Introduction
Malignant astrocytic tumors are the most common
devastating primary neoplasms of the central nervous system with
hallmark characteristic of aggressive biological behavior, such as
rapid proliferation potential and diffused invasive capability
(1). The strong capacity of these
tumors to invade and migrate into surrounding normal brain tissue
in early stage usually prevents complete removal of the tumor
during operation even with radical resection approach and these
tumors inevitably recur with no effective treatment (2). Same as in other solid tumors, genetic
alterations that result in activation of oncogenes and/or
inactivation of tumor suppressor genes are the underlying causes of
glioma (3). Sequential gain of
oncogenes, such as epidermal growth factor receptor (EGFR) and/or
loss of tumor suppressor genes, such as p53, phosphatase and tensin
homolog (PTEN), provides the necessary foundation for step-wise
progression of malignant glioma from initiation to transformation
and aggressive progression (4,5).
Among various identified oncogenes associated with
aggressiveness of glioma, EGFR is best characterized. EGFR is
amplified in a range of 30–50% of high-grade malignant astrocytoma
cases (6). Enhanced EGFR signaling
was proven to contribute to glioma initiation, progression,
treatment-resistance and poor survival (7). In a search for negative regulator of
EGFR signaling, we had previously cloned and characterized the
human leucine-rich repeats and immunoglobulin-like domains 1
(LRIG1) gene (8). LRIG1 belongs to
the LRIG gene family, which also includes LRIG2 and LRIG3 (9). LRIG1 is located at chromosome 3p14.3
(10), a region frequently deleted
in various human cancers (11).
Previous studies demonstrated that LRIG1 was downregulated in
conventional renal cell carcinoma, squamous cell carcinoma of the
skin and lung (12), breast cancer
(13), nasopharyngeal carcinoma
(14) and in various human cancer
cell lines (10,15–17).
LRIG1 negatively regulates EGFR, both by inducing receptor
degradation (18,19) and by inhibiting signaling (20,21).
Thus, ectopic expression of LRIG1 suppresses glioma cell
proliferation (21,22) and invasion (23). However, the exact mechanisms behind
these suppressive effects have not been thoroughly analyzed.
In this study, we investigated the effect of LRIG1
over-expression on the malignant glioma cell line U-251 MG and
analyzed the dynamic cell cycle and screened its potential target
cyclins. Next we investigated the effects of LRIG1 on glioma cell
invasion and production of MMP2 and MMP9. Moreover, we found that
LRIG1 can modulate EGFR downstream signaling pathway-PI3K/AKT
expression. Our findings demonstrate LRIG1 may exert its tumor
suppressor function via targeting EGFR and its down signaling
pathway.
Materials and methods
Cell culture and transfection
The human glioma cell line U-251 MG purchased from
American Type Culture Collection was cultured in Dulbecco’s
modified Eagle’s medium containing 10% (v/v) fetal bovine serum
(Hyclone, USA) in a humidified (37°C, 5% CO2) incubator.
Cells were transfected with expression vector pLRIG1-GFP, encoding
an LRIG1-GFP fusion protein (24)
or, as vector control, pEGFP-N1 (Clontech), encoding GFP, using
Lipofectamine 2000 (Invitrogen, USA) according to the
manufacturer’s instructions. G418-resistant clones that represented
possible stably transfected cells were ring-cloned and expanded for
further experiments.
RNA isolation and qRT-PCR
Total RNAs from cultured cells were isolated using
TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. qRT-PCR was used to evaluate the expression levels of
mRNAs as described before (25).
Oligonucleotide primer sequences used were as follows: MMP2 sense
ATGGATCCTGGCTTTCCC-3′ and antisense 5′-GCTTCCAAACTTCACGCTC-3′; MMP9
sense 5′-TGAC AGCGACAAGAAGTG-3′ and antisense 5′-CAGTGAAGCGG
TACATAGG-3′; LRIG1, 18S primers used were the same as described in
a previously published report (16).
Western blot analysis and
immunohistochemical staining
Cells were lysed and analyzed by Western blotting as
described (26). The following
antibodies were used: LRIG1 (Santa Cruz, USA); anti-EGFR,
anti-phospho-EGFR (Upstate, USA); anti-phospho-ERK, anti-ERK,
anti-AKT, anti-phospho-AKT (Cell Signaling, USA); anti-cyclin A,
anti-cyclin E, GAPDH (Boster, China); anti-cyclin D1 (Neomarker,
USA).
MTT assay and soft agar colony formation
assay
The proliferation rate of various cells were
measured by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium
bromide (MTT) cell viability/proliferation assay as described
(26). Soft agar colony formation
assay was performed as described (27).
Cell cycle synchronization and dynamic
cell cycle analysis
Cells grown to 80% confluency in 10-cm dishes were
synchronized in culture medium containing 250 ng/ml nocodazole for
24 h. Then, detached cells were shaken-off and collected and washed
twice with phosphate buffered saline and then, replated
subconfluently in complete growth medium. Cells were collected at
indicated time-points after nocodazole washout. Dynamic cell cycle
were analyzed by flow cytometry using propidium iodide (PI)
staining and was performed as previously described (28). Cell lysates were collected at
indicated time-points and immunoblotting was performed using
anti-cyclin antibodies.
In vitro invasion assay and cell
migration assay
In vitro invasion capabilities were measured
in transwell chamber assay as previous described (26). Cell migration activities were
examined by two-dimensional wound healing assay or in vitro
scratch assay as previous described (25).
Gelatin zymography
Serum-free conditioned media were used in SDS-PAGE
gelatin-substrate zymography to detect the activity of MMP2 and 9
as described (29). Briefly,
3×105 transfected U-251 MG cells were plated in 6-well
plates, cultured in 1 ml serum-free DMEM and incubated for 24 h.
Twenty microliters of serum-free culture medium per sample were
prepared in non-denaturating loading buffer and were
size-fractionated in 10% SDS-polyacrylamide gel impregnated with
0.1% gelatin. The gels were incubated in a developing buffer for 42
h at 37°C. Direct comparisons between separate gels were not made,
because the intensity of background staining was variable.
Experiments were repeated three times.
In vivo tumor model
Pooled populations of transfected U-251 MG cells
were used for subcutaneous tumor growth experiments. Briefly,
1×106 glioma cells were injected into both flanks of
female BALB/c athymic nude mouse at 4–6 week of age. Tumor volumes
were calculated using the following formula: tumor volume = 1/2 ×
(shortest diameter)2 × longest diameter
(mm3). These experiments were performed in accordance
with the guidelines of the Animal Experimental Committee, China
Institutes for Biological Sciences.
Statistical analysis
Results are expressed as means ± standard deviation
(SD). Statistical analyses were performed using SPSS statistical
software (SPSS Inc., Chicago, IL). Student’s t-test was used.
Significance was defined as *p<0.05 and
**p<0.01.
Results
Transformation of U-251 MG cells with
LRIG1 expression vector
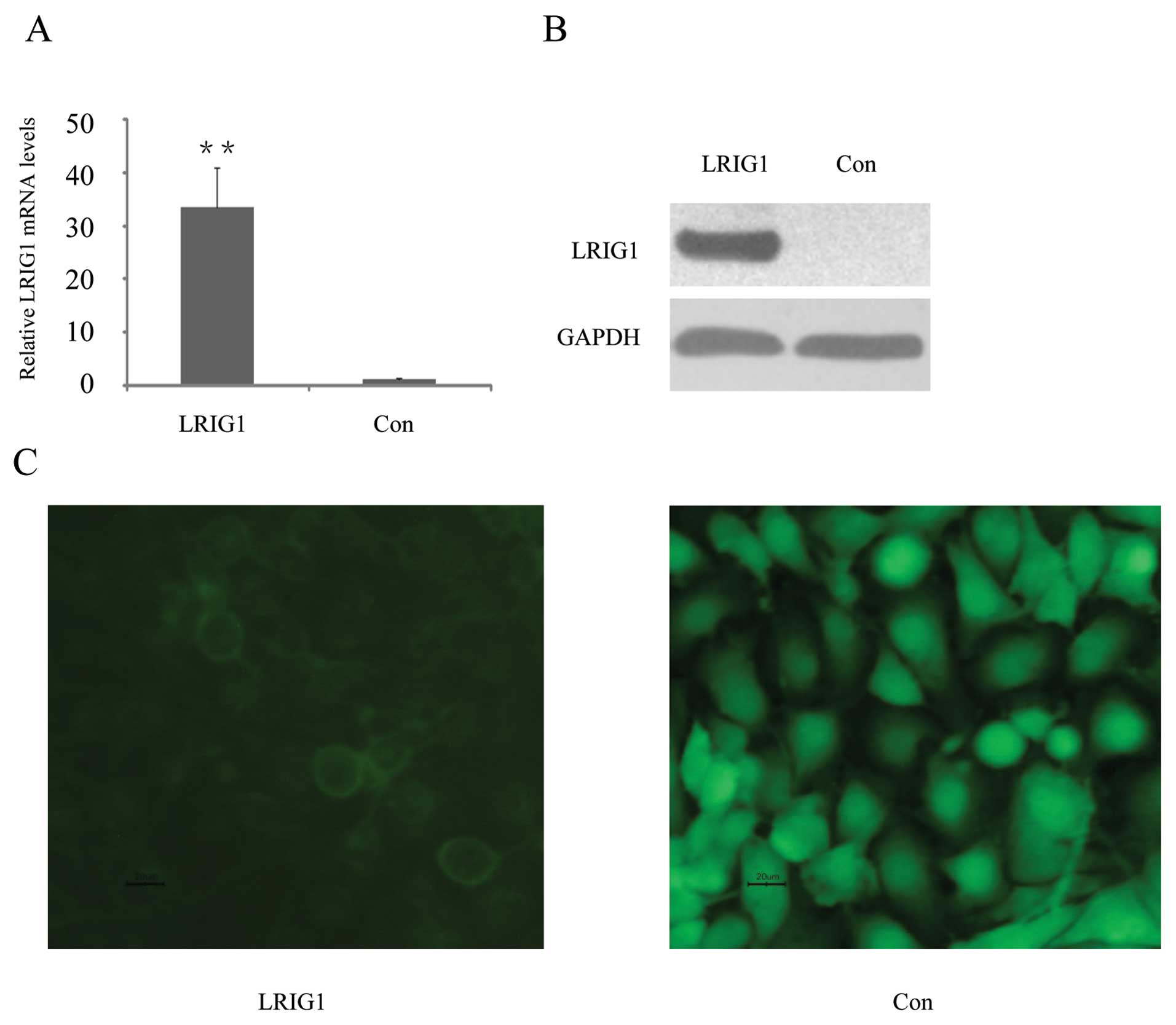
U-251 MG cells were transfected with pLRIG1-GFP,
encoding an LRIG1-GFP fusion protein, or as a vector control, with
pEGFP-N1, encoding GFP (green fluorescent protein). Quantitative
reverse transcriptase PCR (qRT-PCR) showed that LRIG1 mRNA levels
were increased 33-fold in the pLRIG1-GFP transformed cells compared
to the control cells (Fig. 1A).
Western blotting confirmed expression of the LRIG1-GFP fusion
protein in the pLRIG1-GFP transformed cells but not in the control
cells (Fig. 1B). As previously
described (24,30), the LRIG1-GFP fusion protein was
mainly located in the plasma membrane (Fig. 1C).
LRIG1 represses cell proliferation by
delaying cell cycle progression
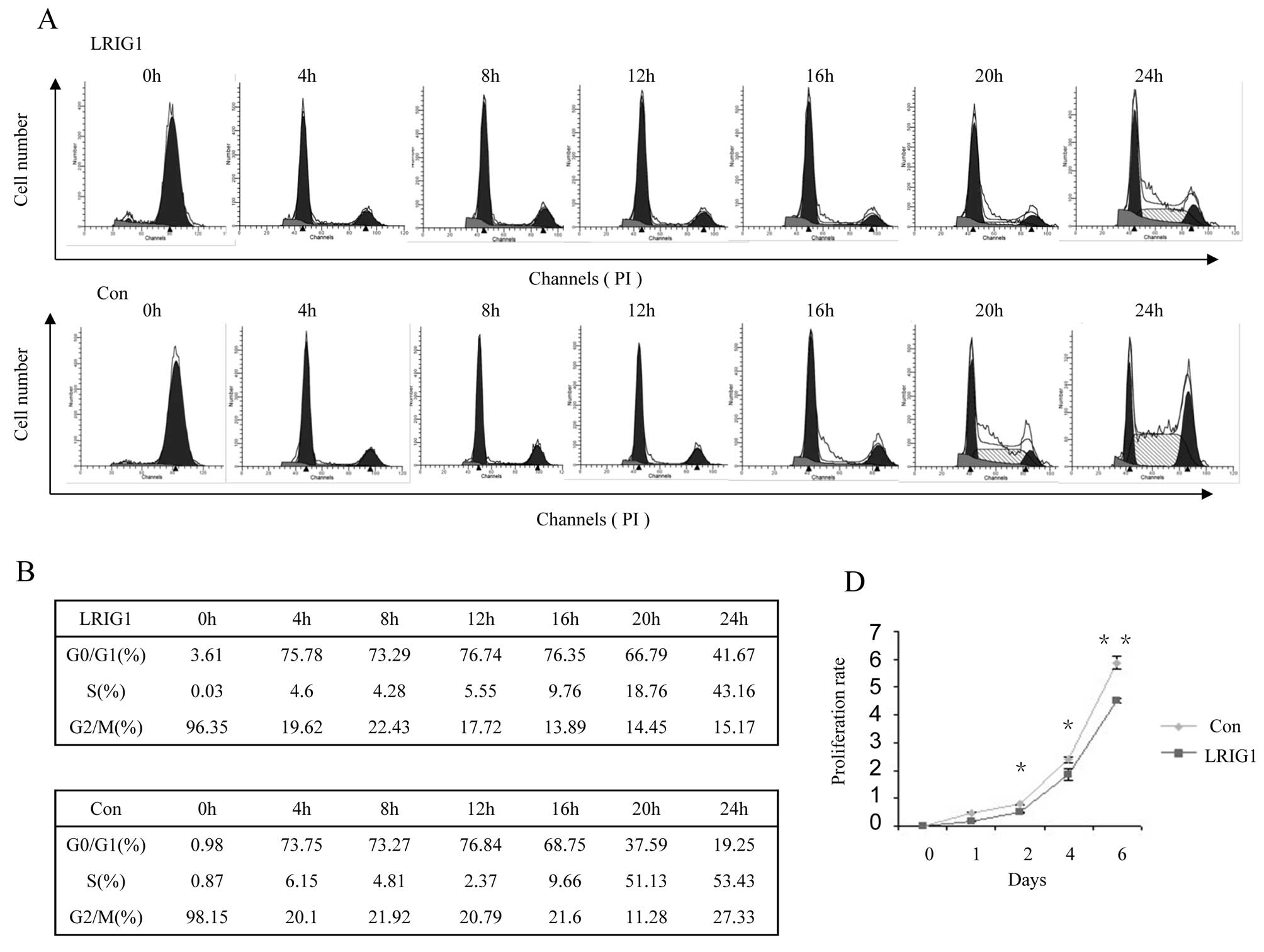
Next, we test dynamic cell cycle change in
synchronized clonal cell line after exogenous LRIG1 introduction.
Most of the control cells left G0/G1 phase and entered S phase at
16-h time-point and entered S phase at 20-h time-point (Fig. 2B, low panel), while most of the
LRIG1 overexpression cells started to leave G0/G1 phase and entered
S phase at 20 h and then, entered S phase at 24 h (Fig. 2B, upper panel). This experiment
demonstrated that exogenous introduction of LRIG1 into U-251 MG
cells caused cell cycle arrest at G0/G1 phase and delayed cell
cycle to enter S phase (Fig. 2A and
B).
As ectopic expression of LRIG1 in U-251 MG cells
delayed cell cycle, we tested whether LRIG1 expression alters the
expression of cyclins. As shown in Fig. 2C, the expression of cyclin D1 lasts
apparently longer in U-251 MG-LRIG1 cells than control cells,
indicating LRIG1 overexpression lengthened the time of the cell
cycle. The peak expression level of cyclin E was at 4-h for control
group, while it was at 12–16-h in LRIG1 overexpressing cells,
indicating the cell cycle was arrested at G0/G1 phase and delayed
to enter S phase. The time course expression pattern of cyclin A
was not significantly altered, indicating the entry to M phase was
not affected by LRIG1 overexpression. These results were consistent
with the dynamic cell cycle analysis shown in Fig. 2A. We next evaluated the effect of
LRIG1 on the growth of glioma cells. The results of cell
proliferation assay showed that ectopic expression of LRIG1 led to
significant inhibition of cell proliferation compared to control
cells (Fig. 2D). These results
indicate a growth-inhibitory role of LRIG1 on glioma cell line
U-251 MG.
LRIG1 inhibits colony formation in vitro
and tumorigenicity in vivo
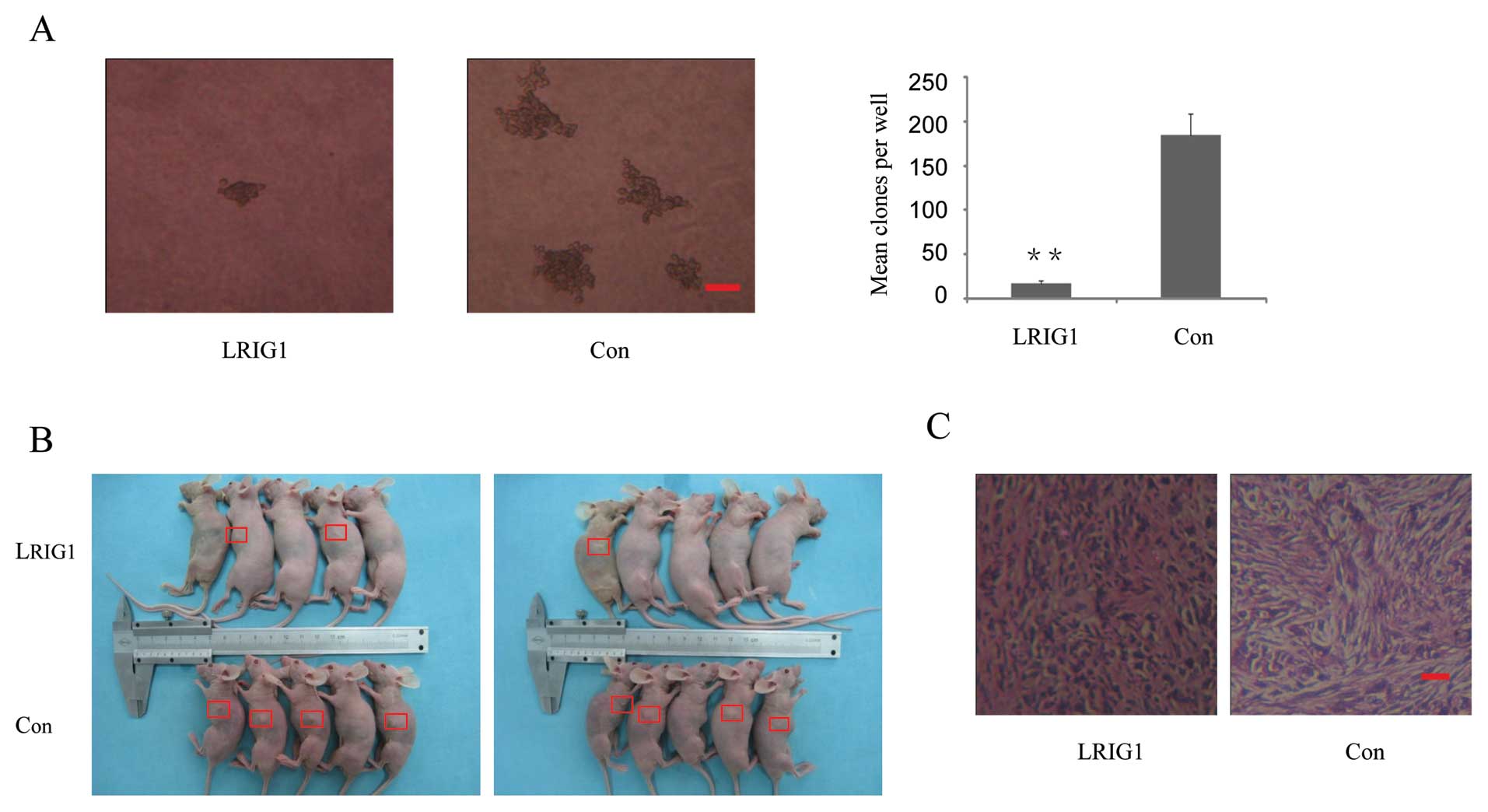
To examine the potential role of LRIG1 in
tumorigenesis, the capacity of colony formation was evaluated on
LRIG1 overexpressing and control cells. Notably, LRIG1
overexpression cells displayed obviously fewer and smaller colonies
compared with control cells (Fig.
3A). This result indicated a growth-inhibitory role of LRIG1 on
glioma cells. To further determine the antitumor effects of LRIG1,
LRIG1 overexpression and control cells were injected into five nude
mice as described. Although tumor volume between LRIG1
overexpression and control groups was not statistically significant
(p= 0.28, data not shown), there was a trend for loss of LRIG1 with
a higher risk with increasing tumor size. The average tumor volume
was 106.4 mm3 in control group vs 6.89 mm3 in
LRIG1 overexpression group at the end of eighth week after
implantation. The tumor incidence was 80% (8 tumors/10 injection
sites) in control group vs 30% (3 tumors/10 injection sites) in
LRIG1 overexpression group (Fig. 3B
and C). The results indicate that overexpression of LRIG1
inhibited tumorigenicity of U-251 MG cells in the nude mouse
xenograft model.
LRIG1 inhibits U-251 MG cell migration
and invasion and decreases secretion and activity of MMP2 and
MMP9
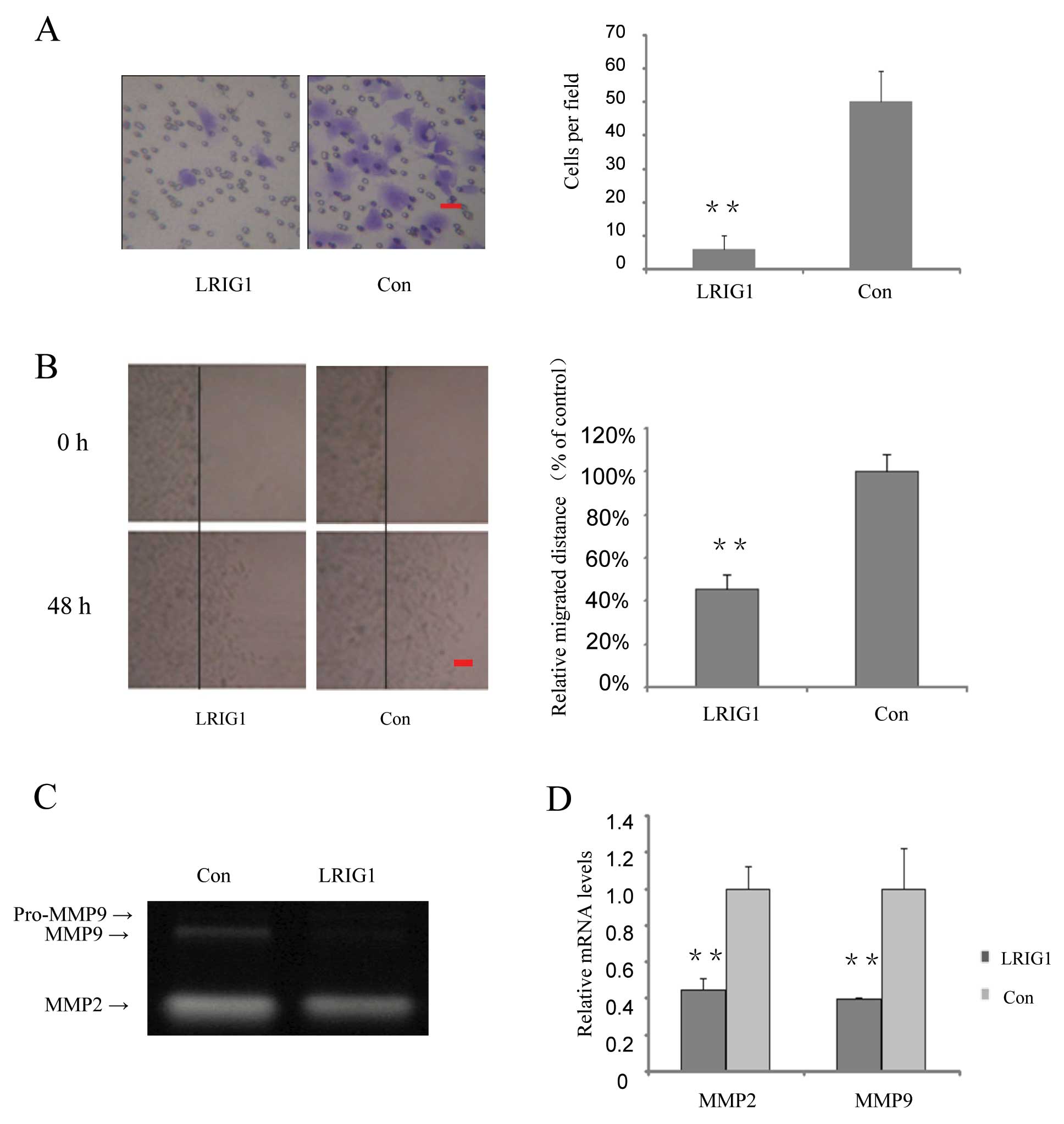
In the following part of this study, we investigated
whether over-expression of LRIG1 could affect cell invasion and
migration. We found that invasion cells in the LRIG1 overexpressing
cells were decreased to 12% of that of the control cells (Fig. 4A). Cell migration capability was
further tested by wound scratch assay. The relative migrated
distances of LRIG1 overexpressing cells were 45% of the distances
migrated by the control cells at 48 h (Fig. 4B).
Gelatin zymography assay was performed to
investigate the effect of the LRIG1 on the activity of MMP2 and
MMP9. After exogenous introduction of LRIG1 into U-251 MG cells,
MMP9 activity was significantly decreased, while MMP2 activity was
modestly decreased (Fig. 4C). To
further confirm the effect of LRIG1 on MMPs, quantitative real-time
PCR results showed that after exogenous introduction of LRIG1 in to
U-251 MG cell, the MMP2 mRNA levels were decreased to 44.8% and
MMP9 mRNA levels were decreased to 39.5%, compared to control group
(Fig. 4D). These results suggest
that LRIG1 plays an important role in suppressing MMP2 and 9
production and is involved in invasion and migration in U-251 MG
cells.
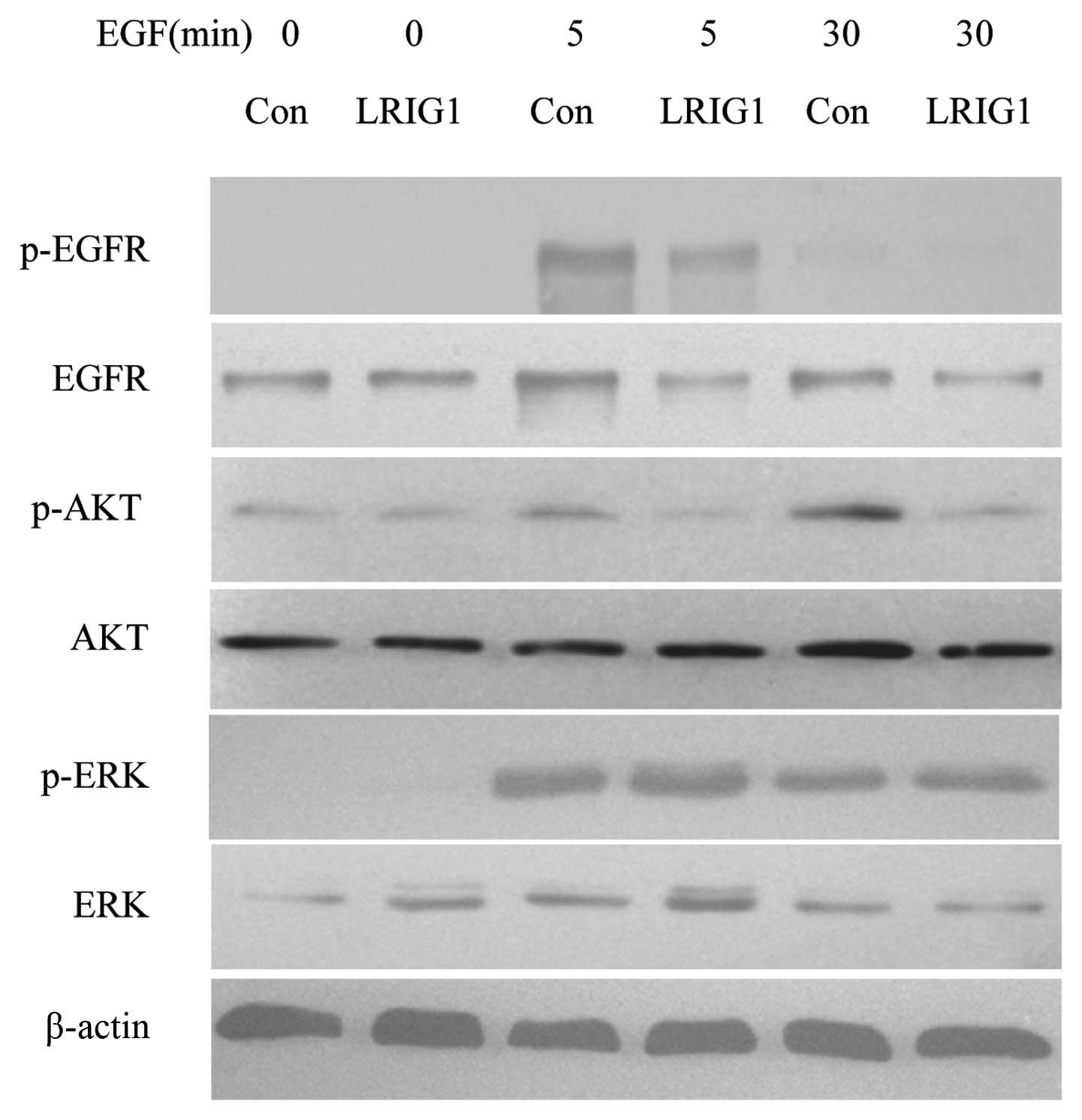
LRIG1 exerts more effect on PI3K/AKT
pathway than MAPK/ERK1/2 pathway
In the next experiments, we confirmed that LRIG1
negatively regulated the levels of EGFR and phosphorylated EGFR in
our experimental system (Fig. 5)
(19). We then analyzed the
expressions and phosphorylation of two key intracellular signal
molecules, AKT and ERK1/2. The basal expression levels of AKT and
ERK were not altered. Further, we showed the levels of
phosphorylated AKT decreased in LRIG1 overexpressing cells, while
the phosphorylated ERK level was not altered, indicating the LRIG1
exerts more influence on intracellular PI3K/AKT pathway than
MAPK/ERK1/2 pathway (Fig. 5).
Discussion
In this study, we provided that LRIG1 functioned as
a negative growth regulator for glioma cells and showed that
over-expression of LRIG1 inhibited glioma to form clonies in
vitro and to develop tumors in vivo. Ectopic expression
of LRIG1 suppressed the invasion and migration of cultured glioma
cells by regulating secreation of MMP2 and MMP9. These observations
provide further understanding of the molecular mechanisms of
inhibitory effects of LRIG1 on glioma cell aggressiveness.
Malignant astrocytic tumors are the most aggressive
and highly lethal type of brain tumor with a strong ability of
rapid proliferation and diffuse invasion into surrounding normal
brain tissue. While the molecular events involved in the initiation
of glioblastoma are not fully understood, it is believed that,
deregulated tumor cell proliferation appears to be a critical early
event in glioma development. To assess the potential role of LRIG1
in glioma cell growth, we over-expressed LRIG1 in glioma cell line
U-251 MG. Our study revealed that LRIG1 overexpression resulted in
decreased tumor cell growth confirmed by MTT and soft agar colony
assay. We further showed that LRIG1 overexpression inhibits glioma
cell proliferation in vivo. Cellular proliferation is
tightly regulated by progression through the cell cycle of DNA
synthesis and mitosis by the formation and activity of
cyclin-cyclin-dependent-kinase (CDK) complexes (31). We demonstrated that enhanced
expression of LRIG1 in glioma cells significantly changed cell
cycle control protein cyclin D1, E expression pattern, resulting in
cell cycle delay at G0/G1. The strong exogenous LRIG1 regulation
over cell cycle control further implies that fundamental
alterations of cyclins and tumor suppressors result in deregulated
cell cycle and unrestrained cell proliferation (32).
In addition to its function as a potent negative
growth regulator, the present and our previous study showed that
LRIG1 overexpression led to a significant decrease of glioblastoma
cell migration and invasion (23),
while the molecular mechanisms of its inhibitory effects on glioma
cell metastasis still remain obscure. Matrix metalloproteinases
(MMPs) are a family of extracellular zinc-dependent endopeptidases
that selectively cleave the protein components of the extracellular
matrix (33). Numerous studies
have demonstrated that malignant glioma cells secrete MMPs to
facilitate their migration and invasion (34). We found that overexpression of
LRIG1 inhibited glioma cell invasion and migration by suppressing
MMP2 and 9 production. Because EGFR activation can promote MMP2
(35) and MMP9 expression
(36,37) and LRIG1 negatively regulated the
level of EGFR, we deduced that LRIG1 regulated glioma cell
metastasis by attenuating EGFR signaling pathway, at least in
part.
Accumulating evidence has identified the EGFR and
its downstream signal networks as commonly deregulated components
in the oncogenesis of glioma, especially primary GBM (1,38).
Once activated and subsequent dimerization, EGFR activates a
variety of intracellular phosphorylation-mediated signal
transduction events (38). The
important events in glioma biology include the RAS/mitogen
activated protein kinase (MAPK) and phosphatidylinositol 3-kinase
(PI3K)/AKT pathways that regulate cell proliferation, survival and
invasion (1,38). Upregulation of LRIG1 influenced
expression levels of p-AKT rather than p-ERK in response to ligand
stimulation. Constitutive activation of PI3K/AKT signaling pathway
plays a pivotal role in the pathogenesis and malignant progression
of several human cancers, including glioblastoma (39). Herein, we found that overexpression
of LRIG1 negatively regulated glioma cell proliferation, migration
and invasion, possibly by inhibiting EGFR activation and its
downstream signaling PI3K/AKT pathway.
Collectively, we identified that LRIG1
overexpression resulted in decreased cell growth in vitro
and in vivo and delay in cell cycle by changing the
expression pattern of cyclins. LRIG1 also attenuated invasion of
glioma cells by decreasing production of MMP2 and 9. Analysis of
the signaling pathways revealed that LRIG1 regulated EGFR signaling
pathway transduction involved in the regulation of tumour cell
proliferation and migration and invasion. It was demonstrated that
LRIG1 exerts its tumor suppressor function and implicates its
potential targets for future brain tumor treatments.
Acknowledgements
This study was supported by National
Natural and Science Foundation of China (No. 81001116).
References
|
1
|
Furnari FB, Fenton T, Bachoo RM, et al:
Malignant astrocytic glioma: genetics, biology and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
MacDonald TJ, Aguilera D and Kramm CM:
Treatment of high-grade glioma in children and adolescents.
Neurooncology. 13:1049–1058. 2011.PubMed/NCBI
|
|
3
|
Sathornsumetee S, Reardon DA, Desjardins
A, Quinn JA, Vredenburgh JJ and Rich JN: Molecularly targeted
therapy for malignant glioma. Cancer. 110:13–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith JS, Tachibana I, Passe SM, et al:
PTEN mutation, EGFR amplification and outcome in patients with
anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer
Inst. 93:1246–1256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reifenberger G and Collins VP: Pathology
and molecular genetics of astrocytic gliomas. J Mol Med.
82:656–670. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frederick L, Wang XY, Eley G and James CD:
Diversity and frequency of epidermal growth factor receptor
mutations in human glioblastomas. Cancer Res. 60:1383–1387.
2000.PubMed/NCBI
|
|
7
|
Heimberger AB, Hlatky R, Suki D, et al:
Prognostic effect of epidermal growth factor receptor and EGFRvIII
in glioblastoma multiforme patients. Clin Cancer Res. 11:1462–1466.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nilsson J, Vallbo C, Guo D, et al:
Cloning, characterization and expression of human LIG1. Biochem
Biophys Res Commun. 284:1155–1161. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Guo D, Holmlund C, Henriksson R and Hedman
H: The LRIG gene family has three vertebrate paralogs widely
expressed in human and mouse tissues and a homolog in Ascidiacea.
Genomics. 84:157–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hedman H, Nilsson J, Guo D and Henriksson
R: Is LRIG1 a tumour suppressor gene at chromosome 3p14.3? Acta
Oncol. 41:352–354. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Knuutila S, Aalto Y, Autio K, et al: DNA
copy number losses in human neoplasms. Am J Pathol. 155:683–694.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boelens MC, van den Berg A, Fehrmann RS,
et al: Current smoking-specific gene expression signature in normal
bronchial epithelium is enhanced in squamous cell lung cancer. J
Pathol. 218:182–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller JK, Shattuck DL, Ingalla EQ, et al:
Suppression of the negative regulator LRIG1 contributes to ErbB2
overexpression in breast cancer. Cancer Res. 68:8286–8294. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sheu JJ, Lee CH, Ko JY, et al: Chromosome
3p12.3–p14.2 and 3q26.2–q26.32 are genomic markers for prognosis of
advanced nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers
Prev. 18:2709–2716. 2009.
|
|
15
|
Tanemura A, Nagasawa T, Inui S and Itami
S: LRIG-1 provides a novel prognostic predictor in squamous cell
carcinoma of the skin: immunohistochemical analysis for 38 cases.
Dermatol Surg. 31:423–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomasson M, Hedman H, Guo D, Ljungberg B
and Henriksson R: LRIG1 and epidermal growth factor receptor in
renal cell carcinoma: a quantitative RT-PCR and
immunohisto-chemical analysis. Br J Cancer. 89:1285–1289. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang WM, Yan ZJ, Ye ZQ and Guo DS: LRIG1,
a candidate tumour-suppressor gene in human bladder cancer cell
line BIU87. BJU Int. 98:898–902. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gur G, Rubin C, Katz M, et al: LRIG1
restricts growth factor signaling by enhancing receptor
ubiquitylation and degradation. EMBO J. 23:3270–3281. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Laederich MB, Funes-Duran M, Yen L, et al:
The leucine-rich repeat protein LRIG1 is a negative regulator of
ErbB family receptor tyrosine kinases. J Biol Chem.
279:47050–47056. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Goldoni S, Iozzo RA, Kay P, et al: A
soluble ectodomain of LRIG1 inhibits cancer cell growth by
attenuating basal and ligand-dependent EGFR activity. Oncogene.
26:368–381. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yi W, Holmlund C, Nilsson J, et al:
Paracrine regulation of growth factor signaling by shed
leucine-rich repeats and immunoglobulin-like domains 1. Exp Cell
Res. 317:504–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stutz MA, Shattuck DL, Laederich MB,
Carraway KL III and Sweeney C: LRIG1 negatively regulates the
oncogenic EGF receptor mutant EGFRvIII. Oncogene. 27:5741–5752.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ye F, Gao Q, Xu T, et al: Upregulation of
LRIG1 suppresses malignant glioma cell growth by attenuating EGFR
activity. J Neurooncol. 94:183–194. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nilsson J, Starefeldt A, Henriksson R and
Hedman H: LRIG1 protein in human cells and tissues. Cell Tissue
Res. 312:65–71. 2003.PubMed/NCBI
|
|
25
|
Song H, Li Y, Lee J, Schwartz AL and Bu G:
Low-density lipoprotein receptor-related protein 1 promotes cancer
cell migration and invasion by inducing the expression of matrix
metalloproteinases 2 and 9. Cancer Res. 69:879–886. 2009.
View Article : Google Scholar
|
|
26
|
Wang B, Han L, Chen R, et al:
Downregulation of LRIG2 expression by RNA interference inhibits
glioblastoma cell (GL15) growth, causes cell cycle redistribution,
increases cell apoptosis and enhances cell adhesion and invasion in
vitro. Cancer Biol Ther. 8:1018–1023. 2009. View Article : Google Scholar
|
|
27
|
Lau YK, Murray LB, Houshmandi SS, Xu Y,
Gutmann DH and Yu Q: Merlin is a potent inhibitor of glioma growth.
Cancer Res. 68:5733–5742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cai M, Han L, Chen R, et al: Inhibition of
LRIG3 gene expression via RNA interference modulates the
proliferation, cell cycle, cell apoptosis, adhesion and invasion of
glioblastoma cell (GL15). Cancer Lett. 278:104–112. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Galis ZS, Muszynski M, Sukhova GK, et al:
Cytokine-stimulated human vascular smooth muscle cells synthesize a
complement of enzymes required for extracellular matrix digestion.
Circ Res. 75:181–189. 1994. View Article : Google Scholar
|
|
30
|
Guo D, Nilsson J, Haapasalo H, et al:
Perinuclear leucine-rich repeats and immunoglobulin-like domain
proteins (LRIG1-3) as prognostic indicators in astrocytic tumors.
Acta Neuropathol. 111:238–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: a review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stamenkovic I: Extracellular matrix
remodelling: the role of matrix metalloproteinases. J Pathol.
200:448–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakada M, Okada Y and Yamashita J: The
role of matrix metalloproteinases in glioma invasion. Front Biosci.
8:e261–269. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kodali R, Hajjou M, Berman AB, et al:
Chemokines induce matrix metalloproteinase-2 through activation of
epidermal growth factor receptor in arterial smooth muscle cells.
Cardiovasc Res. 69:706–715. 2006. View Article : Google Scholar
|
|
36
|
Alper O, Bergmann-Leitner ES, Bennett TA,
Hacker NF, Stromberg K and Stetler-Stevenson WG: Epidermal growth
factor receptor signaling and the invasive phenotype of ovarian
carcinoma cells. J Natl Cancer Inst. 93:1375–1384. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cox G, Jones JL and O’Byrne KJ: Matrix
metalloproteinase 9 and the epidermal growth factor signal pathway
in operable non-small cell lung cancer. Clin Cancer Res.
6:2349–2355. 2000.PubMed/NCBI
|
|
38
|
Huang PH, Xu AM and White FM: Oncogenic
EGFR signaling networks in glioma. Sci Signal. 2:re62009.PubMed/NCBI
|
|
39
|
Knobbe CB, Trampe-Kieslich A and
Reifenberger G: Genetic alteration and expression of the
phosphoinositol-3-kinase/Akt pathway genes PIK3CA and PIKE in human
glioblastomas. Neuropathol Appl Neurobiol. 31:486–490. 2005.
View Article : Google Scholar : PubMed/NCBI
|