Introduction
Acquired chemoresistance is a major cause of
clinical treatment failure and cancer mortality. Chemotherapeutic
agents and death receptor ligands (e.g., tumor necrosis factor, TNF
and Fas), induce cell death through activation of specific
cytotoxic pathways (1–3). Chemotherapeutic-induced cell death
involves activation of death receptor cascades resulting in
programmed cell death (4–6). Resistance to clinical
chemotherapeutics often involves alterations in the death receptor
signaling cascade to promote survival, rather than apoptosis, in
response to these agents (7,8).
Acquired chemoresistance is often accompanied by an increase in
cellular growth pathways and enhanced metastatic potential
(9). In the case of breast cancer,
resistance often correlates with the loss of estrogen receptor
expression and activation of ER (ERα isoform unless otherwise
specified) independent growth pathways, such as PI3K/Akt and
mitogen-activated protein kinases signaling cascades, as well as
the induction of epithelial-to-mesenchymal transition (EMT)
(10–13).
Mitogen-activated protein kinase pathway (MAPK)
consists of Erk1/2, JNK, p38 and Erk5/BMK family members (14). These kinases are activated in
response to upstream MAPK-kinases (MEKs), which phosphorylate MAPKs
in response to specific cellular signals (15–17).
Activation of MAPKs, and p38 in particular, results in
phosphorylation of selective downstream targets leading to
regulation of genes involved in diverse cellular events and have
more recently been intensely studied for their critical roles in
cancer promotion and progression. By regulating gene expression in
response to specific extracellular stimuli, including cytokines
(TNF and TRAIL), and known cytotoxic drugs (18), this signaling kinase also promotes
transcription of genes involved in invasion, metastasis and
survival (19–22). Moreover, p38 activates known
downstream transcriptional regulators, including the NF-κB and ATF2
(21,23,24).
NF-κB, which is known to promote survival and metastasis, can be
directly activated by p38 or phosphorylated by Akt in a
p38-dependent manner (21,25). Our laboratory has previously shown
that increased basal and activated p38 MAPK signaling is critical
to death receptor resistance (26–29).
Given the interaction between the p38 MAPK pathway and TNF-NF-κB
signaling, the role of p38 in acquired apoptosis resistance is of
both biological and therapeutic interest. However, the role of p38
in the development of a mesenchymal phenotypic remains unclear.
MicroRNAs are now well recognized as important
regulators of mRNA processing. However, their pathological role in
disease processes has only recently come under investigation, with
a growing number of studies reporting miRNAs to play roles in
tumorigenesis, metastasis and chemoresistance (30–32),
and the regulation of EMT (33).
Consequently, in this study, we investigated global miRNA
expression changes involved in both death receptor and chemotherapy
resistance, utilizing mesenchymal MCF-7TN-R cells that are
completely resistant to TNF and chemotherapeutics (34–39).
Further, we examined the role of p38 MAPK in EMT progression using
the p38 inhibitor RWJ67657 (40–42).
Given the need for chemoresistant cancer treatment strategies, the
development of novel therapeutic targets is of increasing
importance and here we demonstrate the therapeutic potential of
targeting the p38 MAPK pathway in the treatment of invasive,
chemoresistant breast cancer.
Materials and methods
Cell generation and culture
MCF-7 cells and MCF-7TN-R were cultured as
previously described (43,44). We previously gene rated MCF-7TN-R
cells by exposing MCF-7 cells to increasing concentrations of TNFα
until resistance was established (35). The culture flasks were maintained
in a tissue culture incubator in a humidified atmosphere of 5%
CO2 and 95% air at 37°C.
miRNA microarray analysis
MicroRNA analysis was performed as previously
described (45). Briefly,
MCF-7TN-R cells were plated at a density of 2×106 cells
in 25 cm2 flasks in normal culture media (DMEM media
supplemented with 5% FBS, 1% penicillin/streptomycin, 1% essential
amino acids, 1% non-essential amino acids and 1% sodium pyruvate),
and then allowed to adhere overnight at 37°C, 5% CO2 and
air. The following day the media were changed to phenol red-free
media (supplemented as above) and 5% CS-FBS. Cells were harvested
in PBS, collected by centrifugation, and total-RNA, including small
RNA, extracted using the miRNeasy kit (Qiagen, Valencia, CA)
according to manufacturer’s protocol, although miRNA enrichment was
not performed. Quantity and quality of RNA was determined by
absorbance (260, 280 nm), and 5 μg total-RNA was used for
microarray analysis. Microarray was used to determine miRNA
expression, using three biological replicates for each cell line.
Low intensity probes (signal <100 in more than half samples)
were excluded from the analysis. Raw data was log-transformed and
normalized by IQR (spell out). Clustering of miRNA expression data
was performed using cluster analysis (20), with filtering to remove
inconsistencies between replicates. For clustering, we first
log-transformed the data and median-centered the array and genes,
followed by average linkage clustering, with clustering results
visualized by TreeView (http://rana.lbl.gov/EisenSoftware.htm). Significant
array changes are shown in Table
I.
 | Table ImicroRNA changes associated with
acquired TNF resistance. |
Table I
microRNA changes associated with
acquired TNF resistance.
Downregulated
| Upregulated
|
|---|
| microRNA | Expression (fold
MCF-7) | P-value | microRNA | Expression (fold
MCF-7) | P-value |
|---|
| hsa-miR-21 | −75.47 | 3.29E-05 | hsa-miR-30a-5p | 2.02 | 1.07E-02 |
| hsa-let-7a | −3.37 | 2.24E-03 | hsa-miR-100 | 5.43 | 3.84E-04 |
| hsa-let-7a | −3.34 | 6.35E-03 | hsa-miR-100 | 6.03 | 2.76E-04 |
| hsa-let-7b | −2.34 | 2.44E-02 | hsa-miR-106a | 3.70 | 5.55E-04 |
| hsa-let-7c | −2.34 | 3.44E-02 | hsa-miR-106a | 3.76 | 5.65E-03 |
| hsa-let-7d | −2.73 | 2.38E-02 | hsa-miR-106b | 2.15 | 2.73E-02 |
| hsa-let-7e | −2.62 | 1.24E-02 | hsa-miR-10a | 7.89 | 2.19E-04 |
| hsa-let-7f | −4.43 | 4.68E-03 | hsa-miR-10a | 7.90 | 1.60E-04 |
| hsa-let-7f | −3.31 | 2.68E-02 | hsa-miR-10b | 6.52 | 2.34E-04 |
| hsa-let-7g | −5.70 | 2.85E-04 | hsa-miR-10b | 6.57 | 2.34E-04 |
| hsa-let-7g | −4.88 | 1.05E-03 | hsa-miR-130a | 5.20 | 7.49E-05 |
| hsa-let-7i | −6.52 | 1.77E-04 | hsa-miR-130a | 5.45 | 3.16E-04 |
| hsa-let-7i | −4.72 | 6.66E-04 | hsa-miR-130b | 4.54 | 5.82E-07 |
| hsa-miR-125a | −2.49 | 4.22E-02 | hsa-miR-130b | 4.91 | 1.28E-04 |
| hsa-miR-125a | −2.46 | 1.06E-02 | hsa-miR-148a | 2.06 | 1.07E-02 |
| hsa-miR-141 | −9.28 | 4.75E-05 | hsa-miR-17-5p | 4.29 | 2.56E-04 |
| hsa-miR-141 | −8.66 | 1.28E-04 | hsa-miR-17-5p | 4.47 | 5.68E-04 |
| hsa-miR-185 | −2.05 | 1.95E-02 | hsa-miR-18a | 5.04 | 7.29E-04 |
| hsa-miR-193b | −2.35 | 4.30E-04 | hsa-miR-18a | 5.34 | 5.25E-04 |
| hsa-miR-193b | −2.32 | 2.18E-03 | hsa-miR-18b | 4.11 | 1.08E-03 |
| hsa-miR-200a | −6.71 | 6.85E-04 | hsa-miR-18b | 4.89 | 4.91E-04 |
| hsa-miR-200a | −6.36 | 1.77E-05 | hsa-miR-19a | 4.72 | 1.21E-03 |
| hsa-miR-200b | −12.57 | 8.40E-06 | hsa-miR-19a | 9.06 | 1.66E-04 |
| hsa-miR-200b | −11.79 | 1.90E-04 | hsa-miR-19b | 7.00 | 7.51E-04 |
| hsa-miR-200c | −14.38 | 3.23E-05 | hsa-miR-19b | 7.44 | 9.85E-04 |
| hsa-miR-200c | −10.24 | 2.56E-03 | hsa-miR-20a | 6.05 | 3.40E-04 |
| hsa-miR-202 | −3.74 | 4.27E-04 | hsa-miR-20a | 6.87 | 7.08E-04 |
| hsa-miR-202 | −2.94 | 3.31E-03 | hsa-miR-20b | 5.21 | 1.34E-04 |
| hsa-miR-203 | −6.11 | 4.54E-03 | hsa-miR-20b | 5.87 | 1.03E-03 |
| hsa-miR-203 | −5.47 | 2.90E-04 | hsa-miR-221 | 12.97 | 1.76E-04 |
| hsa-miR-206 | −3.44 | 4.89E-08 | hsa-miR-221 | 13.85 | 6.05E-04 |
| hsa-miR-206 | −2.17 | 3.54E-03 | hsa-miR-222 | 12.61 | 3.52E-04 |
| hsa-miR-21 | −68.35 | 6.86E-06 | hsa-miR-222 | 12.69 | 1.70E-04 |
| hsa-miR-210 | −3.85 | 1.39E-03 | hsa-miR-223 | 5.21 | 1.52E-03 |
| hsa-miR-210 | −3.35 | 4.18E-04 | hsa-miR-223 | 7.97 | 4.48E-04 |
| hsa-miR-23a | −5.00 | 2.74E-03 | hsa-miR-30a-5p | 2.18 | 6.57E-03 |
| hsa-miR-23a | −4.96 | 1.17E-02 | hsa-miR-30b | 2.20 | 2.92E-02 |
| hsa-miR-23b | −4.83 | 1.39E-02 | hsa-miR-30d | 2.07 | 2.82E-02 |
| hsa-miR-23b | −4.75 | 3.09E-03 | hsa-miR-30d | 2.18 | 1.60E-02 |
| hsa-miR-24 | −4.17 | 3.21E-03 | hsa-miR-31 | 2.02 | 1.53E-02 |
| hsa-miR-24 | −3.86 | 5.66E-04 | hsa-miR-34a | 2.07 | 8.92E-04 |
| hsa-miR-27a | −5.64 | 1.90E-04 | hsa-miR-34a | 2.09 | 3.52E-03 |
| hsa-miR-27a | −5.37 | 6.21E-05 | hsa-miR-9 | 2.13 | 9.83E-03 |
| hsa-miR-27b | −6.74 | 7.10E-05 | hsa-miR-92 | 2.26 | 1.76E-03 |
| hsa-miR-27b | −6.30 | 6.34E-04 | hsa-miR-92b | 2.24 | 1.52E-03 |
| hsa-miR-29a | −2.02 | 3.98E-03 | hsa-miR-99a | 4.59 | 5.48E-04 |
| hsa-miR-29a | −2.00 | 2.18E-02 | | | |
| hsa-miR-29b | −2.59 | 1.01E-02 | | | |
| hsa-miR-29b | −2.52 | 1.11E-02 | | | |
| hsa-miR-335 | −2.10 | 3.27E-03 | | | |
| hsa-miR-342 | −8.25 | 2.77E-03 | | | |
| hsa-miR-342 | −7.16 | 9.04E-03 | | | |
| hsa-miR-345 | −2.27 | 1.04E-03 | | | |
| hsa-miR-345 | −2.19 | 1.35E-03 | | | |
| hsa-miR-425-5p | −2.37 | 2.02E-02 | | | |
| hsa-miR-425-5p | −2.00 | 3.57E-02 | | | |
| hsa-miR-487b | −2.07 | 8.62E-04 | | | |
| hsa-miR-574 | −2.37 | 1.95E-02 | | | |
| hsa-miR-675 | −2.20 | 4.40E-05 | | | |
| hsa-miR-7 | −3.01 | 7.11E-03 | | | |
| hsa-miR-98 | −2.92 | 5.55E-03 | | | |
Real-time RT-PCR
Real-time RT-PCR was performed similarly to
previously reported studies (38,46).
In brief, total cellular RNA was extracted using RNeasy mini
columns (Qiagen), following the manufacturer’s instructions.
Reverse transcription (RT) was performed using SuperScript
First-Strand (Invitrogen). Gene transcript levels were determined
using the iQ5 real-time quantitative PCR detection system (BioRad
Inc., Hercules, CA). Quantification and relative gene expression
were calculated with internal controls using the 2−ΔΔCt
method (47), with the ratio
between these values used to determine relative gene expression
levels. Primer sequences are available upon request.
Western blot analysis
Protein expression analysis was performed similarly
to previously published methods (39,48).
MCF-7TN-R cells were plated at 5×105 cells in T-25
culture flasks and treated as indicated. Cells were harvested,
total protein transferred to membranes, and membranes blocked with
PBS-Tween (0.05%)-5% low-fat dry milk solution at room temperature
for 1 h. The membranes were subsequently probed with the
phosphorylated p38 (1:1,000; Cell Signaling, Boston, MA) or
phosphorylated ATF-2 (1:1,000; Cell Signaling). Following
incubation, blots were washed in PBS-Tween (0.05%) solution and
incubated with goat-anti-rabbit antibodies (1:10,000 dilution) for
2 h at room temperature. Following four washes, immunoreactive
proteins were detected using the ECL chemiluminescence system
(Amersham/GE Healthcare, Pittsburgh, PA) and recorded by
fluorography on Hyperfilm (Amersham/GE Healthcare), according to
manufacturer’s instructions.
Reporter gene assays
Reporter gene assays were performed as previously
described (49,50). MCF-7TN-R cells were plated at
50,000 cells per 24-well plate and allowed to attach overnight. The
next morning, cells were transfected with 50 ng of Gal-4-luciferase
(Stratagene, La Jolla, CA) in combination with 25 ng of Gal-4-CHOP
(Stratagene) using Effectene reagent for 5 h, according to the
manufacturer’s protocol. For NF-κB-luciferase experiments, cells
were transfected with 10 ng of pFC-NF-κB-luciferase plasmid
(Stratagene). For all luci ferase assays, cells were then incubated
for 18–24 h in DMEM with 10% FBS in the presence of vehicle, RWJ or
TNF as previously described (22).
Following treatment, the media were removed and 150 μl of 1X
lysis buffer (Promega, Madison, WI) was added to each well for 1 h
on a rocker at room temperature. Luciferase activity for 25
μl of cell extracts was determined using Promega Luciferase
System (Promega) in a Berthold AutoLumat Plus luminometer (Wildbad,
Germany). For each experiment, changes in luciferase activity were
determined as relative light units (RLU) and represented as percent
transcriptional activation normalized to vehicle control
samples.
Clonogenic survival assay
As previously described (34,39).
MCF-7TN-R cells were plated in 6-well plates at 1×103
cells per well in complete media (10% DMEM). Twenty-four hours
later the cells were pre-treated with vehicle (DMSO) or the MAPK
inhibitor RWJ67657 (Johnson and Johnson Pharmaceutical Research
& Development, L.L.C., Raritan, NJ), as indicated. The cells
were then monitored microscopically for colony growth and 7–14 days
later were fixed by adding glutaraldehyde (2.5% final
concentration) directly to the well. Following fixation for 30 min,
the plates were washed and stained with a 0.4% solution of crystal
violet in 20% methanol for 30 min, washed and allowed to dry.
Colonies greater than 50 were counted and data normalized as
percent clonogenic survival from vehicle control cell. For the
transfection-based clonogenic survival assay, MCF-7TN-R cells were
plated as above. The following day the cells were transfected using
the Effectene method (Qiagen) with increasing concentrations of a
DI-P38α or β expression vector (0 to 300 ng/well) with total DNA
concentrations increasing to 300 ng/well with empty vector.
Twenty-four hours later the cells were treated as indicated in the
figure legends. The cells were then monitored microscopically for
colony growth and harvested and quantitated as above for clonogenic
survival.
Animal studies
Xenograft tumor studies were conducted as previously
described (42,51). Immune-compromised female
ovariectomized mice (29–32 days old) were obtained from Charles
River Laboratories (Wilmington, MA). The animals were allowed a
period of adaptation in a sterile and pathogen-free environment
with food and water ad libitum. MCF-7 and MCF-7TN-R cells
were harvested in the exponential growth phase and viable cells
mixed with Matrigel Reduced Factors (BD Biosciences, San Jose, CA).
Injections (5×106 cells/injection) were made bilaterally
into the mammary fat pad. All the procedures in animals were
carried out under anesthesia using a mix of isofluorane and oxygen
delivered by mask. Tumor sizes were measured twice weekly using a
digital caliper, with tumor volumes calculated using the following
formula: 4/3πLM2, where L is the larger radius, and M is
the smaller radius. At necropsy on day 21, animals were euthanized
by cervical dislocation after CO2 exposure. Tumors were
removed and either frozen in liquid nitrogen or fixed in 10%
formalin for further analysis. All animal procedures were conducted
in compliance with State and Federal laws, standards of the US
Department of Health and Human Services, and guidelines established
by the Tulane University Animal Care and Use Committee. The
facilities and laboratory animal programs of Tulane University are
accredited by the Association for the Assessment and Accreditation
of Laboratory Animal Care.
Statistical analysis
Studies involving more than 2 groups were analyzed
by one-way ANOVA with Tukey’s post-test using the Graph Pad Prism
V.4 software program (Horsham, PA). All others were subjected to
unpaired Student’s t-test. A value of p<0.05 was considered
statistically significant (52–54).
Results
Death receptor resistance promotes
hormone independent tumorigenesis
Death receptor-resistant and ER(−) MCF-7TN-R cells
exhibit reduced apoptosis, increased survival and multi-drug
chemoresistance, as compared to their parental ER(+) MCF-7 cells
(35,55). Given the clinical association of
chemo-resistance with hormone independence, we investigated the
ability of MCF-7TN-R cells to form tumors in the absence of
estrogen (56–58). MCF-7TN-R and MCF-7 cells were
injected subcutaneously into the flanks of NOD-SCID mice and
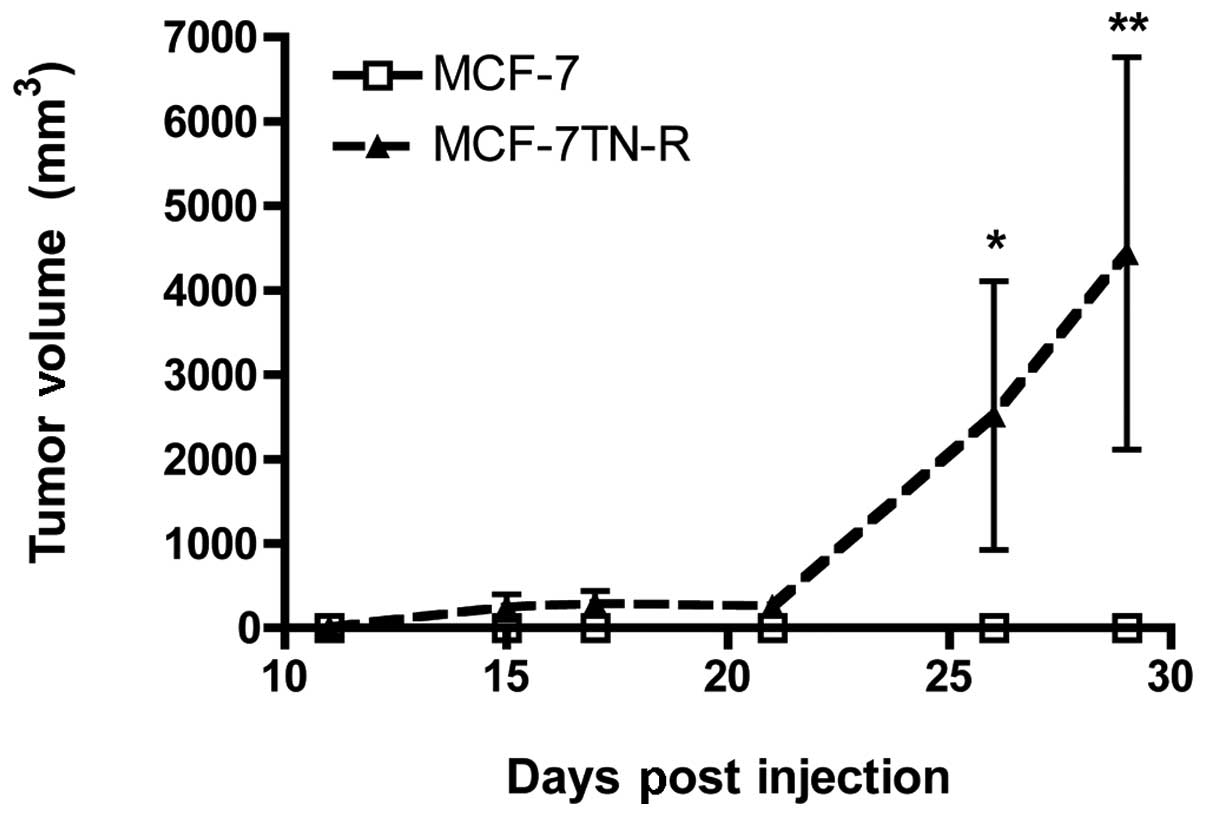
measured for tumor growth. As seen in Fig. 1, MCF-7TN-R cells showed increased
tumor formation and tumor growth compared to MCF-7 cells. The lack
of MCF-7 tumor formation in the absence of estrogen correlates with
previously published studies (59). These results demonstrate that TNF
resistance increased hormone-independent tumorigenesis in
vivo.
Global microRNA profiling associated with
death receptor resistance
We next investigated the mechanism of the increased
tumorigenesis seen in MCF-7TN-R cells. miRNAs are small RNAs that
target mRNAs for degradation or prevent translation. Several miRNAs
have been found to be involved in breast cancer tumorigenesis,
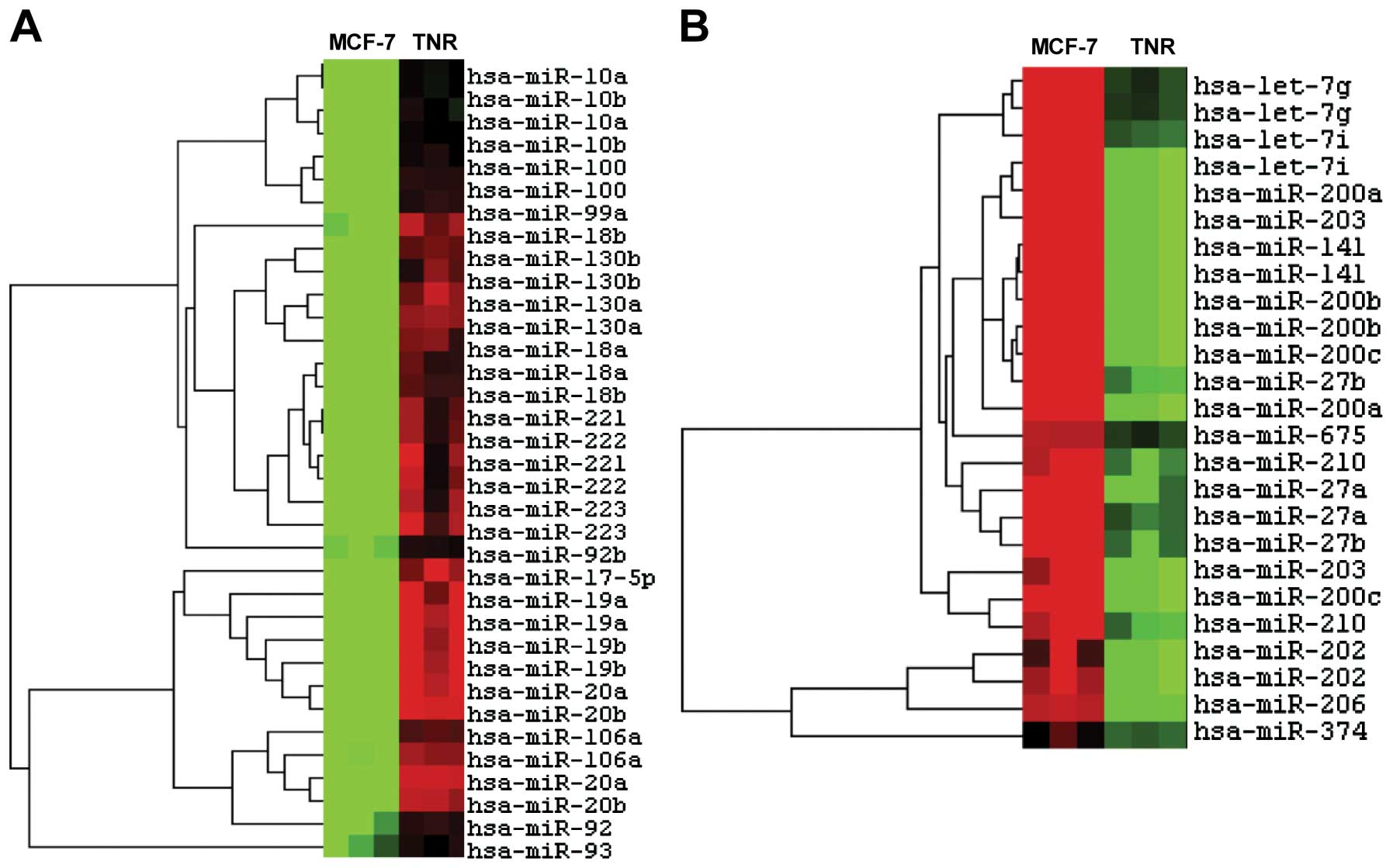
including miR-221/222 and the miR-200 family (60). Heatmap analysis demonstrated
differential microRNA expression between MCF-7 and its daughter
MCF-7TN-R cells (Fig. 2). Specific
microRNA expression changes (Table
I) demonstrated downregulation of several miRNA associated with
tumor suppression, including miR-16, miR-424, miR-15 and miR-19.
There was also decreased expression of miR-200, which is strongly
associated with breast cancer tumorigenesis. miR-182, miR-183,
miR-23 and miR-27, which all target the tumorigenic MEF2C
transcription factor, were also downregulated. Furthermore, we
noted downregulation of miR-203, a recently identified tumor
suppressor (61). These data
demonstrate miRNA expression profiles indicative of enhanced
tumorigenesis and a more aggressive phenotype in death
receptor-resistant cells vs. their parental MCF-7 cells.
We recently demonstrated that chemoresistant
MCF-7TN-R cells exhibit increased EMT, resulting in a mesenchymal
phenotype (36). As miRNA
dysregulation contributes to human breast cancer development
(60,62,63),
we hypothesized that changes in miRNA expression may also
contribute to EMT progression. Microarray results were analyzed for
changes in expression of levels of miRNAs whose targets are
associated with EMT. Results showed downregulation of several
miRNAs known to regulate EMT, including miR-203 and miR-26, which
target Slug and Lef, respectively, and miR-200, which targets ETS1,
ZEB1 and ZEB2. The metastasis-promoting miR-10b, by contrast
(associated with the EMT transcription factor Twist), was markedly
upregulated (30).
Our laboratory has previously shown increased basal
and stimulated p38 MAPK signaling in MCF-7TN-R cells. As p38 MAPK
is known to promote breast cancer hormone independence and EMT
progression (24), we hypothesized
that changes in miRNA expression may increase p38 signaling in
these resistant cells. Therefore, we next analyzed our microarray
findings for alterations in miRNA associated with p38 MAPK
(64–67). Results revealed increased
expression of miRNA associated with increase p38 activity,
including miR-34 (a p53 target), miR-17, miR-9, miR-199 (a tumor
suppressor that targets c-Met), miR-125 (previously found to be a
tumor suppressor in breast cancer), which associate with increased
p38 MAPK activity (68–72). Furthermore, compared to parental
MCF-7 cells, miR-21 was markedly decreased, in inverse correlation
with p38 signaling (73).
Together, these results suggest that the increased p38 MAPK
activity in MCF-7TN-R cells may in part result from differential
microRNA expression.
RWJ67657 inhibits p38 activation and
blocks downstream MAPK signaling
The microRNA profile above correlates with increased
MAPK signaling previously observed in these cells (27). Given the role of p38 signaling in
breast cancer promotion and progression, we further investigated
the therapeutic potential of targeting this pathway by determining
whether p38 pharmacological inhibition could reverse the neoplastic
changes found in these cells. To that end, we used the p38
inhibitor RWJ67657 for investigating suppression of p38 activity.
We first determined RWJ67657 blockage of p38 signaling in
chemoresistant breast cancer cells. MCF-7TN-R cells, exposed to
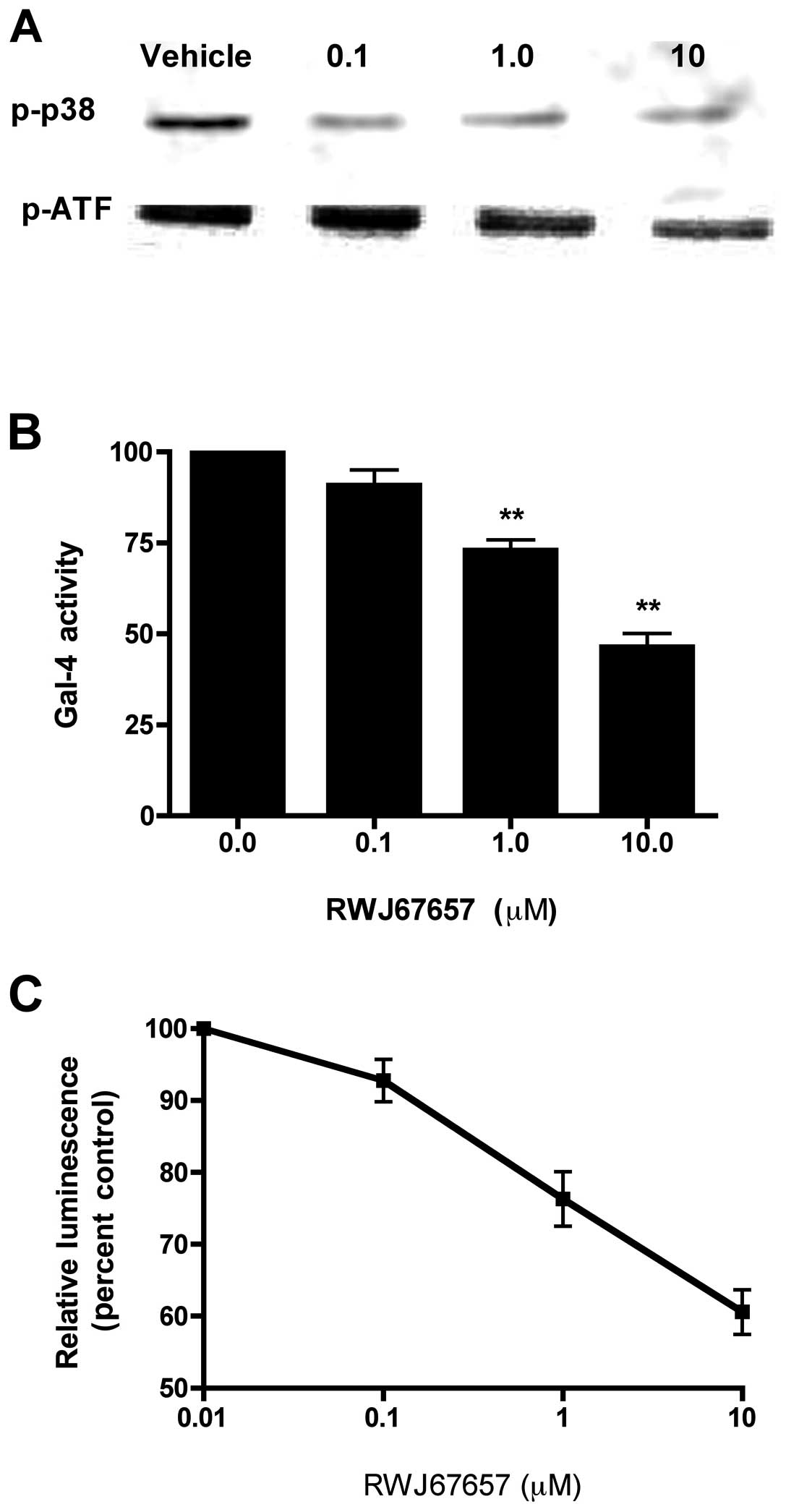
increasing concentrations of RWJ67657, were analyzed for
phosphorylated p38 by western blot analysis (Fig. 3A). In comparison to vehicle treated
cells, treatment with RWJ67657 resulted in a dose-dependent
decrease phosphorylation of p38 MAPK. Exposure to RWJ67657 had no
effect on total levels of p38 (data not shown), suggesting that
RWJ67657 blocks activation, but not basal expression, of p38 and
ATF, a downstream effector of p38 signaling.
We next determined whether inhibition of p38 by
RWJ67657 might also decrease pathway-downstream transcriptional
activators. MCF-7TN-R cells were treated with RWJ67657 and
activation of the p38 pathway was determined by measurement of
Gal-CHOP transactivation (Fig.
3B). RWJ67657 decreased p38 transcriptional activity in a
dose-dependent manner, suggesting functional blockage of p38 MAPK
signal transduction. Additionally, we also analyzed the ability of
RWJ67657 to block that NF-κB signaling, a known downstream target
of p38 (21,24,27,74).
As shown in Fig. 3C,
dose-dependent RWJ67657 treatment decreased NF-κB transcriptional
activity. Taken together, these findings provide proof-of-principle
that RWJ67657 blocks p38 MAPK signaling found in MCF-7TN-R
cells.
Inhibition of p38 suppresses clonogenic
survival and in vivo tumor growth
Our laboratory has previously demonstrated increased
colony formation and long-term survival of MCF-7TN-R cells, as
compared to their parental MCF-7 (35). Therefore, to determine whether p38
MAPK inhibition could suppress in vitro survival of
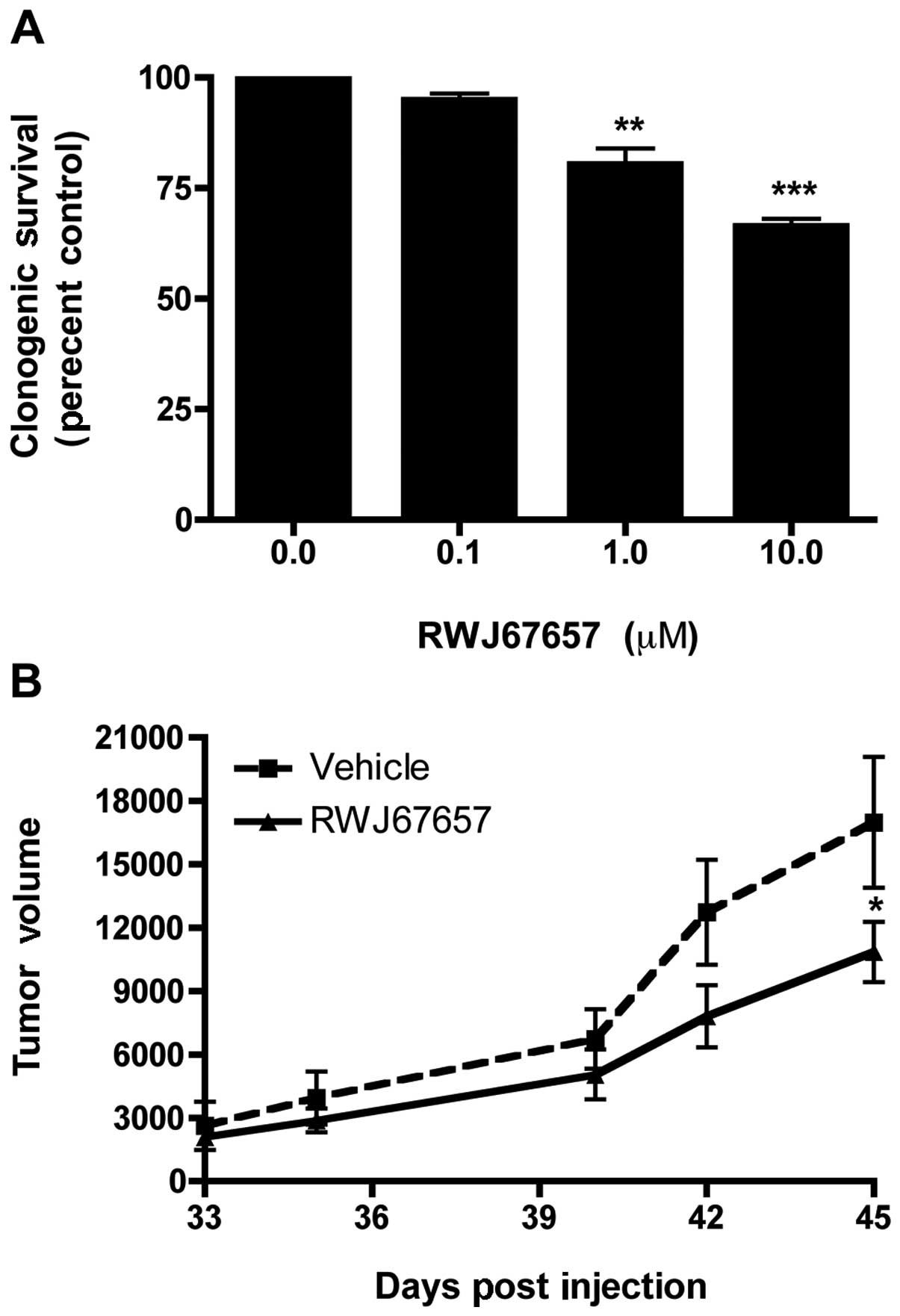
MCF-7TN-R cells, cells treated with increasing concentrations of
RWJ67657 were analyzed for long-term colony formation. As shown in
Fig. 4A, treatment with RWJ67657
inhibited clonogenic survival in a dose-dependent manner. We next
sought to validate our clonogenic survival results using a
xenograft animal tumor model. MCF-7TN-R cells were implanted into
the mammary fat pad of female immune-compromised, treated with
either vehicle control or RWJ67657, and monitored for tumor
formation. Fig. 4B illustrates
that treatment with RWJ67657 resulted in a statistically
significant decrease in tumor volume compared to vehicle-treated
animals in our chemoresistant xenograft model. These results
suggest that targeting p38 may be therapeutically relevant in the
treatment of death ligand-resistant breast cancer.
p38 inhibition reverses
epithelial-to-mesenchymal transition
Our above microRNA findings suggest an increased EMT
phenotype in MCF-7TN-R cells, consistent with previously published
findings (36). Given the ability
of RWJ67657 to inhibit MCF-7TN-R survival and tumor growth, we next
determined whether inhibition of p38 could reverse the EMT changes
found in this chemoresistant cell model. MCF-7TN-R cells were
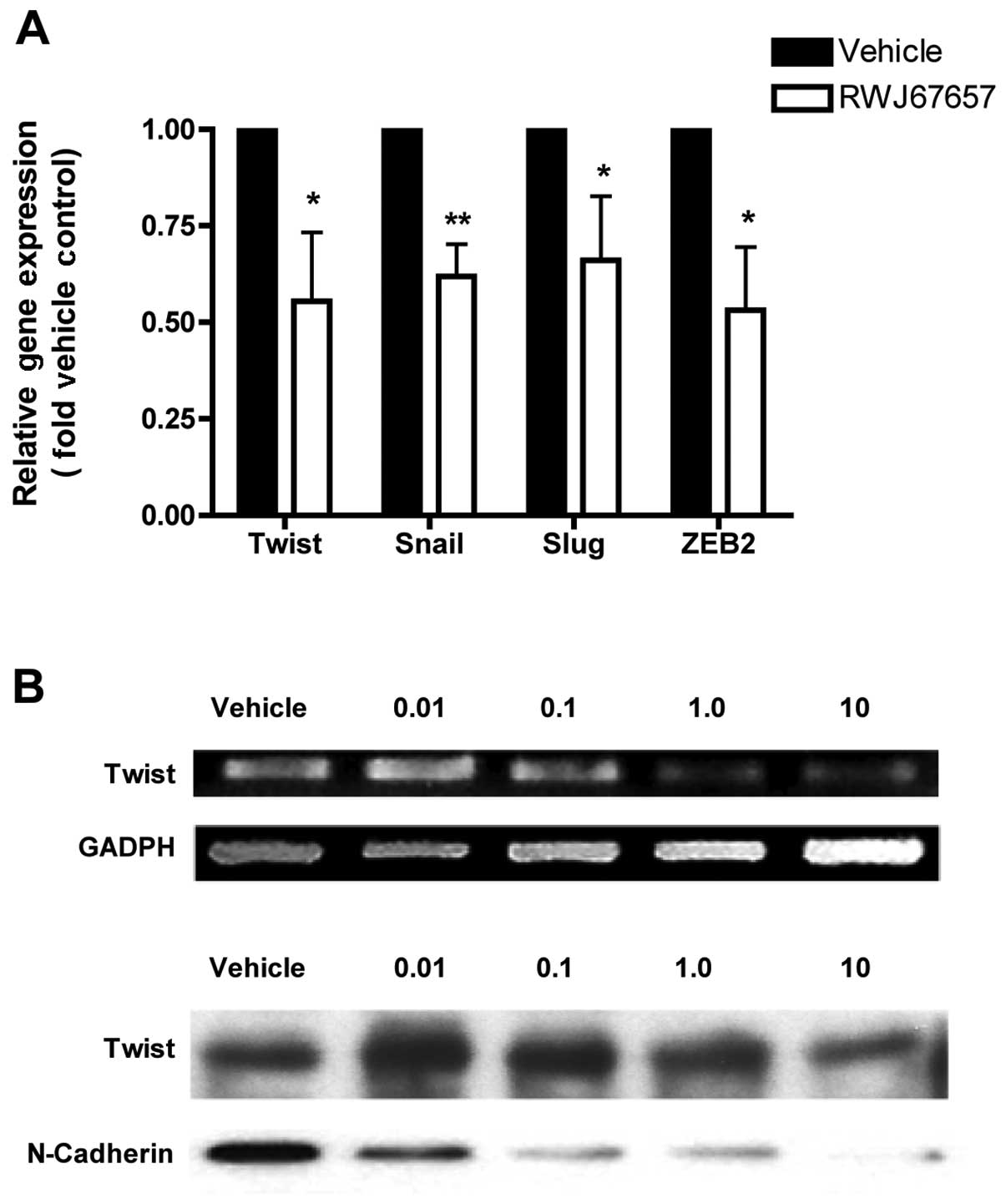
treated with RWJ67657 and measured for expression of EMT markers
Twist, Snail, Slug and ZEB2, chosen because MCF-7TN-R exhibited
increased expression of these EMT genes (36). Treatment with RWJ67657 decreased
mRNA expression of all four EMT markers (Fig. 5A), suggesting that EMT in
chemoresistant cells is at least partially p38-dependent. We next
sought to validate these results using RT-PCR and western blot
analysis for Twist. Our microRNA analysis above showed a
significant increase in pro-metastasis miR-10b in MCF-7TN-R cells,
possible association with Twist expression (30,75,76).
Treatment with RWJ67657 dose-dependently decreased Twist mRNA and
protein expression (Fig. 5B). A
hallmark of EMT in breast cancer cells is expression of the
mesenchymal surface protein N-cadherin, which is regulated by Twist
(77,78). To confirm possible anti-EMT
activity, RWJ67657-treated cells were examined for the EMT marker
N-cadherin. Expression of N-cadherin was indeed decreased by
dose-dependent RWJ67657. Taken together, these results suggest that
pharmacological inhibition of p38 may inhibit the EMT, an advanced
cancer phenotype, in chemoresistant MCF-7TN-R cells.
RWJ67657 alters endogenous microRNA
profiling
Given the differential microRNA expression found in
MCF-7TN-R, we next determined whether p38 suppression might alter
microRNAs functional in breast cancer cells. Consequently, vehicle-
or RWJ67657-treated MCF-7TN-R cells were analyzed for microRNA
expression changes. Heatmap analysis demonstrated differential
microRNA expression associated with RWJ67657 treatment (Fig. 6). Specific microRNA expression
changes are shown in Table II.
Treatment with RWJ67657 resulted in changes in expression of
several microRNAs known to be involved in cancer chemotherapy and
drug resistance. Of note, we found down-regulation of miR-200 and
miR-303, which are involved in resistance to doxorubicin (79), as was a known mediator of
paclitaxel resistance, miR-302 (80,81).
We also found changes in microRNA associated with endocrine therapy
resistance, including RWJ67657-altered expression of miR-199, a
known effector of fulvestrant resistance, and miR-328, associated
with resistance to both fulvestrant and mitoxantrone (81,82).
Thus, MCF-7TN-R-misexpressed microRNAs likely contribute to
resistance to doxorubicin, paclitaxel and fulvestrant, in addition
to their more general alterations associated with the anticancer
effects of RWJ67657.
| Table IImicroRNA changes associated with
RWJ67657. |
Table II
microRNA changes associated with
RWJ67657.
| microRNA | Expression (fold
vehicle control) | P-value |
|---|
| hsa-miR-143 | 1.62 | 4.81E-02 |
| hsa-miR-153 | −2.38 | 3.11E-03 |
| hsa-miR-190 | −1.76 | 1.43E-02 |
| hsa-miR-199b | −2.82 | 4.32E-02 |
| hsa-miR-199b | −2.76 | 2.86E-02 |
| hsa-miR-200a | −2.91 | 1.84E-03 |
| hsa-miR-200a | −3.14 | 4.92E-03 |
| hsa-miR-200a | −1.83 | 2.01E-02 |
| hsa-miR-200b | −1.94 | 2.70E-02 |
| hsa-miR-200b | −2.39 | 2.09E-02 |
| hsa-miR-217 | −2.67 | 4.24E-02 |
| hsa-miR-219 | −2.04 | 2.08E-02 |
| hsa-miR-300 | 2.07 | 1.64E-02 |
| hsa-miR-301 | −1.94 | 4.47E-02 |
| hsa-miR-302a | −3.97 | 3.08E-02 |
| hsa-miR-302a | −2.81 | 4.32E-02 |
| hsa-miR-302a | −3.05 | 2.27E-03 |
| hsa-miR-302b | −2.50 | 3.71E-02 |
| hsa-miR-302c | −2.11 | 4.79E-02 |
| hsa-miR-324-3p | −2.09 | 2.58E-02 |
| hsa-miR-324-5p | −1.74 | 2.69E-02 |
| hsa-miR-325 | −2.72 | 1.00E-02 |
| hsa-miR-328 | −2.68 | 4.36E-02 |
| hsa-miR-33 | −2.41 | 4.06E-02 |
| hsa-miR-335 | −1.50 | 3.12E-02 |
| hsa-miR-362 | −3.03 | 2.78E-03 |
| hsa-miR-363 | −2.46 | 8.92E-03 |
| hsa-miR-488 | −3.63 | 1.45E-02 |
| hsa-miR-492 | 1.75 | 1.27E-02 |
| hsa-miR-492 | 1.83 | 1.33E-02 |
| hsa-miR-520g/h | −2.79 | 6.26E-03 |
| hsa-miR-521 | −3.05 | 3.59E-02 |
| hsa-miR-522 | −3.52 | 1.15E-02 |
| hsa-miR-549 | −2.25 | 4.64E-02 |
| hsa-miR-550 | −2.91 | 2.41E-02 |
| hsa-miR-551b | −2.32 | 3.17E-02 |
| hsa-miR-570 | −1.81 | 2.50E-02 |
| hsa-miR-574 | −3.23 | 4.71E-02 |
| hsa-miR-592 | −2.19 | 4.63E-02 |
| hsa-miR-592 | −4.27 | 1.17E-02 |
| hsa-miR-593 | −2.24 | 4.81E-02 |
| hsa-miR-595 | −2.14 | 4.43E-02 |
| hsa-miR-596 | −2.48 | 2.62E-02 |
| hsa-miR-607 | 1.68 | 4.75E-02 |
| hsa-miR-616 | −2.48 | 1.97E-02 |
| hsa-miR-620 | −1.58 | 2.69E-02 |
| hsa-miR-620 | −1.58 | 2.69E-02 |
| hsa-miR-627 | −1.59 | 4.11E-02 |
| hsa-miR-628 | −1.53 | 3.28E-02 |
| hsa-miR-663 | 1.74 | 2.88E-02 |
| hsa-miR-871 | −3.21 | 3.39E-02 |
| hsa-miR-871 | −5.42 | 1.45E-02 |
| hsa-miR-886-5p | −1.81 | 7.97E-04 |
| hsa-miR-888 | −1.66 | 4.44E-02 |
| hsa-miR-891b | 1.80 | 3.41E-02 |
| hsa-miR-922 | −2.41 | 3.90E-02 |
| hsa-miR-922 | −5.42 | 3.14E-03 |
| hsa-miR-923 | 1.68 | 9.30E-03 |
Discussion
Tumor cell progression to an invasive and
chemotherapeutic-resistant phenotype represents a significant
obstacle that must be overcome to successfully treat and cure
cancer. To investigate mechanisms by which cancer cells progress to
a resistant phenotype, we employed a previously established in
vitro model of acquired resistance (27,35,37,39,83),
the cell line MCF-7TN-R, which is resistant to both death receptors
and chemotherapeutic drugs that depend on p38 MAPK and NF-κB
signaling (27,37,55,84).
Further, we investigated whether progression to apoptotic
resistance also associated with increased hormone-independent tumor
formation and p38 signaling. To delineate possible mechanisms of
resistance, we investigated whether alternatively expressed
microRNAs allow progression from the chemo-sensitive MCF-7 cell
line to the chemoresistant cell line MCF-7TN-R.
Given the increasing evidence that microRNAs play a
significant role in cancer, we examined the miRNA profile of
parental MCF-7 and resistant MCF-7TN-R cell lines by micro-array
analysis. Our results revealed differential expression of several
miRNAs involved in the progression to a mesenchymal phenotype,
including miR-200 and miR-10b. Specifically, miR-200 is a key
regulator of EMT in numerous cancers, promoting an epithelial
phenotype by inhibiting several EMT genes, including ZEB1 and ZEB2
(33). Further, we also
demonstrated a significant increase in miR-10, which associates
with expression of the EMT gene Twist (30,75,76,85).
Twist is known to upregulate ER and N-cadherin expression to
promote EMT (77,78). The gain of mesenchymal properties
via EMT permits cells to detach from one another and migrate
through the basement membrane. Our findings here also concur with
previously published results demonstrating increased EMT in
resistant MCF-7TN-R cells, and further suggest that microRNA
changes may promote a mesenchymal phenotype.
Recent studies have linked tumor growth, invasion,
and chemoresistance to miRNA alterations (86). Of particular interest, a recent
microarray analysis by Chen et al comparing a
doxorubicin-resistant MCF-7 cell line to the doxorubicin-sensitive
MCF-7 parental cell line revealed differential miRNA expression,
suggesting that specific miRNAs may modulate chemoresistance in
breast cancer (32). The miRNA
changes described by Chen et al are consistent with the
miR-21 and miR-34 changes seen in our MCF-7TN-R cells.
Additionally, a recent study by Zhu et al showed that
molecular inhibition of miR-21 in MDA-MB-231 cells results in
suppression of invasion and metastasis (87). We found inhibition of p38-altered
expression of miRNAs known to promote both endocrine therapy- and
chemo-resistance in these cells, including miR-200, miR-303,
miR-302, miR-199 and miR-328 (79–81,82).
In light of the recent evidence supporting miRNA changes in
aggressive breast cancer EMT, the differential miRNA expression
found in our microarray likely contributes to the apoptotic
resistance of these cells.
Having previously identified p38-NF-κB-signaling as
the mediator of chemoresistance in MCF-7TN-R cells, we hypothesized
that this pathway may be responsible for the EMT and increased
tumorigenesis seen in the MCF-7TN-R cells (27). To test this hypothesis, we
attempted to reverse the changes seen in the MCF-7TN-R cell line by
inhibiting this pathway with RWJ67657, a pharmacologic inhibitor of
p38. Western blot analysis and reporter gene assays following
exposure to the inhibitor revealed decreased p38 activation and
downstream signaling, demonstrating that RWJ67657 functionally
blocked p38 signaling in our resistant cells. We then evaluated the
role of p38 in EMT through utilization of this p38 inhibitor.
Treatment with RWJ67657 decreased mRNA expression of the EMT
markers Twist, Snail, Slug, and ZEB2. Overall, loss of these
markers indicates loss of a mesenchymal markers in MCF-7TN-R cells
when treated with RWJ67657. These results agree with recent
findings that suggest a role for p38 in EMT, strongly supporting
the therapeutic potential of targeting this pathway in EMT breast
cancer (88–90). Pharmacological inhibition of p38
was also associated with decreased clonogenic survival in MCF-7TN-R
cells in vitro and a statistically significant reduction in
tumor volume, as compared to vehicle mice, thus indicating that
RWJ67657 MAPK inhibition can biologically reverse the aggressive
properties seen in MCF-7TN-R cells, in addition to the molecular
reversals described above.
Taken together, our findings suggest that p38
MAPK-microRNA signaling is required for the maintenance of an
aggressive phenotype in MCF-7TN-R cells, stimulating increased
clonogenic survival and tumor growth, along with EMT indicators.
Specifically, p38 downstream modulation of EMT-regulator Twist and
microRNAs in the p38 pathway appears to play an important role in
this process. Further research is warranted to determine the exact
mechanism by which these effectors interact. Finally, disruption of
the p38 MAPK signaling pathway by the pharmacological inhibitor
RWJ67657 reverses this aggressive phenotype, implicating the
signaling pathway as a therapeutic target for aggressive breast
cancers.
Acknowledgements
This study was supported by NIH grants
CA085289 and CA113001. We thank Dr C. Balch for helpful
discussion.
References
|
1
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Seidman AD: Chemotherapy for advanced
breast cancer: a current perspective. Semin Oncol. 23:55–59.
1996.PubMed/NCBI
|
|
3
|
Hsu H, Xiong J and Goeddel DV: The TNF
receptor 1-associated protein TRADD signals cell death and NF-kappa
B activation. Cell. 81:495–504. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herrnring C, Reimer T, Jeschke U, et al:
Expression of the apoptosis-inducing ligands FasL and TRAIL in
malignant and benign human breast tumors. Histochem Cell Biol.
113:189–194. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zyad A, Benard J, Tursz T, Clarke R and
Chouaib S: Resistance to TNF-alpha and adriamycin in the human
breast cancer MCF-7 cell line: relationship to MDR1, MnSOD, and TNF
gene expression. Cancer Res. 54:825–831. 1994.PubMed/NCBI
|
|
6
|
Zyad A, Branellec D, Mahe Y, Tursz T and
Chouaib S: The development of human tumor-cell resistance to
TNF-alpha does not confer resistance to cytokine-induced cellular
cytotoxic mechanisms. Int J Cancer. 52:953–958. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gatti L and Zunino F: Overview of tumor
cell chemoresistance mechanisms. Methods Mol Med. 111:127–148.
2005.PubMed/NCBI
|
|
8
|
Gottesman MM: Mechanisms of cancer drug
resistance. Annu Rev Med. 53:615–627. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huber MA, Azoitei N, Baumann B, et al:
NF-kappaB is essential for epithelial-mesenchymal transition and
metastasis in a model of breast cancer progression. J Clin Invest.
114:569–581. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Radisky DC and Bissell MJ: NF-kappaB links
oestrogen receptor signalling and EMT. Nat Cell Biol. 9:361–363.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park S, Song J, Joe CO and Shin I: Akt
stabilizes estrogen receptor alpha with the concomitant reduction
in its transcriptional activity. Cell Signal. 20:1368–1374. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frigo DE, Vigh KA, Struckhoff AP, et al:
Xenobiotic-induced TNF-alpha expression and apoptosis through the
p38 MAPK signaling pathway. Toxicol Lett. 155:227–238. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thomas RS, Sarwar N, Phoenix F, Coombes RC
and Ali S: Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is
important for estrogen receptor-alpha activity. J Mol Endocrinol.
40:173–184. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Santen RJ, Song RX, McPherson R, et al:
The role of mitogen-activated protein (MAP) kinase in breast
cancer. J Steroid Biochem Mol Biol. 80:239–256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ballif BA and Blenis J: Molecular
mechanisms mediating mammalian mitogen-activated protein kinase
(MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ.
12:397–408. 2001.PubMed/NCBI
|
|
16
|
Geh E, Meng Q, Mongan M, et al:
Mitogen-activated protein kinase kinase kinase 1 (MAP3K1)
integrates developmental signals for eyelid closure. Proc Natl Acad
Sci USA. 108:17349–17354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
18
|
Guo YL, Kang B, Han J and Williamson JR:
p38beta MAP kinase protects rat mesangial cells from
TNF-alpha-induced apoptosis. J Cell Biochem. 82:556–565. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bulavin DV, Saito S, Hollander MC, et al:
Phosphorylation of human p53 by p38 kinase coordinates N-terminal
phosphorylation and apoptosis in response to UV radiation. EMBO J.
18:6845–6854. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Driggers PH, Segars JH and Rubino DM: The
proto- oncoprotein Brx activates estrogen receptor beta by a p38
mitogen-activated protein kinase pathway. J Biol Chem.
276:46792–46797. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Madrid LV, Mayo MW, Reuther JY and Baldwin
AS Jr: Akt stimulates the transactivation potential of the RelA/p65
subunit of NF-kappa B through utilization of the Ikappa B kinase
and activation of the mitogen-activated protein kinase p38. J Biol
Chem. 276:18934–18940. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zimmermann J, Lamerant N, Grossenbacher R
and Furst P: Proteasome- and p38-dependent regulation of ERK3
expression. J Biol Chem. 276:10759–10766. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Waas WF, Lo HH and Dalby KN: The kinetic
mechanism of the dual phosphorylation of the ATF2 transcription
factor by p38 mitogen-activated protein (MAP) kinase alpha.
Implications for signal/response profiles of MAP kinase pathways. J
Biol Chem. 276:5676–5684. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bradham C and McClay DR: p38 MAPK in
development and cancer. Cell Cycle. 5:824–828. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maemura M, Iino Y, Koibuchi Y, Yokoe T and
Morishita Y: Mitogen-activated protein kinase cascade in breast
cancer. Oncology. 57(Suppl 2): 37–44. 1999. View Article : Google Scholar
|
|
26
|
Weldon CB, Burow ME, Rolfe KW, Clayton JL,
Jaffe BM and Beckman BS: NF-kappa B-mediated chemoresistance in
breast cancer cells. Surgery. 130:143–150. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Weldon CB, Parker AP, Patten D, et al:
Sensitization of apoptotically-resistant breast carcinoma cells to
TNF and TRAIL by inhibition of p38 mitogen-activated protein kinase
signaling. Int J Oncol. 24:1473–1480. 2004.
|
|
28
|
Weldon CB, Scandurro AB, Rolfe KW, et al:
Identification of mitogen-activated protein kinase kinase as a
chemoresistant pathway in MCF-7 cells by using gene expression
microarray. Surgery. 132:293–301. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Frigo DE, Tang Y, Beckman BS, et al:
Mechanism of AP-1-mediated gene expression by select
organochlorines through the p38 MAPK pathway. Carcinogenesis.
25:249–261. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Si ML, Zhu S, Wu H, Lu Z, Wu F and Mo YY:
miR-21-mediated tumor growth. Oncogene. 26:2799–2803. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen GQ, Zhao ZW, Zhou HY, Liu YJ and Yang
HJ: Systematic analysis of microRNA involved in resistance of the
MCF-7 human breast cancer cell to doxorubicin. Med Oncol.
27:406–415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Antoon JW, Liu J, Gestaut MM, Burow ME,
Beckman BS and Foroozesh M: Design, synthesis, and biological
activity of a family of novel ceramide analogues in chemoresistant
breast cancer cells. J Med Chem. 52:5748–5752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Struckhoff AP, Bittman R, Burow ME, et al:
Novel ceramide analogs as potential chemotherapeutic agents in
breast cancer. J Pharmacol Exp Ther. 309:523–532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou C, Nitschke AM, Xiong W, et al:
Proteomic analysis of tumor necrosis factor-alpha resistant human
breast cancer cells reveals a MEK5/Erk5-mediated
epithelial-mesenchymal transition phenotype. Breast Cancer Res.
10:R105:2008.
|
|
37
|
Antoon JW, Lai R, Struckhoff AP, et al:
Altered death receptor signaling promotes epithelial-to-mesenchymal
transition and acquired chemoresistance. Sci Rep. 2:5392012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Antoon JW, White MD, Burow ME and Beckman
BS: Dual inhibition of sphingosine kinase isoforms ablates
TNF-induced drug resistance. Oncol Rep. 27:1779–1786.
2012.PubMed/NCBI
|
|
39
|
Antoon JW, White MD, Slaughter EM, et al:
Targeting NFκB mediated breast cancer chemoresistance through
selective inhibition of sphingosine kinase-2. Cancer Biol Ther.
11:678–689. 2011.
|
|
40
|
Wadsworth SA, Cavender DE, Beers SA, et
al: RWJ 67657, a potent, orally active inhibitor of p38
mitogen-activated protein kinase. J Pharmacol Exp Ther.
291:680–687. 1999.PubMed/NCBI
|
|
41
|
Antoon JW, Bratton MR, Guillot LM, et al:
Pharmacology and anti-tumor activity of RWJ67657, a novel inhibitor
of p38 mitogen activated protein kinase. Am J Cancer Res.
2:446–458. 2012.PubMed/NCBI
|
|
42
|
Antoon JW, Bratton MR, Guillot LM,
Wadsworth S, Salvo VA and Burow ME: Inhibition of p38-MAPK alters
SRC coactivation and estrogen receptor phosphorylation. Cancer Biol
Ther. 13:1026–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Antoon JW, Liu J, Ponnapakkam AP, Gestaut
MM, Foroozesh M and Beckman BS: Novel D: -erythro N-octanoyl
sphingosine analogs as chemo- and endocrine-resistant breast cancer
therapeutics. Cancer Chemother Pharmacol. 65:1191–1195. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Antoon JW and Beckman BS:
Anti-proliferative effects of the novel ceramide analog
(S)-2-(benzylideneamino)-3-hydroxy-N-tetrade-cylpropanamide in
chemoresistant cancer. Bioorg Med Chem Lett. 22:2624–2628. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rhodes LV, Nitschke AM, Segar HC, et al:
The histone deacetylase inhibitor trichostatin A alters microRNA
expression profiles in apoptosis-resistant breast cancer cells.
Oncol Rep. 27:10–16. 2012.
|
|
46
|
Antoon JW, White MD, Driver JL, Burow ME
and Beckman BS: Sphingosine kinase isoforms as a therapeutic target
in endocrine therapy resistant luminal and basal-A breast cancer.
Exp Biol Med (Maywood). 237:832–844. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schmittgen TD, Zakrajsek BA, Mills AG,
Gorn V, Singer MJ and Reed MW: Quantitative reverse
transcription-polymerase chain reaction to study mRNA decay:
comparison of endpoint and real-time methods. Anal Biochem.
285:194–204. 2000. View Article : Google Scholar
|
|
48
|
Antoon JW, Meacham WD, Bratton MR, et al:
Pharmacological inhibition of sphingosine kinase isoforms alters
estrogen receptor signaling in human breast cancer. J Mol
Endocrinol. 46:205–216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bratton MR, Antoon JW, Duong BN, et al:
Gαo potentiates estrogen receptor α activity via the ERK signaling
pathway. J Endocrinol. 214:45–54. 2012.
|
|
50
|
Collins-Burow BM, Antoon JW, Frigo DE, et
al: Antiestrogenic activity of flavonoid phytochemicals mediated
via the c-Jun N-terminal protein kinase pathway. Cell-type specific
regulation of estrogen receptor alpha. Journal Steroid Biochem Mol
Biol. 132:186–193. 2012. View Article : Google Scholar
|
|
51
|
Antoon JW, White MD, Meacham WD, et al:
Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor
ABC294640. Endocrinology. 151:5124–5135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Payton-Stewart F, Schoene NW, Kim YS, et
al: Molecular effects of soy phytoalexin glyceollins in human
prostate cancer cells LNCaP. Mol Carcinog. 48:862–871. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Rhodes LV, Antoon JW, Muir SE, Elliott S,
Beckman BS and Burow ME: Effects of human mesenchymal stem cells on
ER-positive human breast carcinoma cells mediated through
ER-SDF-1/CXCR4 crosstalk. Mol Cancer. 9:2952010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rhodes LV, Short SP, Neel NF, et al:
Cytokine receptor CXCR4 mediates estrogen-independent
tumorigenesis, metastasis, and resistance to endocrine therapy in
human breast cancer. Cancer Res. 71:603–613. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Walker CH, Drew BA, Antoon JW, Kalueff AV
and Beckman BS: Neurocognitive effects of chemotherapy and
endocrine therapies in the treatment of breast cancer: recent
perspectives. Cancer Invest. 30:135–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Clarke R, Thompson EW, Leonessa F, et al:
Hormone resistance, invasiveness, and metastatic potential in
breast cancer. Breast Cancer Res Treat. 24:227–239. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nakanishi H, Taylor RM, Chrest FJ, et al:
Progression of hormone-dependent adenocarcinoma cells to
hormone-independent spindle carcinoma cells in vitro in a clonal
spontaneous rat mammary tumor cell line. Cancer Res. 55:399–407.
1995.
|
|
58
|
Murphy LC and Dotzlaw H: Variant estrogen
receptor mRNA species detected in human breast cancer biopsy
samples. Mol Endocrinol. 3:687–693. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rhodes LV, Muir SE, Elliott S, et al:
Adult human mesenchymal stem cells enhance breast tumorigenesis and
promote hormone independence. Breast Cancer Res Treat. 121:293–300.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Di Leva G, Gasparini P, Piovan C, et al:
MicroRNA cluster 221–222 and estrogen receptor alpha interactions
in breast cancer. J Natl Cancer Inst. 102:706–721. 2010.
|
|
61
|
Sonkoly E, Lovén J, Xu N, et al:
MicroRNA-203 functions as a tumor suppressor in basal cell
carcinoma. Oncogenesis. 1:e32012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Burk U, Schubert J, Wellner U, et al: A
reciprocal repression between ZEB1 and members of the miR-200
family promotes EMT and invasion in cancer cells. EMBO Rep.
9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
le Sage C, Nagel R, Egan DA, et al:
Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222
promotes cancer cell proliferation. EMBO J. 26:3699–3708.
2007.PubMed/NCBI
|
|
64
|
Massarweh S, Osborne CK, Creighton CJ, et
al: Tamoxifen resistance in breast tumors is driven by growth
factor receptor signaling with repression of classic estrogen
receptor genomic function. Cancer Res. 68:826–833. 2008. View Article : Google Scholar
|
|
65
|
Massarweh S and Schiff R: Unraveling the
mechanisms of endocrine resistance in breast cancer: new
therapeutic opportunities. Clin Cancer Res. 13:1950–1954. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Obrero M, Yu DV and Shapiro DJ: Estrogen
receptor-dependent and estrogen receptor-independent pathways for
tamoxifen and 4-hydroxytamoxifen-induced programmed cell death. J
Biol Chem. 277:45695–45703. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
St-Laurent V, Sanchez M, Charbonneau C and
Tremblay A: Selective hormone-dependent repression of estrogen
receptor beta by a p38-activated ErbB2/ErbB3 pathway. J Steroid
Biochem Mol Biol. 94:23–37. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Cannell IG, Kong YW, Johnston SJ, et al:
p38 MAPK/MK2-mediated induction of miR-34c following DNA damage
prevents Myc-dependent DNA replication. Proc Natl Acad Sci USA.
107:5375–5380. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yang F, Yin Y, Wang F, et al: miR-17-5p
promotes migration of human hepatocellular carcinoma cells through
the p38 mitogen-activated protein kinase-heat shock protein 27
pathway. Hepatology. 51:1614–1623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rajaram MV, Ni B, Morris JD, et al:
Mycobacterium tuberculosis lipomannan blocks TNF biosynthesis by
regulating macrophage MAPK-activated protein kinase 2 (MK2) and
microRNA miR-125b. Proc Natl Acad Sci USA. 108:17408–17413. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Akhtar N, Rasheed Z, Ramamurthy S,
Anbazhagan AN, Voss FR and Haqqi TM: MicroRNA-27b regulates the
expression of matrix metalloproteinase 13 in human osteoarthritis
chondrocytes. Arthritis Rheum. 62:1361–1371. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ben-Hamo R and Efroni S: Gene expression
and network-based analysis reveals a novel role for hsa-miR-9 and
drug control over the p38 network in glioblastoma multiforme
progression. Genome Med. 3:772011. View
Article : Google Scholar
|
|
73
|
Zaman MS, Shahryari V, Deng G, et al:
Up-regulation of microRNA-21 correlates with lower kidney cancer
survival. PloS One. 7:e310602012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Alam J, Wicks C, Stewart D, et al:
Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7
mammary epithelial cells. Role of p38 kinase and Nrf2 transcription
factor. J Biol Chem. 275:27694–27702. 2000.PubMed/NCBI
|
|
75
|
Liu Z, Zhu J, Cao H, Ren H and Fang X:
miR-10b promotes cell invasion through RhoC-AKT signaling pathway
by targeting HOXD10 in gastric cancer. Int J Oncol. 40:1553–1560.
2012.PubMed/NCBI
|
|
76
|
Li G, Wu Z, Peng Y, et al: MicroRNA-10b
induced by Epstein-Barr virus-encoded latent membrane protein-1
promotes the metastasis of human nasopharyngeal carcinoma cells.
Cancer Lett. 299:29–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ng YH, Zhu H and Leung PC: Twist modulates
human trophoblastic cell invasion via regulation of N-cadherin.
Endocrinology. 153:925–936. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Alexander NR, Tran NL, Rekapally H,
Summers CE, Glackin C and Heimark RL: N-cadherin gene expression in
prostate carcinoma is modulated by integrin-dependent nuclear
translocation of Twist1. Cancer Res. 66:3365–3369. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kovalchuk O, Filkowski J, Meservy J, et
al: Involvement of microRNA-451 in resistance of the MCF-7 breast
cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther.
7:2152–2159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yang H, Kong W, He L, et al: MicroRNA
expression profiling in human ovarian cancer: miR-214 induces cell
survival and cisplatin resistance by targeting PTEN. Cancer Res.
68:425–433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ma J, Dong C and Ji C: MicroRNA and drug
resistance. Cancer Gene Ther. 17:523–531. 2010. View Article : Google Scholar
|
|
82
|
Xin F, Li M, Balch C, et al: Computational
analysis of microRNA profiles and their target genes suggests
significant involvement in breast cancer antiestrogen resistance.
Bioinformatics. 25:430–434. 2009. View Article : Google Scholar
|
|
83
|
Burow ME, Weldon CB, Melnik LI, et al:
PI3-K/AKT regulation of NF-kappaB signaling events in suppression
of TNF-induced apoptosis. Biochem Biophys Res Commun. 271:342–345.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Frigo DE, Basu A, Nierth-Simpson EN, et
al: p38 mitogen-activated protein kinase stimulates
estrogen-mediated transcription and proliferation through the
phosphorylation and potentiation of the p160 coactivator
glucocorticoid receptor-interacting protein 1. Mol Endocrinol.
20:971–983. 2006. View Article : Google Scholar
|
|
85
|
Bourguignon LY, Wong G, Earle C, Krueger K
and Spevak CC: Hyaluronan-CD44 interaction promotes c-Src-mediated
twist signaling, microRNA-10b expression, and RhoA/RhoC
up-regulation, leading to Rho-kinase-associated cytoskeleton
activation and breast tumor cell invasion. J Biol Chem.
285:36721–36735. 2010. View Article : Google Scholar
|
|
86
|
Croce CM: Causes and consequences of
microRNA dysregulation in cancer. Nat Rev Genet. 10:704–714. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhu S, Wu H, Wu F, Nie D, Sheng S and Mo
YY: MicroRNA-21 targets tumor suppressor genes in invasion and
metastasis. Cell Res. 18:350–359. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Grund EM, Kagan D, Tran CA, et al: Tumor
necrosis factor-alpha regulates inf lammatory and mesenchymal
responses via mitogen-activated protein kinase kinase, p38, and
nuclear factor kappaB in human endometriotic epithelial cells. Mol
Pharmacol. 73:1394–1404. 2008. View Article : Google Scholar
|
|
89
|
Kolosova I, Nethery D and Kern JA: Role of
Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal
transition of pulmonary epithelial cells. J Cell Physiol.
226:1248–1254. 2011.
|
|
90
|
Strippoli R, Benedicto I, Foronda M, et
al: p38 maintains E-cadherin expression by modulating TAK1-NF-kappa
B during epithelial-to-mesenchymal transition. J Cell Sci.
123:4321–4331. 2010. View Article : Google Scholar : PubMed/NCBI
|