
×400 magnification, and images were acquired with Confocal
Microscopy Software Release 3.0 (510 META, Carl Zeiss).
Induction of CM1-mediated signaling
For immobilization, anti-CM1 or MOPC21 (IgG1κ,
isotype control antibody, Sigma-Aldrich) antibodies (50
μg/ml in PBS) were incubated overnight at 4°C on a 96-well
culture plate (0.1 ml/well; washed with PBS before use). Each
antibody was used at various concentrations (0.625, 1.25, 2.5, 5
and 10 μg/ml). A549 and HCC827 cells (5×105
cells/well) were incubated in plates coated with antibodies at 37°C
for 2, 4, 8 and 12 h. In some cases, cells were pretreated with
z-VAD-fmk as a broad caspase inhibitor (20 μM, Calbiochem,
La Jolla, CA, USA), z-DEVD-fmk
(N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone, 20
μM in DMSO, a caspase-3 inhibitor), and z-IETD-fmk
(N-benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone, 20
μM in DMSO, a caspase-8 inhibitor) from Calbiochem for 2 h
before stimulation with anti-CM1 antibody. In some cases, NAC, a
ROS inhibitor (10 mM, Sigma-Aldrich) or ZB4, anti-Fas antibody (0.5
μg/ml, Abcam, Cambridge, UK), was added 1 h before
stimulation with anti-CM1 antibodies. Cells were washed of all
chemicals and antibodies before CM1 stimulation.
Proliferation measurement by AlamarBlue
assay
The evaluation of cell growth was determined using
an AlamarBlue assay. A549 and HCC827 cells (5×104
cells/well) were cultured in medium containing 10% FBS in 96-well
flat bottom plates and treated with 10 μg/ml anti-CM1
antibodies or MOPC (isotype control antibodies) for 48 h before
adding AlamarBlue solution (Serotec Ltd, Kidlington, UK).
AlamarBlue was added (10% by volume) to each well and the relative
fluorescence was determined 7 h later by Fluorometer (Synergy HT;
Bio-Tek Instruments Inc., Winnoski, VT, USA; excitation, 570 nm;
emission, 600 nm). Experiments were performed in triplicate.
Detection of CM1-mediated apoptosis
To evaluate the apoptosis-inducing effect of CM1,
cells were analyzed for Annexin V expression by flow cytometry.
Following treatment, cell were collected and washed twice with PBS
and resuspended in 100 μl of Annexin V binding buffer (10 mM
of HEPES/NaOH pH 7.4, 140 mM of NaCl, 2.5 mM of CaCl2).
After 2 μl of FITC or PE-conjugated Annexin V (BD
Pharmingen) was added, cells were incubated in the dark at RT for
15 min with gentle vortexing. Finally, 400 μl of Annexin V
binding buffer was added to each tube and cells were analyzed using
FACSCalibur (BD Pharmingen).
Measurement of mitochondrial membrane
potential and ROS generation
Cells were pretreated with 10 μM of 2′,
7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Molecular Probes,
Eugene, OR) for 30 min and ROS levels were assessed by the
conversion of DCFH to the highly fluorescent dichlorofluorescein
(DCF) in the presence of intracellular ROS. Cells were washed twice
with cold PBS and then incubated with immobilized mouse anti-human
CM1 antibody or the isotype control antibody. To measure
mitochondrial membrane potentials, cells were collected and
incubated in 100 μl of PBS containing 20 nM of
3,3′-dihexyloacarbocyanine iodide (DiOC6, Molecular
Probes) at 37°C for 15 min. Cells were then collected and washed
with cold PBS twice, and ROS levels and mitochondrial membrane
potential were detected by FACSCalibur (BD Pharmingen).
Reverse transcription polymerase chain
reaction
Cells were washed three times with PBS. RNA was
extracted using RNeasy mini kit (Qiagen, Hilden, Germany) and cDNA
was produced using RT premix (Bioneer, Daejeon, Korea). FasL cDNA
was then amplified using 5′-GGT CCA TGC CTC TGG AAT GG-3′ as a
forward primer and 5′-CAC ATC TGC CCA GTA GTG CA-3′ as a reverse
primer. PCR amplification was also performed using specific primer
sets (Bioneer) for Bcl-2 (upstream primer, 5′-GGA TTG TGG CCT TCT
TTG AG; downstream primer, 5′-CAG CCA GGA GAA ATC AAA CAG, 209-bp
product), Bax (upstream primer, 5′-CCA AGA AGC TGA GCG AGT GT;
downstream primer, 5′-CAG CCC ATG ATG GTT CTG AT, 250-bp product)
and Bad (upstream primer, 5′-CGA GTG AGC AGG AAG ACT CC; downstream
primer, 5′-CTG TGC TGC CCA GAG CTT, 299-bp product). For control, a
specific primer set for β-actin (upstream primer, 5′-ATC CAC GAA
ACT ACC TTC AA; downstream primer, 5′-ATC CAC ACG GAG TAC TTG C)
was used, which yielded a 200-bp product. PCR (25 cycle; 20 sec at
94°C, 10 sec at 60°C, 30 sec at 72°C) was performed using AccuPower
PCR premix (Bioneer). PCR products were analyzed by agarose gel
electrophoresis and visualized with ethidium bromide under UV light
using Multiple Gel-DOC system (Fujifilm, Tokyo, Japan).
Western blot analysis
Cells were lysed in 50 mM Tris-Cl (pH 7.5)
containing 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS
and a protease inhibitor (Sigma-Aldrich) on ice for 10 min. Lysates
were clarified by centrifugation at 14,000 × g for 15 min at 4°C.
Protein concentrations were determined by the Bradford method.
Sample-loading buffer was added, the mixture was boiled for 5 min
and the proteins were then separated by electrophoresis on 10%
polyacrylamide-SDS gels before being transferred to the
nitrocellulose membrane by electroblotting. Blots were blocked
using overnight incubation with 5% skim milk in TBS, and then
incubated for 1 h at RT with primary antibodies, followed by
horseradish peroxidaseconjugated secondary antibodies (Amersham
Biosciences). The following primary antibodies were used:
anti-caspase-8, anti-caspase-3, Bid, phospho-ERK1/2
(Thr202/Tyr204), ERK1/2, phospho-Akt
(Ser473), Akt, caspase-9, Bcl-XL, phospho-JNK
(Thr183/Tyr185), JNK and β-actin antibodies
from Cell Signaling Technology (Beverly, MA); phospho-c-jun and
c-jun from Santa Cruz Biotechnology; PARP [poly(ADP-ribose)
polymerase] from Upstate Biotechnology (Lake Placid, NY). Data were
analyzed using ImageJ 1.38 software.
Results
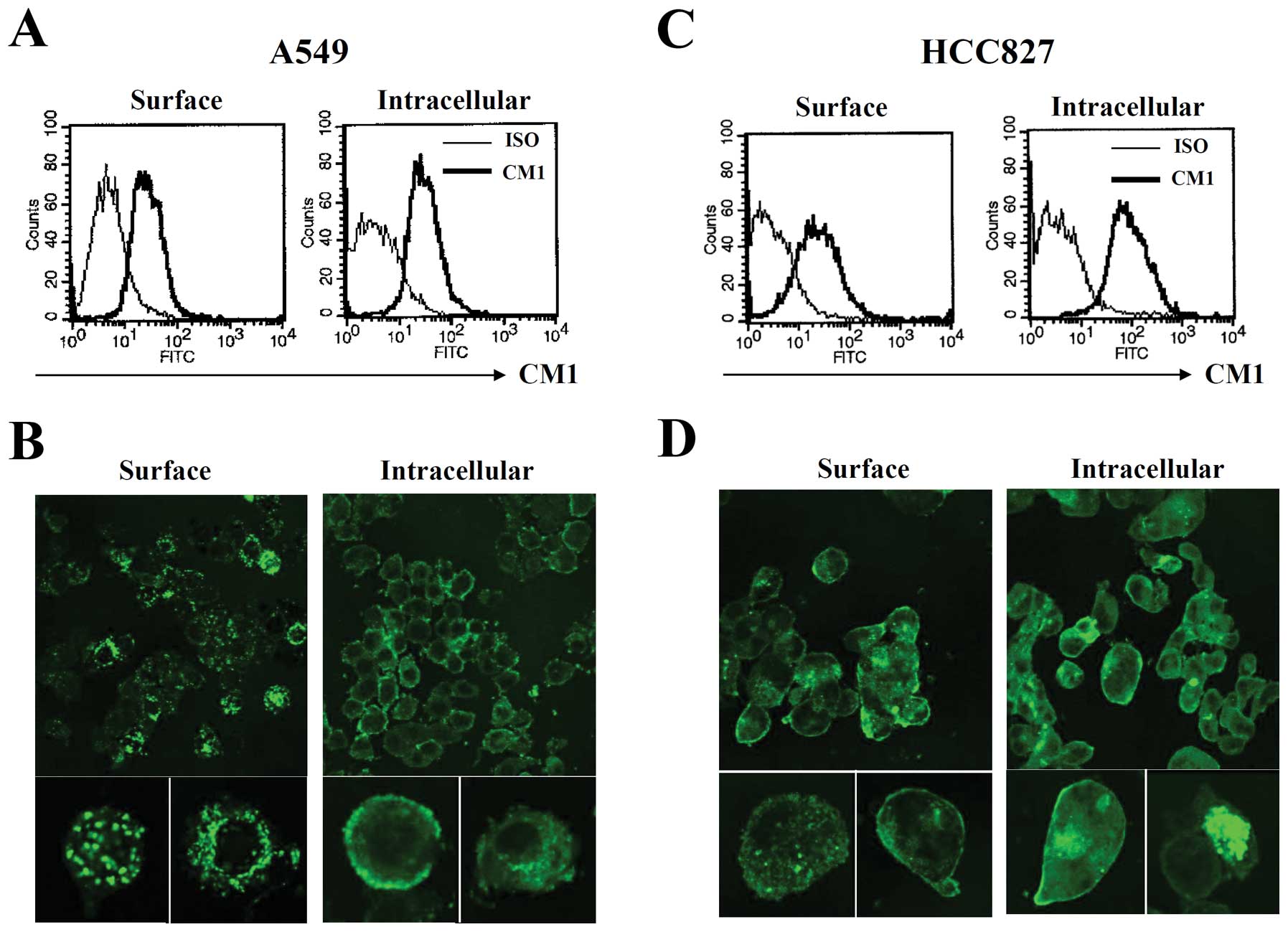
CM1 expression on the surface of lung
cancer cells
CM1 expression was evaluated on A549 and HCC827 lung
cancer cells using flow cytometry and confocal microscopy. Flow
cytometric analysis results showed that both A549 and HCC827 cells
expressed CM1 molecules on their cell surface and intracellularly.
Moreover, the CM1 expression pattern was also confirmed by confocal
microscopy. Interestingly, the expression pattern of CM1 differed
in the two lines: CM1 expression was clustered on the cell surface
in A549 cells, but more dispersed on the cell surface in HCC827
cells (Fig. 1).
Cross-linking of CM1 inhibits the growth
of lung cancer cells
We first determined the anti-proliferative effects
of anti-CM1 mAb on both lung cancer cell lines using AlamarBlue
assay after treatment with anti-CM1 mAb at various concentrations.
Growth inhibition of both lung cancer cell lines was detectable
following 48 h treatment using anti-CM1 mAb concentrations starting
at 1 μg/ml (data not shown). As shown in Fig. 2A and C, approximately 40–60% growth
inhibition was achieved at a 10 μg/ml concentration in both
A549 and HCC827 lung cancer cells.
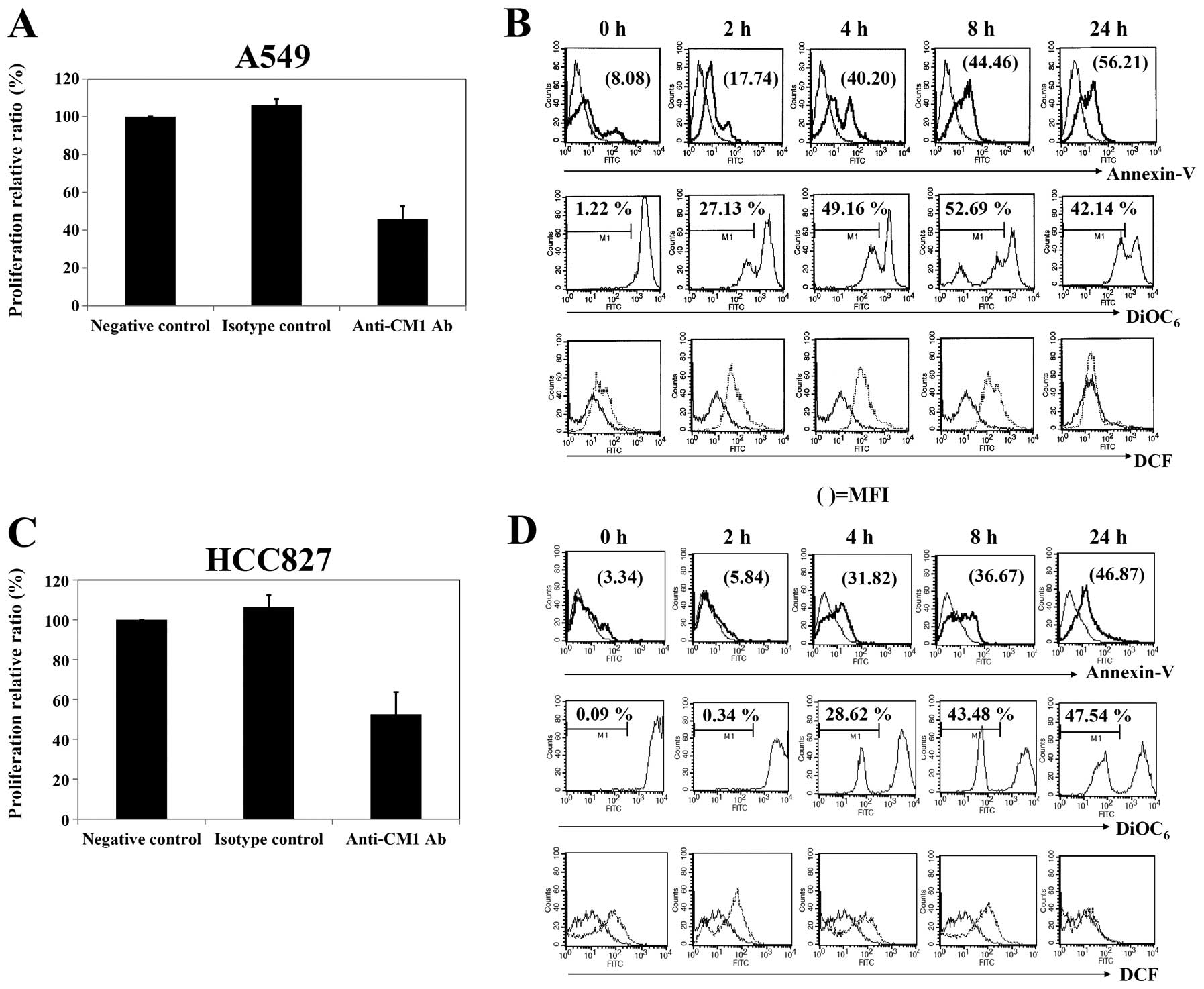 | Figure 2.Proliferation and apoptosis in A549
and HCC827 lung cancer cells after ligation of CM1. (A and C) To
measure the extent of proliferation, cells were incubated
(5×104 cells/well, 200 μl) in an anti-CM1 or MOPC
antibody-coated (2.5 μg/well) plate for 48 h, and then
determined by an alamarBlue assay. (B and D) To evaluate apoptosis,
ROS generation and mitochondrial membrane potential disruption,
cells were incubated (5×105 cells/well, 200 μl)
in an anti-CM1 or MOPC antibody-coated (2.5 μg/well) pate
for 2, 4, 8 and 24 h. Cells were collected at the indicated times
and washed with PBS. Twenty-four hours later, cells were stained
with FITC-conjugated Annexin V as described in Materials and
methods and analyzed by flow cytometry. The indicated number
represents the apoptotic cell proportion (B and D, first row). The
number of DiOC6 represents the percentage of
mitochondrial membrane potential disruption of cells. The thin-line
histograms represent the apoptosis of MOPC (isotype control for CM1
antibody) treated cells, and thick-line histograms represent that
of anti-CM1 antibody-treated cells (B and D, second row). To
measure ROS generation, cells were pre-incubated with DCFH-DA
before being treated with anti-CM1 or MOPC antibody. Thin-line and
dotted histograms represent ROS level of isotype controlled and
anti-CM1 antibody-treated cells, respectively. The number in DCF
histograms indicates the MFI (B and D, third row). Results are
representative of four independent experiments. |
CM1 stimulation induces apoptosis in both
A549 and HCC827 cells
To determine whether CM1 stimulation could induce
apoptosis of CM1 expressing A549 and HCC827 cells, as described in
previous studies (7,8), A549 and HCC827 cells were stimulated
with anti-CM1 mAb for 2, 4, 8 and 24 h. Cells were stained with
FITC-labeled Annexin V and analyzed by flow cytometry. The ligation
of surface CM1 molecules with anti-CM1 antibody increased the
number of Annexin V positive apoptotic cells in both A549 and
HCC827 cell lines. The increase of apoptotic cells in A549 was
detected earlier than in HCC827 cells (Fig. 2B and D, first row).
We next characterized the molecular mechanisms
underlying CM1-mediated apoptosis in these lung cancer cells. The
ligation of surface CM1 molecules with anti-CM1 antibody generated
ROS and disrupted mitochondrial membrane potential in both A549 and
HCC827 lung cancer cells. Induction of mitochondrial membrane
potential disruption in A549 cells was earlier than that in HCC827
cells (Fig. 2B and D, second and third
rows). The ROS level was restored to normal in about 24 h in
both cell types.
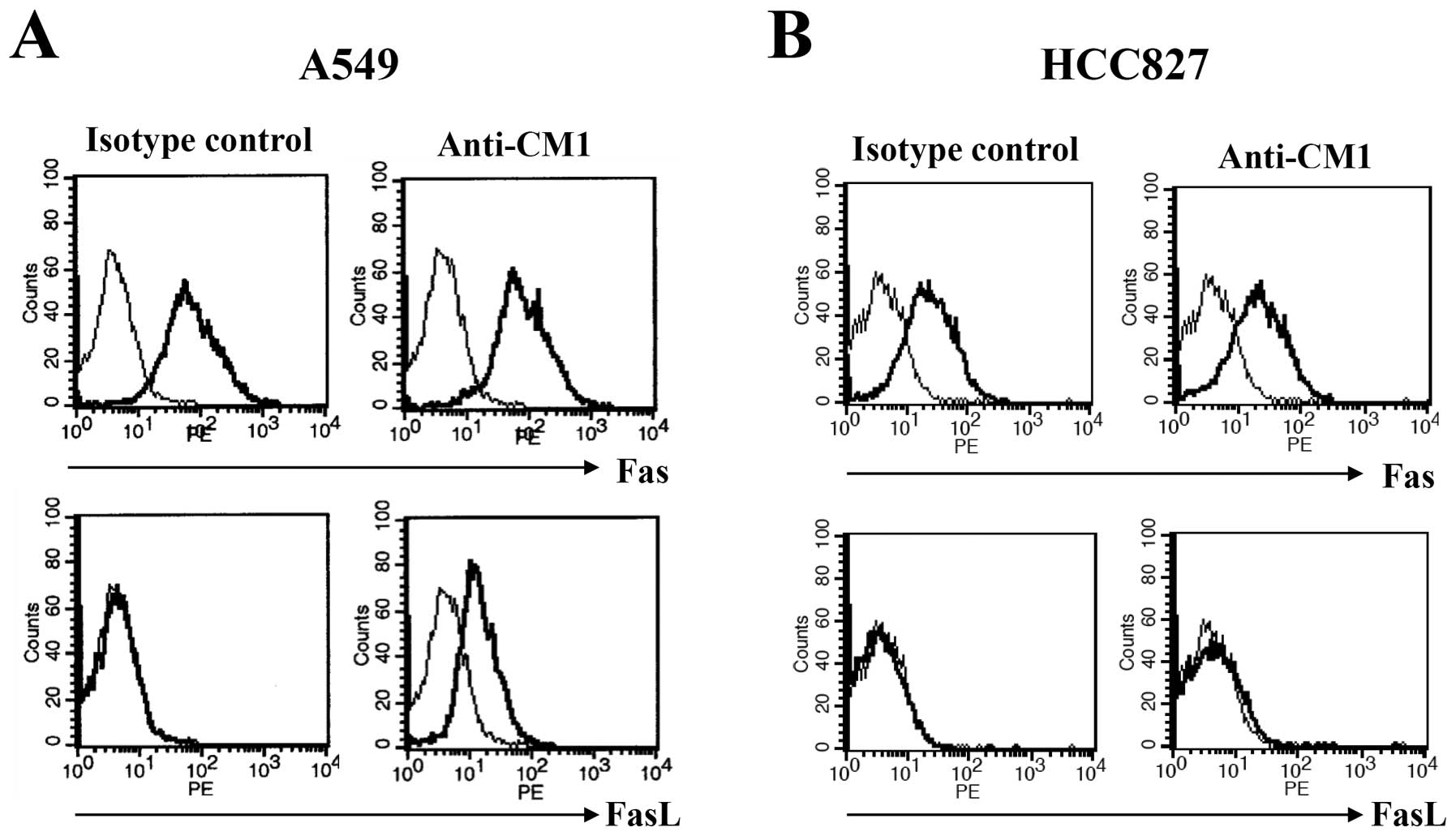
CM1 ligation induces Fas ligand
expression only on A549 cells
To examine surface molecules associated with
apoptosis after CM1 ligation, flow cytometric analysis was
performed for surface Fas (CD95) and Fas ligand (FasL, CD178). A549
and HCC827 lung cancer cells were incubated with immobilized
anti-CM1 or MOPC antibodies (5 μg/ml) for 2 h. Cells were
washed and stained using FITC conjugated anti-Fas and FasL
antibodies. Fas was expressed constitutively on both A549 and
HCC827 cells and was unaffected by CM1 ligation. However, FasL was
not expressed on unstimulated lung cancer cells, but was induced in
A549 but not HCC827 cells after CM1 ligation (Fig. 3). To confirm induction of FasL on
A549 cells after CM1 ligation, cells were incubated with
immobilized anti-CM1 or MOPC antibody and analyzed by reverse
transcription polymerase chain reaction (RT-PCR) for FasL message.
FasL mRNA was increased after CM1 ligation on A549 cells (Fig. 5A, first row). We also performed
these experiments in HCC827 cells, but FasL mRNA was not increased
in the same conditions (data not shown).
CM1 ligation induces Fas-mediated
apoptosis on A549 cells
We wondered if FasL induction after CM1 ligation
would trigger apoptosis in a Fas/FasL mechanism in the A549 lung
cancer cells. To confirm that CM1-induced FasL would interact with
constitutively expressed Fas on the cell surface, A549 cells were
pre-incubated with ZB4, an antagonistic (blocking) anti-Fas
antibody, for 1 h. Cells were washed thrice and then incubated on
immobilized anti-CM1 or MOPC antibody for 2 h. ZB4 blocked the
inhibition of growth effectively in A549 cells but not in HCC827
cells (Fig. 4B and E). ZB4
pretreatment also effectively blocked Annexin V-positive apoptotic
cells in A549 cells but not HCC827 cells (Fig. 4C and F). Thus, the Fas-FasL
interaction appeared to be active only in the A549 cells following
CM1 ligation.
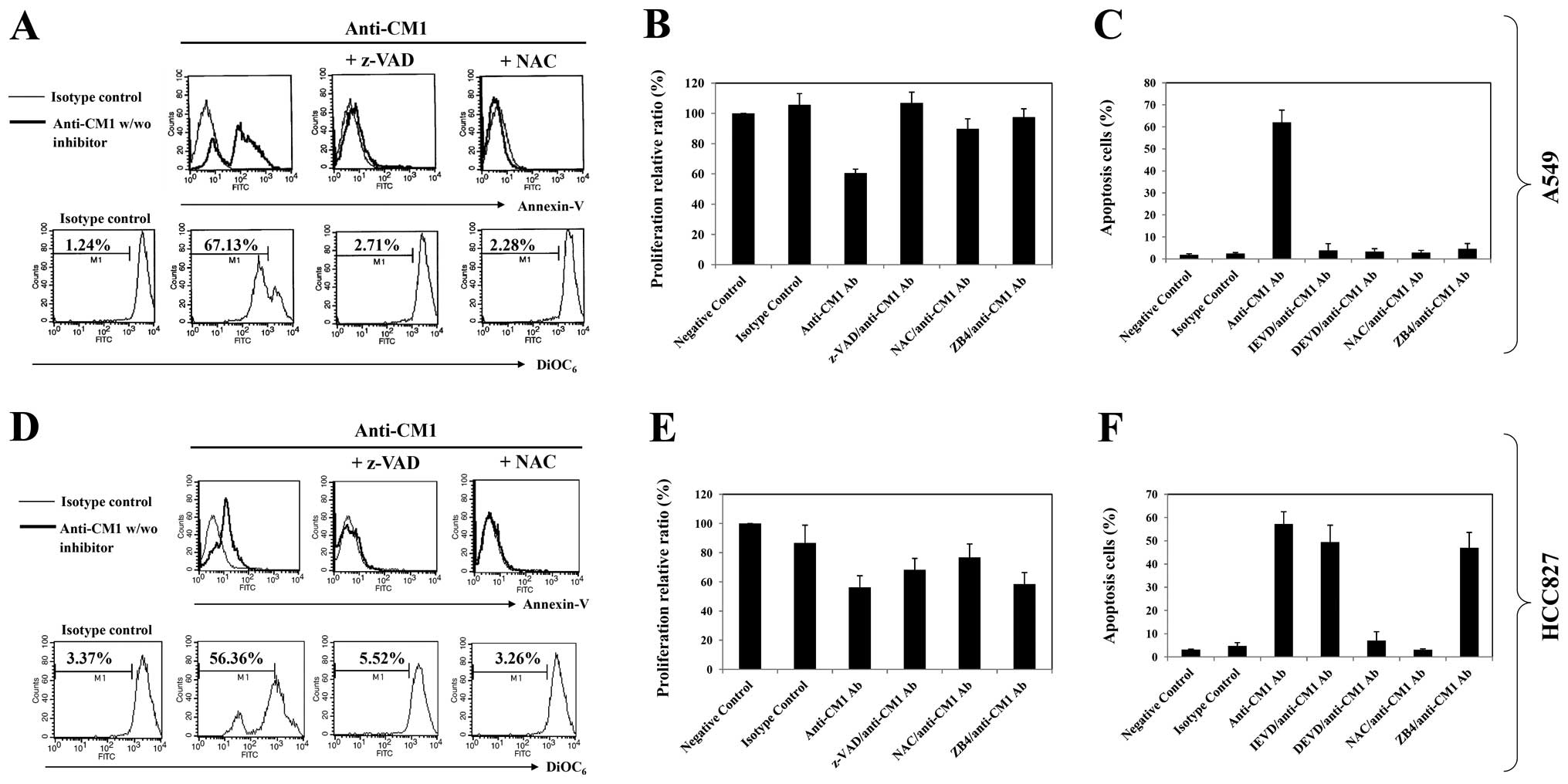 | Figure 4.The effects of inhibition on
CM1-mediated apoptosis of A549 and HCC827 lung cancer cells. Cells
were pre-incubated with z-VAD-fmk (20 μM, 2 h), NAC (10 mM,
1 h), z-IETD fmk (20 μM, 2 h), z-DEVD-fmk (20 μM, 2
h) or ZB4 (a Fas-blocking antibody, 500 μg/ml, 1 h). Cells
were then washed with PBS and incubated in an anti-CM1 or MOPC
antibody-coated (2.5 μg/well) plate for 2 h. Cells were
harvested and further incubated in RPMI-1640 medium. Sixteen hours
later, the number of Annexin V-positive cells and mitochondrial
membrane potential disruption were obtained as described in
Materials and methods. The thin-line histogram represents the
isotype control (MOPC). (A and D) The indicated percentage is the
cell proportion in each bar. Results are representative of four
independent experiments. (B and E) Proliferation assay and (C and
F) apoptosis assay were performed as described previously. (C and
F) The percentage value of apoptotic cells represents the mean
value of three independent experimental results using flow
cytometric analysis. |
ROS and caspase participates in
CM1-mediated apoptosis
Both A549 and HCC827 cells were pre-incubated with
z-VAD-fmk, a broad caspase inhibitor, and NAC, a scavenger of
reactive oxygen species, for 2 h before ligation of CM1. Both
z-VAD-fmk and NAC pretreatment blocked effectively the growth
inhibition, induction of Annexin V positive apoptotic cells and
mitochondrial membrane potential disruption in both lung cancer
cells (Fig. 4A, B, D and E). The
more selective caspase inhibitors (z-DEVD-fmk, caspase-3 inhibitor
or z-IETD-fmk, a caspase-8 inhibitor) blocked CM1-mediated
apoptosis in A549 cells, however, only z-DEVD-fmk blocked apoptosis
of HCC827 cells (Fig. 4C and
F).
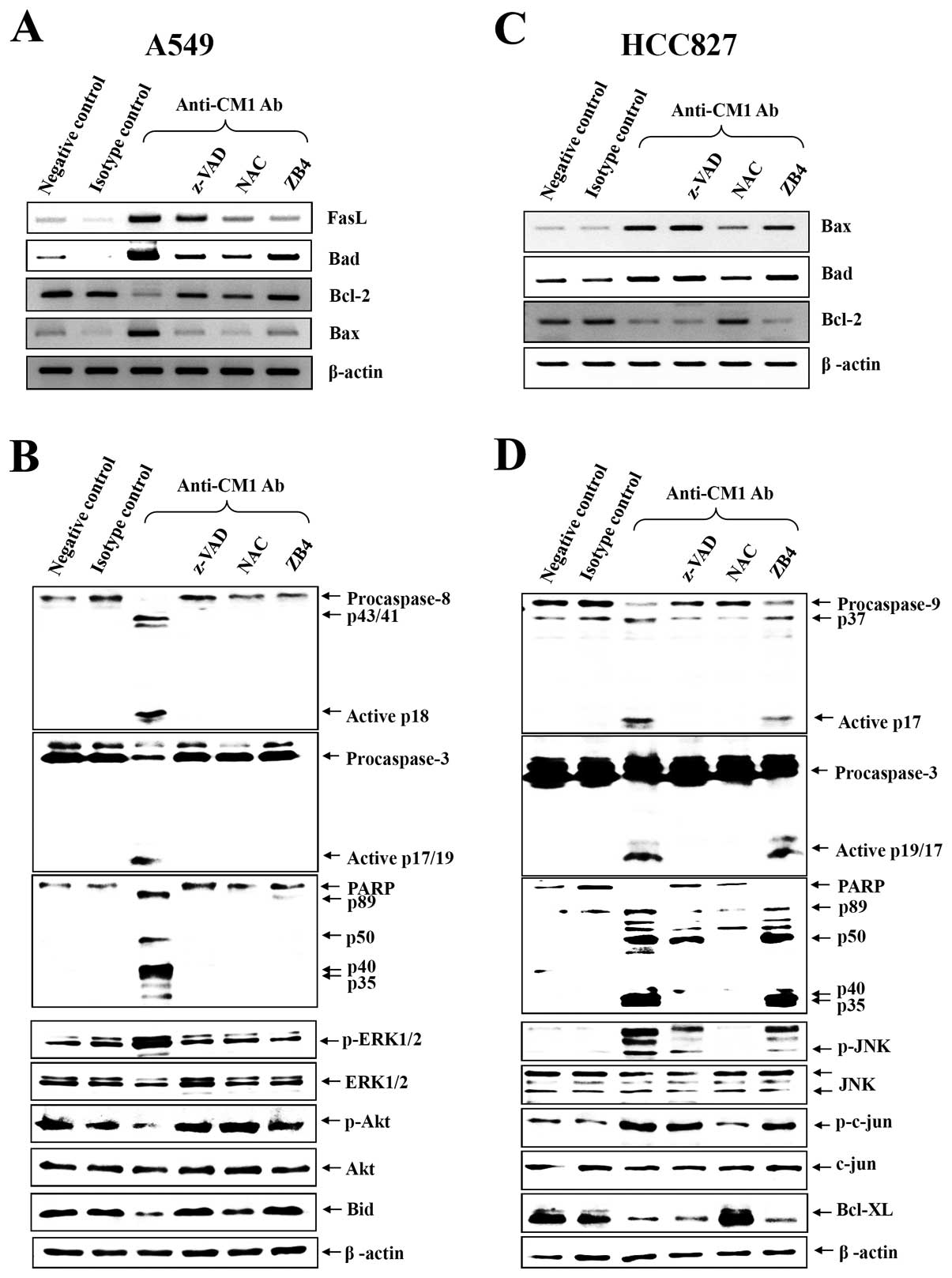
CM1-induced apoptosis activates different
caspases in the two cell lines
To elucidate the role of the different caspases in
induction of apoptosis via CM1 ligation, caspase-3, -8, -9 and
poly-[ADP]-ribose]-polymerase (PARP) were analyzed by western blot
analysis. As shown in Fig. 5B and
D, CM1 ligation induced caspase -3, -8 and PARP processing in
A549 cells, however, caspase -3, -9 and PARP were processed in
HCC827 cells. Furthermore, pretreatment with z-VAD-fmk, NAC and
anti-Fas antagonist (ZB4) completely prevented caspase and PARP
processing in A549 cells but only ZB4 blocked effectively caspase
and PARP processing in HCC827 cells (Fig. 5B and D).
CM1 ligation induces changes in
pro-apoptotic and anti-apoptotic gene expression
To investigate the mechanism of CM1-mediated
apoptosis by mitochondrial membrane disruption, some candidate
signaling molecules were studied. Expression of genes associated
with apoptosis was studied by RT-PCR in A549 and HCC827 lung cancer
cells after anti-CM1 mAb or MOPC (isotype control antibody)
treatment. At baseline, BCL-2 mRNA was constitutively and strongly
expressed in both A549 and HCC827 cells, but BAX and BAD were
slightly expressed on these cells. Following CM1 ligation, BCL-2
decreased and BAX and BAD increased. Consistent with the previous
results, z-VAD-fmk, NAC and ZB4 also almost completely blocked the
change on the expression of apoptosis-associated genes by CM1
ligation in A549 cells. But only NAC blocked effectively the change
of the expression of apoptosis-associated genes by CM1 ligation in
HCC827 cells (Fig. 5A and C).
CM1 ligation induces the changes of MAPK
phosphorylation by different mechanisms in A549 and HCC827
cells
Because apoptosis-related genes can be regulated by
upstream kinases, we next examined phosphorylation of kinases after
CM1 ligation in A549 and HCC827 cells. CM1 ligation induced the
phosphorylation of ERK1/2, whereas it downregulated the Akt and Bid
phosphorylation in A549 cells (Fig.
5B). However, CM ligation induced the phosphorylation of JNK
and its major substrate c-jun, whereas it downregulated the
Bcl-XL expression in HCC827 cells (Fig. 5D). Furthermore, we investigated the
blocking effects of various inhibitors. Z-VAD-fmk, NAC and ZB4
almost completely blocked CM1-induced phosphorylation of MAPKs in
A549 cells. NAC only blocked effectively CM1-induced
phosphorylation of MAPKs and downregulation of Bcl-XL
expression in HCC827 cells (Fig. 5B
and D).
Cytochrome c, AIF and endoG are released
from mitochondria to the cytoplasm by CM1 ligation
CM1 ligation induced disruption of the mitochondrial
membrane potential, so we next investigated the translocation of
proapoptotic proteins in mitochondria using confocal microscopy. At
baseline, in both A549 and HCC827 cells, cytochrome c, AIF
and endoG were located within mitochondria (Fig. 6A and B, 1st and 2nd columns). After
CM1 ligation in both A549 and HCC827 cells, cytochrome c and
AIF were released from the mitochondria to the cytosol and endoG
and AIF were translocated into the nucleus (Fig. 6A and B, 3rd column). In accord with
previous results, in A549 cells, z-VAD-fmk, NAC and ZB4 almost
completely blocked CM1-induced release of pro-apopotic proteins
from the mitochondria (Fig. 6A,
4th–6th column) whereas in HCC827 cells, release was blocked
only by NAC (Fig. 6B, 4th and 5th
column).
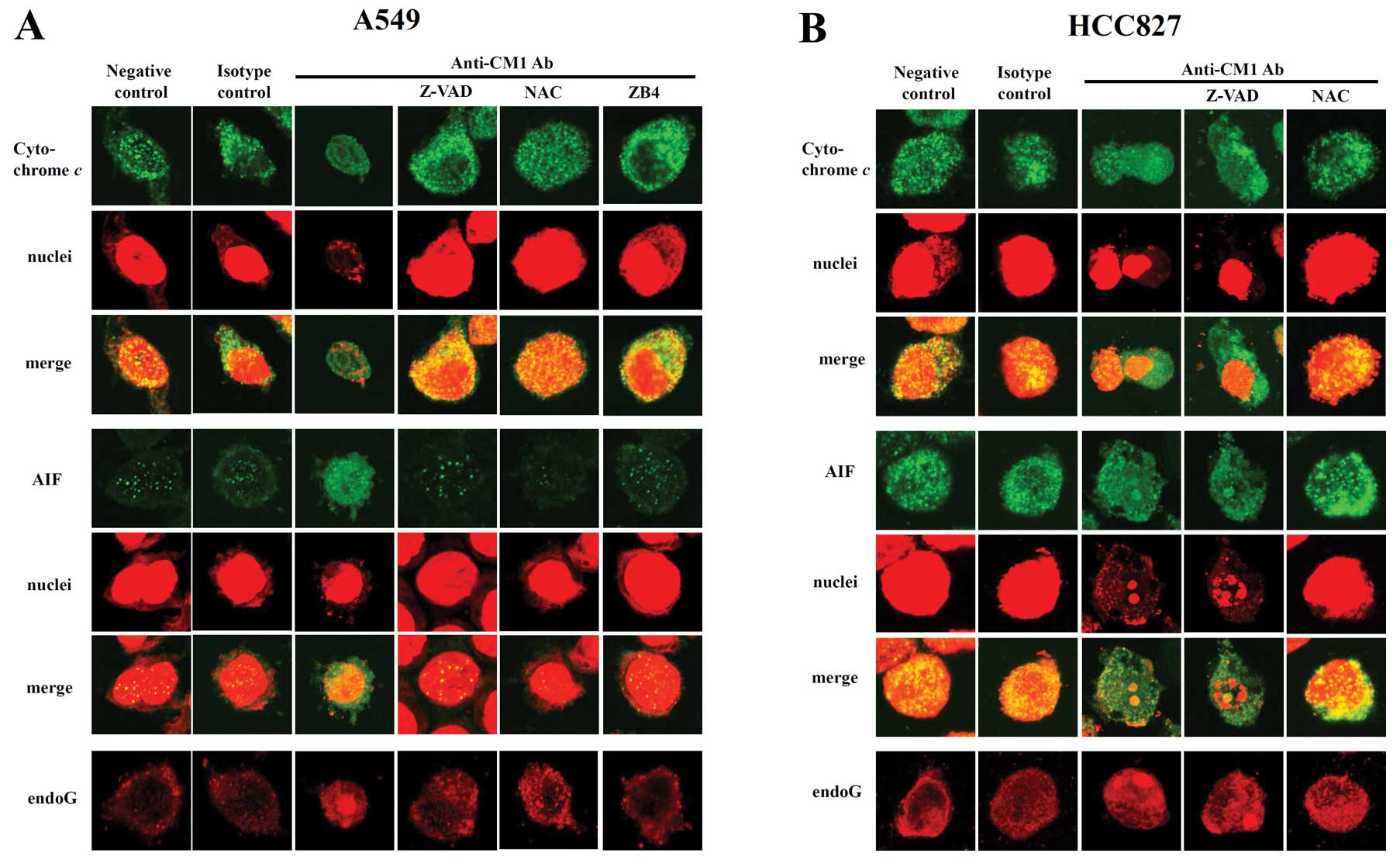 | Figure 6.Subcellular distribution of
cytochrome c, AIF and endoG in (A) A549 and (B) HCC827 lung
cancer cells after ligation of CM1. Cells were pretreated with
z-VAD-fmk (20 μM, 2 h), antagonistic blocking anti-fas Ab
ZB4 (0.5 μg/ml, 1 h) or NAC (10 μM, 1 h). Cells were
washed and incubated with anti-CM1 mAb (5 μg/ml, 40 min) or
MOPC21 (isotype control antibody, 5 μg/ml, 40 min) and goat
anti-mouse IgG (2 μg/ml, 15 min). Cells were harvested and
further incubated in RPMI-1640 medium for 16 h. Cells were
permeabilized with 0.1% saponin in PBS. Intracellular staining was
performed using anti-cytochrome c (mouse IgG2b), AIF (mouse
IgG2b) or endoG (goat polyclonal IgG) Ab and FITC-conjugated goat
anti-mouse IgG or FITC conjugated rabbit anti-goat IgG. The nucleus
was stained with PI. Cells were observed under a confocal
microscope (×400 magnification). The procedure is described in
detail in Materials and methods. Green fluorescence indicates
cytochrome c or AIF, respectively, and red fluorescence
indicates nucleus or endoG (last row). |
Discussion
CM1 was newly defined as a centroblast (or
centrocyte) cell marker, but mainly identified as an apoptosis
triggering molecule in several B lymphoma cell lines and
EBV-transformed B cells (14–16).
Interestingly, both flow cytometric and confocal microscopic
results showed that CM1 was expressed on the cell surface in A549
and HCC827 lung cancer cells in this study. These results suggest
that CM1 could be developed as a candidate marker of lung cancer
for diagnosis and/or prognostic application.
The role of CM1 expressed on two lung cancer cell
lines was investigated using an anti-CM1 antibody. As shown in
Fig. 2, the ligation of CM1 using
immobilized anti-CM1 antibody inhibited proliferation and induced
the apoptosis of both A549 and HCC827 cells. CM1-mediated apoptosis
involved mitochondria membrane potential disruption and
intra-cellular reactive oxygen species (ROS) generation. ROS are
important messengers of intracellular signaling, transcription
activation, proliferation and apoptosis (17). It has long been recognized that ROS
are generated by external oxidative stress or by the byproducts of
altered cellular metabolism involving several oxidases such as
NAD(P)H-oxidase, mitochondrial respiration or cytoskeletal
organization (18,19). However, the precise mechanism of
ROS generation remains unclear. ROS can modulate MAP protein
kinases, cytoskeletal metabolism and intracellular Ca2+,
and influence the mitochondrial membrane directly or indirectly
(20). These studies supported
that ROS had a close relationship with the mitochondrial membrane
potential disruption in the mechanism of CM1-mediated apoptosis on
lung cancer cells.
To evaluate the relationship with Fas-FasL signaling
in CM1-mediated apoptosis, flow cytometric analysis was performed
for the changes of Fas and FasL expression after the ligation of
CM1. Fas (CD95) was constitutively expressed on both A549 and
HCC827 cells, but FasL (CD137) was not expressed before stimulation
of CM1. The ligation of CM1 using immobilized anti-CM1 antibody
induced FasL expression in only A549 cells as shown in Fig. 3A. RT-PCR for FasL transcripts
confirmed the fact that CM1 ligation induced FasL mRNA expression
on A549 cells in Fig. 5A.
To clarify that FasL induced by CM1 ligation would
interact with Fas on adjacent cells resulting in apoptosis,
antagonistic anti-Fas antibody, ZB4, was used. Pretreatment of ZB4
almost completely blocked CM1-mediated apoptosis. NAC pretreatment
completely blocked FasL transcription after CM1 ligation in
Fig. 5A. This result indicated
that FasL expression was strongly related to ROS generated by the
ligation of CM1. These results suggested that ligation of CM1
induced ROS generation, and ROS triggered Fas ligand expression in
A549 cells. It has already been reported that
H2O2 induces upregulation of Fas and FasL
expression in the nerves is linked to modulation by cAMP (21). This study also supports that
CM1-mediated ROS generation leads to FasL expression.
Interestingly, ligation of CM1 did not induce Fas
ligand expression in HCC827 cells despite apoptosis induction by
ligation of CM1. This (Fig. 3B)
result indicated that apoptosis signaling by ligation of CM1 could
be triggered through different mechanisms from Fas-Fas ligand
pathway in HCC827 cells. Recently, it was demonstrated that
knockdown of Fas specifically enhanced cell death induced by the
EGFR tyrosine kinase inhibitor in EGFR-mutant lung cancer cells
(10) and Fas signaling promotes
tumor growth through the JNK and Jun pathway (22). These studies also support that
ligation of CM1 could induce apoptosis of HCC827 cells through
another pathway rather than Fas-Fas ligand pathway.
To identify the intracellular signaling mechanism of
CM1-mediated apoptosis on A549 and HCC827 lung cancer cells,
further experiments using some inhibitors were performed.
z-VAD-fmk, as a broad caspase inhibitor, was pre-incubated before
stimulation of CM1 because caspases are commonly linked to
pro-apoptotic molecules released from disrupted mitochondria, and
it effectively blocked CM1-mediated apoptosis of cells and
mitochondrial membrane potential disruption as expected in both
A549 and HCC827 cells. Pre-treatment with NAC also completely
blocked CM1-mediated apoptosis of cells and mitochondrial membrane
potential disruption (Fig. 4A and
D). Proliferation assay showed the same effects as the
apoptosis assay results. These results suggested that both caspase
activation and ROS generation was directly related to CM1-mediated
apoptosis in both lung cancer cells. Additionally, z-DEVD-fmk, an
executor caspase-3 inhibitor, and z-IEVD-fmk, a caspase-8
inhibitor, restored CM1-mediated apoptosis, and cleavage of
procaspase-8, procaspase-3 and PARP were found after ligation of
CM1 in A549 cells (Fig. 5B).
However, in HCC827 cells only z-DEVD-fmk restored CM1-mediated
apoptosis, and cleavage of procaspase-9, procaspase-3 and PARP were
found after ligation of CM1 (Fig.
5D). Based on these results, we concluded that CM1-mediated
apoptosis resulted from activation of caspases in both A549 and
HCC827 lung cancer cells, but the participating caspases
differed.
Bcl-2 family proteins exert many of their effects
when they locate to the mitochondrial outer membrane (23). Overexpression of Bcl-2 or
Bcl-XL inhibits apoptosis by blocking the release of
cytochrome c(24,25). On the contrary, Bax targeted to
mitochondria can trigger rapid release of cytochrome
c(26). Bad can
heterodimerize with Bcl-XL at mitochondrial membrane
sites to promote cell death (27).
Therefore, the expression of Bcl-2, Bax, Bad mRNA was investigated
in CM1-mediated apoptosis. As expected, the expression of both Bax
and Bad mRNA increased, but Bcl-2 mRNA was repressed in both lung
cancer cells. Consistent with the previous results, NAC, z-VAD-fmk
and ZB4 effectively blocked the changes of Bcl-2 family mRNA
expression by ligation of CM1 in A549 cells, however, only NAC
effectively blocked these changes in HCC827 cells (Fig. 5A and C). Thus, we concluded that
the expression of the Bcl-2 family also related to CM1-mediated
apoptosis in lung cancer cells.
The mitogen-activated protein kinase (MAPK) family
proteins are known as the important regulators in cell apoptosis
(28,29). Therefore, we evaluated whether JNK,
ERK and Akt were involved in CM1-mediated apoptosis. In general,
p38 MAPK and JNK are involved in cell death, whereas ERK1/2 is
associated with cell proliferation. In particular, p38 MAPK is
known to play a critical role in the transmission of apoptotic
signals (30). In this study,
ligation of CM1 induced apoptosis of A549 cells through
phosphorylation of ERK1/2, whereas it induced apoptosis of HCC827
cells mainly through the phosphorylation of JNK and its major
substrate c-jun. We investigated immunoblotting for p38MAPK,
ERK1/2, Akt and JNK in both A549 and HCC827 cells simultaneously
(data not shown partly), but we showed only positive
phosphorylation results. Although the studies on correlation
between Fas-Fas ligand upregulation and ERK1/2 phosphorylation are
controversial, some studies show that Fas and Fas ligand proteins
can be upregulated via p38 MARK/ERK activation (31,32).
In our study, ligation of CM1 induced Fas ligand expression and ERK
phosphorylation simultaneously in A549 cells, so we concluded that
Fas ligand expression was related to ERK phosphorylation. On the
other hand, ligation of CM1 induced JNK and c-jun phosphorylation
independent of Fas/Fas ligand pathway in HCC827 cells. It is
reported that EGFR inhibitor, AG1478, induced apoptosis via
caspase-3 and JNK activation in PC-9 non-small lung cancer cells
(33). Interestingly, ligation of
CM1 induced apoptosis of HCC827 cells with mutated EGFR via
caspase-3 and JNK activation. These results supported that CM1
could be developed as potential therapeutic target to lung cancer
cells independent of EGFR mutation. Consistent with the previous
results, z-VAD-fmk, NAC and ZB4 completely blocked CM1 induced the
phosphorylation of MAPKs in A549, while only the NAC effectively
blocked the changes of MAPKs activation by ligation of CM1 in
HCC827 cells.
To determine the direct relationship between
mitochondrial events and apoptosis, we investigated whether
proapoptotic molecules that were released from the mitochondria and
caspases were directly involved in apoptosis after ligation of CM1.
We showed that proapoptotic molecules, such as cytochrome c,
AIF and endoG, were released from the mitochondria into the cytosol
after ligation of CM1 in both A549 and HCC827 cells. In addition,
translocation into the nuclei of both AIF and endoG, known as
executors of caspase-independent apoptosis (34), was investigated. As expected,
z-VAD-fmk, NAC, ZB4 pretreatment completely blocked the release of
cytochrome c, AIF and endoG after CM1 ligation in A549
cells, while only NAC blocked the release of these molecules in
HCC827 cells. These results also support that CM1-mediated
apoptosis was controlled at the mitochondrial level.
Based on these results, we conclude that ligation of
CM1 induced apoptosis of A549 cells through ROS generation, FasL
expression, caspase-3 and -8, mitochondrial, Bcl-2 family
molecules, ERK1/2 and the Akt kinase-dependent pathways, whereas
ligation of CM1 induced apoptosis of HCC827 cells was induced
through ROS generation, mitochondrial, Bcl-2 family molecules,
caspase-3 and -8, and the JNK and c-jun-dependent pathways. These
results suggest that CM1 can be developed as one candidate for
therapeutic targeting of lung cancer regardless of mutant EGFR.
Abbreviations:
|
CM1
|
centrocyte/-blast marker 1
|
|
EGFR
|
epidermal growth factor receptor
|
Acknowledgements
This study was supported by a grant of
the Korea Healthcare Technology R&D Project, Ministry of Health
and Welfare, Republic of Korea (A084802), and 2008 Inje University
Research Grant.
References
|
1.
|
Matthews JI and Blanton HM: Lung cancer.
An update. Prim Care. 12:267–281. 1985.
|
|
2.
|
Landi MT, Consonni D, Rotunno M, Bergen
AW, Goldstein AM, Lubin JH, Goldin L, Alavanja M, Morgan G, Subar
AF, Linnoila I, Previdi F, Corno M, Rubagotti M, Marinelli B,
Albetti B, Colombi A, Tucker M, Wacholder S, Pesatori AC, Caporaso
NE and Bertazzi PA: Environment and genetics in lung cancer
etiology (EAGLE) study: an integrative population-based
case-control study of lung cancer. BMC Public Health. 8:2032008.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Youlden DR, Cramb SM and Baade PD: The
international epidemiology of lung cancer: geographical
distribution and secular trends. J Thorac Oncol. 3:819–831. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bareschino MA, Schettino C, Rossi A,
Maione P, Sacco PC, Zeppa R and Gridelli C: Treatment of advanced
non small cell lung cancer. J Thorac Dis. 3:122–133.
2011.PubMed/NCBI
|
|
5.
|
Petty RD, Nicolson MC, Kerr KM,
Collie-Duguid E and Murray GI: Gene expression profiling in
non-small cell lung cancer: from molecular mechanisms to clinical
application. Clin Cancer Res. 10:3237–3248. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Mendelsohn J and Baselga J: The EGF
receptor family as targets for cancer therapy. Oncogene.
19:6550–6565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Normanno N, Campiglio M, De LA, Somenzi G,
Maiello M, Ciardiello F, Gianni L, Salomon DS and Menard S:
Cooperative inhibitory effect of ZD1839 (Iressa) in combination
with trastuzumab (Herceptin) on human breast cancer cell growth.
Ann Oncol. 13:65–72. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Doody JF, Wang Y, Patel SN, Joynes C, Lee
SP, Gerlak J, Rolser RL, Li Y, Steiner P, Bassi R, Hicklin DJ and
Hadari YR: Inhibitory activity of cetuximab on epidermal growth
factor receptor mutations in non small cell lung cancers. Mol
Cancer Ther. 6:2642–2651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Camidge DR: The potential of death
receptor 4- and 5-directed therapies in the treatment of lung
cancer. Clin Lung Cancer. 8:413–419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Bivona TG, Hieronymus H, Parker J, Chang
K, Taron M, Rosell R, Moonsamy P, Dahlman K, Miller VA, Costa C,
Hannon G and Sawyers CL: FAS and NF-κB signalling modulate
dependence of lung cancers on mutant EGFR. Nature. 471:523–526.
2011.
|
|
11.
|
Mukohara T, Engelman JA, Hanna NH, Yeap
BY, Kobayashi S, Lindeman N, Halmos B, Pearlberg J, Tsuchihashi Z,
Cantley LC, Tenen DG, Johnson BE and Jänne PA: Differential effects
of gefitinib and cetuximab on non-small-cell lung cancers bearing
epidermal growth factor receptor mutations. J Natl Cancer Inst.
97:1185–1194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lieber M, Smith B, Szakal A, Nelson-Rees W
and Todaro G: A continuous tumor-cell line from a human lung
carcinoma with properties of type II alveolar epithelial cells. Int
J Cancer. 17:62–70. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Amann J, Kalyankrishna S, Massion PP, Ohm
JE, Girard L, Shigematsu H, Peyton M, Juroske D, Huang Y, Stuart
Salmon J, Kim YH, Pollack JR, Yanagisawa K, Gazdar A, Minna JD,
Kurie JM and Carbone DP: Aberrant epidermal growth factor receptor
signaling and enhanced sensitivity to EGFR inhibitors in lung
cancer. Cancer Res. 65:226–235. 2005.PubMed/NCBI
|
|
14.
|
Hur DY, Kim S, Kim YI, Min HY, Kim DJ, Lee
DS, Cho D, Hwang YI, Hwang DH, Park SH, Ahn HK, Chang KY, Kim YB
and Lee WJ: CM1, a possible novel activation molecule on human
lymphocytes. Immunol Lett. 74:95–102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Kim D, Hur DY, Kim YS, Lee K, Lee Y, Cho
D, Kang JS, Kim YI, Hahm E, Yang Y, Yoon S, Kim S, Lee WB, Park HY,
Kim YB, Hwang YI, Chang KY and Lee WJ: CM1 ligation initiates
apoptosis in a caspase 8-dependent manner in Ramos cells and in a
mitochondria-controlled manner in Raji cells. Hum Immunol.
63:576–587. 2002. View Article : Google Scholar
|
|
16.
|
Kim YS, Park GB, Choi YM, Kwon OS, Song
HK, Kang JS, Kim YI, Lee WJ and Hur DY: Ligation of
centrocyte/centroblast marker 1 on Epstein-Barr virus-transformed B
lymphocytes induces cell death in a reactive oxygen
species-dependent manner. Hum Immunol. 67:795–807. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Gamaley IA and Klyubin IV: Roles of
reactive oxygen species: signaling and regulation of cellular
functions. Int Rev Cytol. 188:203–255. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Moldovan L and Moldovan NI: Oxygen free
radicals and redox biology of organelles. Histochem Cell Biol.
122:395–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Ushio-Fukai M and Alexander RW: Reactive
oxygen species as mediators of angiogenesis signaling: role of
NAD(P)H oxidase. Mol Cell Biochem. 264:85–97. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Andreyev AY, Kushnareva YE and Starkov AA:
Mitochondrial metabolism of reactive oxygen species. Biochemistry
(Mosc). 70:200–214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Facchinetti F, Furegato S, Terrazzino S
and Leon A: H(2) O(2) induces upregulation of Fas and Fas ligand
expression in NGF-differentiated PC12 cells: modulation by cAMP. J
Neurosci Res. 69:178–188. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chen L, Park SM, Tumanov AV, Hau A, Sawada
K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E and Peter ME:
CD95 promotes tumour growth. Nature. 465:492–496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Jia L, Macey MG, Yin Y, Newland AC and
Kelsey SM: Subcellular distribution and redistribution of Bcl-2
family proteins in human leukemia cells undergoing apoptosis.
Blood. 93:2353–2359. 1999.PubMed/NCBI
|
|
24.
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Vander Heiden MG, Chandel NS, Williamson
EK, Schumacker PT and Thompson CB: Bcl-xL regulates the membrane
potential and volume homeostasis of mitochondria. Cell. 91:627–637.
1997.PubMed/NCBI
|
|
26.
|
Rossé T, Olivier R, Monney L, Rager M,
Conus S, Fellay I, Jansen B and Borner C: Bcl-2 prolongs cell
survival after Bax-induced release of cytochrome c. Nature.
391:496–499. 1998.PubMed/NCBI
|
|
27.
|
Yang E, Zha J, Jockel J, Boise LH,
Thompson CB and Korsmeyer SJ: Bad, a heterodimeric partner for
Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell.
80:285–291. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Reddy KB, Nabha SM and Atanaskova N: Role
of MAP kinase in tumor progression and invasion. Cancer Metastasis
Rev. 22:395–403. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Yu C, Rahmani M, Almenara J, Sausville EA,
Dent P and Grant S: Induction of apoptosis in human leukemia cells
by the tyrosine kinase inhibitor adaphostin proceeds through a
RAF-1/MEK/ERK- and AKT-dependent process. Oncogene. 23:1364–1376.
2004. View Article : Google Scholar
|
|
30.
|
Van Laethem A, Van Kelst S, Lippens S,
Declercq W, Vandenabeele P, Janssens S, Vandenheede JR, Garmyn M
and Agostinis P: Activation of p38 MAPK is required for Bax
translocation to mitochondria, cytochrome c release and apoptosis
induced by UVB irradiation in human keratinocytes. FASEB J.
18:1946–1948. 2004.PubMed/NCBI
|
|
31.
|
Liu WH and Chang LS: Piceatannol induces
Fas and FasL up-regulation in human leukemia U937 cells via
Ca2+/p38alpha MAPK-mediated activation of c-Jun and
ATF-2 pathways. Int J Biochem Cell Biol. 42:1498–1506. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Duan SG, Cheng L, Li DJ, Zhu J, Xiong Y,
Li XW and Wang SG: The role of MAPK-ERK pathway in 67-kDa laminin
receptor-induced FasL expression in human cholangiocarcinoma cells.
Dig Dis Sci. 55:2844–2852. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Takeuchi K, Shin-ya T, Nishio K and Ito F:
Mitogen-activated protein kinase phosphatase-1 modulated JNK
activation is critical for apoptosis induced by inhibitor of
epidermal growth factor receptor-tyrosine kinase. FEBS J.
276:1255–1265. 2009. View Article : Google Scholar
|
|
34.
|
Bröker LE, Kruyt FA and Giaccone G: Cell
death independent of caspases: a review. Clin Cancer Res.
11:3155–3162. 2005.
|